
Abstract
Background
Overexpression/activation of focal adhesion kinase (FAK) in human malignancies has led to its evaluation as a therapeutic target. We report the first-in-human phase I study of BI 853520, a novel, potent, highly selective FAK inhibitor.
Objective
Our objectives were to identify the maximum tolerated dose (MTD), and to evaluate safety, pharmacokinetics (PK), pharmacodynamics (PD), biomarker expression, and preliminary activity.
Patients and Methods
The study comprised a standard 3 + 3 dose-escalation phase followed by an expansion phase in patients with selected advanced, nonhematologic malignancies.
Results
Thirty-three patients received BI 853520 in the dose-escalation phase; the MTD was 200 mg once daily (QD). Dose-limiting toxicities included proteinuria and fatigue, both of which were grade 3. Preliminary PK data supported QD dosing. In the expansion cohort, 63 patients received BI 853520 200 mg QD. Drug-related adverse events (AEs) in > 10% of patients included proteinuria (57%), nausea (57%), fatigue (51%), diarrhea (48%), vomiting (40%), decreased appetite (19%), and peripheral edema (16%). Most AEs were grade 1–2; grade 3 proteinuria, reported in 13 patients (21%), was generally reversible upon treatment interruption. Nineteen patients underwent dose reduction due to AEs, and three drug-related serious AEs were reported, none of which were fatal. Preliminary PD analysis indicated target engagement. Of 63 patients, 49 were evaluable; 17 (27%) achieved a best response of stable disease (4 with 150 + days), and 32 (51%) patients had progressive disease.
Conclusions
BI 853520 has a manageable and acceptable safety profile, favorable PK, and modest antitumor activity at an MTD of 200 mg QD in patients with selected advanced nonhematologic malignancies.
ClinicalTrials.gov identifier
NCT01335269.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This was the first-in-human study of BI 853520 in patients with nonhematologic malignancies. |
A maximum tolerated dose of 200 mg once daily was identified; dose-limiting toxicities included grade 3 proteinuria and fatigue. |
Preliminary pharmacokinetic findings support once-daily dosing, which was associated with a manageable safety profile and some evidence of antitumor activity. |
1 Introduction
Cellular interactions with the extracellular matrix play a pivotal role in tumor initiation, progression, and metastasis [1]. Focal adhesion kinase [FAK; also known as protein tyrosine kinase 2 (PTK2)] is a widely expressed, nonreceptor, cytoplasmic tyrosine kinase [1,2,3] that is a key component of the focal adhesion complex, which plays an important role in integrating signals from integrins and growth factor receptors in both normal and cancer cells [1, 4], among other functions. A role for FAK in cancer development is suggested by its amplification, overexpression, and/or activation in a variety of human malignancies [1, 3, 5], including breast [6, 7], colorectal [7, 8], ovarian [9], prostate [10], and sarcoma [7, 11]. Research suggests that FAK signaling is involved in cellular proliferation, migration, angiogenesis, invasion, survival, and epithelial-mesenchymal transition, via a variety of different pathways [4]. In addition, FAK appears to play a role in tumor metabolism, and can promote glucose consumption, lipogenesis, and glutamine dependency to promote cancer cell proliferation, motility and survival [12]. FAK has been shown to be a central driver of the fibrotic and immunosuppressive tumor microenvironment that protects pancreatic ductal adenocarcinomas from immune surveillance [13]. Furthermore, through its effects on chemokine transcription in the cell nucleus, FAK can promote antitumor immune evasion [14].
Given its putative role in tumor development and progression, FAK has become a logical therapeutic target, and numerous attempts have been made to inhibit the oncogenic activity of FAK [4]. Preclinical studies have demonstrated antitumor activity with FAK blockade [15,16,17,18,19,20], and several orally bioavailable, adenosine triphosphate (ATP)-competitive, small-molecule inhibitors of FAK are undergoing evaluation in early-phase clinical trials in patients with cancer, including VS-6063, GSK2256098, and PF-00562271 [21,22,23,24].
BI 853520 is a novel, potent, highly selective, ATP-competitive inhibitor of FAK that has demonstrated activity in a variety of preclinical human tumor xenograft models [25]. In vitro, BI 853520 inhibited recombinant FAK with a half maximal (50%) inhibitory concentration (IC50) of 1 nM [26], which is largely comparable to the IC50 values reported for VS-6063 (0.6 nM) [23], PF-00562271 (1.5 nM) [18], and GSK2256098 (2–15 nM) [27]. BI 853520 has also demonstrated high selectivity; IC50 for the related kinase proline-rich tyrosine kinase 2, PYK2, was > 50,000 nmol/L [26]. The antitumor activity of BI 853520 was found to vary widely across a diverse panel of 16 murine subcutaneous adenocarcinoma xenograft models, from complete tumor inhibition to an absolute lack of sensitivity [26]. Biomarker analysis suggests that the in vivo efficacy of BI 853520 in these models is linked to a mesenchymal tumor phenotype characterized by low E-cadherin messenger RNA (mRNA) and protein levels, and by low expression of the microRNA hsa-miR-200c-3p, an epithelial-specific microRNA that promotes E-cadherin expression [26]. All xenograft models that were highly sensitive to BI 853520, including kidney, lung, ovary, pancreas and prostate adenocarcinomas, were found to lack E-cadherin expression or to express low levels of E-cadherin [26]. Conversely, of five BI 853520-resistant models, three were E-cadherin positive. In murine breast cancer models, BI 853520 most effectively prevented the establishment of metastases in tumors in which E-cadherin was either deficient or downregulated [28].
We report here the first-in-human phase I study of BI 853520, which comprised a dose-escalation phase followed by an expansion phase in patients with advanced nonhematologic cancers.
2 Methods
2.1 Patients
Patients aged ≥ 18 years with a confirmed diagnosis of advanced, measurable or evaluable, nonresectable and/or metastatic nonhematologic malignancy that was progressive within 6 months prior to study entry, as demonstrated by serial imaging, and refractory to standard therapy, or for which no effective standard treatment was available, were eligible for enrollment. Recruitment to the expansion phase was restricted to patients with metastatic pancreatic adenocarcinoma, metastatic esophageal carcinoma, metastatic soft tissue sarcoma, or metastatic platinum-resistant ovarian carcinoma. This selection was based on preclinical efficacy and prevalence data on E-cadherin loss in these tumor types. The complete inclusion/exclusion criteria are listed in the electronic supplementary material (ESM).
The trial was approved by the Institutional Review Board at each participating institution, and conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and applicable regulatory requirements (including International Conference on Harmonization guidelines). The Centrale Commissie Mensgebonden Onderzoek, Den Haag, The Netherlands approved the trial, and all patients provided written informed consent.
2.2 Study Design and Treatment
This open-label, phase I trial was conducted at five sites in The Netherlands and Canada between July 2011 and December 2015 (NCT01335269). The primary objective was to evaluate the safety and tolerability of BI 853520, determining the maximum tolerated dose (MTD), and identifying a recommended dose for further clinical development. Secondary objectives included pharmacokinetics (PK), pharmacodynamics (PD), assessment of predictive biomarkers, and efficacy.
Patients received oral BI 853520 in a continuous once daily (QD) dosing schedule in 28-day cycles. In the dose-escalation phase, sequential cohorts of three to six patients received escalating doses of BI 853520 (starting dose of 10 mg) in a standard 3 + 3 design. MTD was defined based on the occurrence of dose-limiting toxicities (DLTs) in the dose-escalation phase during the first cycle only. The MTD was defined as the dose of BI 853520 that was one dose level below the dose at which two or more of six patients experienced a DLT. Following determination of the MTD, additional patients were enrolled into four expansion cohorts with selected tumor types. Criteria for DLTs, dose reductions, and discontinuations are listed in the ESM.
2.3 Safety
The primary endpoint was the MTD of BI 853520. Safety was assessed by monitoring adverse events (AEs; National Cancer Institute Common Terminology Criteria for Adverse Events [CTCAE] version 4.03 [29]) and clinical laboratory parameters. Patients were included in the safety analysis if they had taken at least one dose of trial medication, and were evaluable for DLT if they had been observed for one treatment cycle and had undergone all safety assessments.
2.4 Pharmacokinetics
The PK profile of BI 853520 in plasma and urine was determined in all eligible patients (both the escalation and expansion phase) after a single oral dose and after repeat dosing (steady state). Full details of PK methods are listed in the ESM.
2.5 Pharmacodynamics and Analysis of E-Cadherin Loss as a Potential Predictive Biomarker
Levels of phosphorylated and total FAK in tumor biopsies were determined by enzyme-linked immunosorbent assay (ELISA), and E-cadherin expression was assessed by immunohistochemistry. Full details of the PD methods are listed in the ESM.
2.6 Efficacy
Patients were evaluable if they had at least one tumor assessment after baseline; the first was planned after 8 weeks of treatment, at the end of cycle 2. The following efficacy endpoints were evaluated: investigator-assessed objective response rate and disease control rate [complete response, partial response, or stable disease (SD)] according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 [30], duration of disease control, and tumor shrinkage. Tumors were assessed by computed tomography/magnetic resonance imaging (CT/MRI) scans at screening (baseline), and at the end of every two cycles until treatment discontinuation.
2.7 Statistical Analyses
All analyses in this trial were exploratory and data were analyzed using descriptive statistics. The overall analysis of safety and efficacy was based on patients who had received at least one dose of BI 853520 (treated set).
3 Results
3.1 Dose-Escalation Phase
A total of 41 patients provided informed consent and 33 patients were treated with BI 853520 in the dose-escalation phase. Among the treated patients, 58% were female, median age was 60 years (range 33–82), and 9%/91% had an Eastern Cooperative Oncology Group performance status (ECOG PS) score of 0/1. Patients had various advanced solid tumors, including colorectal cancer (n = 10, 30%) and soft tissue or bone sarcoma (n = 4, 12%). Full baseline demographics for the dose-escalation phase are listed in Electronic Supplementary Table 1.
Patients were treated at the following dose levels: 10 mg QD (n = 3), 25 mg QD (n = 3), 50 mg QD (n = 3), 100 mg QD (n = 6), 200 mg QD (n = 11), and 300 mg QD (n = 7). Median duration of treatment among patients in the dose-escalation phase was 35 days (range 6–281). Twenty-five patients were evaluable for determination of the MTD.
Three patients experienced DLT during cycle 1: two of four evaluable patients in the 300-mg QD cohort (grade 3 proteinuria and grade 3 renal disorder; grade 3 fatigue), and one of nine evaluable patients in the 200-mg QD cohort (grade 3 proteinuria). The MTD of BI 853520 was therefore determined to be 200 mg QD.
Thirty-two (97%) patients experienced drug-related AEs, the most frequent of which were nausea (64%), fatigue (46%), diarrhea (36%), vomiting (36%), and decreased appetite (21%). Drug-related AEs were all CTCAE grade 1 or 2, with the exception of the DLTs mentioned above. Except for proteinuria (15% grade 1–2, 6% grade 3), no notable findings for laboratory assessments and vital signs were observed.
Response data in the escalation phase are described in Table 1.
3.2 Expansion Phase
3.2.1 Patients
Sixty-three patients were included in the expansion phase. Baseline characteristics by tumor type are summarized in Table 2. All eligible patients had a comprehensive history of previous anticancer therapies; 23 (37%) patients had received three or more prior systemic therapies.
3.2.2 Treatment Exposure
All 63 patients received at least one dose of BI 853520 200 mg QD. At the time of data cut-off (29 October 2015), 61 patients had discontinued treatment due to progressive disease (n = 47; 75%), refusal to continue with the medication (n = 8; 13%), AEs (n = 5; 8%), or failure to comply with the study protocol (n = 1; 2%), and two patients remained on treatment. These two patients both discontinued within 6 weeks of database lock due to progressive disease. The maximum treatment duration for one of these patients, both of whom had pancreatic cancer, was 251 days. Among all 63 patients, the median number of cycles received was two (range 1–8), and the mean dose intensity was 83% (Table 3). Seven (11%) patients had received more than four cycles of treatment.
3.2.3 Safety
Drug-related AEs were reported in 61 (97%) patients, most commonly nausea, proteinuria, fatigue, diarrhea, and vomiting (Table 4). The majority of drug-related AEs were grade 1 or 2. Grade 3 drug-related AEs were reported in 22 (35%) patients, and included grade 3 proteinuria in 13 (21%) patients (Table 4). In all but one case, grade 3 proteinuria improved to grade 1 upon treatment interruption, and treatment could be reinitiated at a reduced dose of 100 mg. In the other case, proteinuria was reduced to grade 2. Two patients required a second dose reduction due to the recurrence of grade 3 proteinuria, and two patients underwent a kidney biopsy to further evaluate the proteinuria. Briefly, dysjunction of podocytes from the glomerular basement membrane and moderate-to-marked podocyte effacement were observed (details are provided in the ESM). No drug-related grade 4 or 5 events were observed.
With regard to the laboratory assessments, no notable findings were observed, except for proteinuria (drug-related in 57% of patients). Serious drug-related AEs were reported in three (5%) patients [nausea (n = 2), vomiting (n = 3), and decreased appetite (n = 1)].
Nineteen (30%) patients underwent dose reduction due to AEs. Four (6%) patients had drug-related AEs leading to permanent discontinuation: grade 2 depressed mood (n = 1), grade 3 fatigue (n = 1), grade 2 vomiting (n = 1), and grade 2 abdominal pain combined with grade 1 diarrhea (n = 1).
3.2.4 Pharmacokinetics
The PK profile of BI 853520 was determined in 95 patients who received single and repeated oral doses of 10–300 mg. BI 853520 was rapidly absorbed, followed by at least bi-exponential disposition; maximum plasma concentrations (Cmax values) were observed at a median of 2 h (Fig. 1). Cmax and exposure (area under the curve values) increased with increasing doses. Dose-normalized geometric mean PK parameters suggested a trend toward an overproportional increase of exposure with dose; however, interpatient variability was moderate to high (Fig. 2). For unbound BI 853520, a low-to-moderate interpatient variability of exposure PK parameters was detected over all dose groups. Table 5 summarizes the PK parameters for BI 853520, and unbound BI 853520, after single- and multiple-dose administration (200 mg QD; see Electronic Supplementary Tables 2–5 for all dose groups).

Plasma concentration–time profiles for BI 853520 after single- and multiple-dose administration in the first cycle. Pharmacokinetic profiles after single- and multiple-dose administration were assessed during the first treatment cycle, i.e. after the first dose on day 1 and after repeated dosing on day 28

Individual (circle) and geometric mean (cross) PK parameters for BI 853520 after single- and multiple-dose administration in the first cycle. PK profiles after single- and multiple-dose administration were assessed during the first treatment cycle, i.e. after the first dose on day 1 and after repeated dosing on day 28. AUC area under the plasma concentration–time curve, Cmax maximum plasma concentration, gMean geometric mean, PK pharmacokinetic
3.2.5 Pharmacodynamics
Paired tumor biopsies for analysis of FAK target engagement were available in 21 patients, however only eight of these patients had a complete set of data that allowed calculation of phosphorylated FAK (pFAK) and total FAK levels, both at baseline and during treatment. The lack of a complete data set for the remaining 13 patients was primarily due to FAK and pFAK levels being below the limits of quantification, partly due to limited amounts of tissue being available for both timepoints.
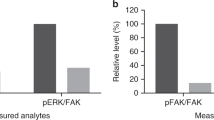
In five of the eight cases for whom complete sets of data were available [soft tissue sarcoma (n = 3), metastatic adenocarcinoma of the pancreas (n = 1), and esophageal cancer (n = 1)], the ratio of pFAK/total FAK was substantially decreased at the end of the first cycle compared with baseline (Fig. 3). There was a non-significant association between durable SD and decrease of the pFAK/total FAK ratio in one patient, while two patients with SD lasting two cycles also had a decrease of the pFAK/total FAK ratio. Given the limited number of patients with complete PD data and the absence of objective responses, it is not possible to draw a sound conclusion regarding correlation between decrease of the pFAK/total FAK ratio and efficacy.

Change from baseline to end of the first cycle in levels of phosphorylated FAK/total FAK in tumor tissue (expansion cohort; 200 mg QD). FAK focal adhesion kinase, QD once daily
3.2.6 Efficacy
Of 63 patients treated in the expansion phase, 17 (27%) achieved a best response of SD [pancreatic adenocarcinoma (n = 6), platinum-resistant ovarian carcinoma (n = 5), soft tissue sarcoma (n = 4), and esophageal carcinoma (n = 2)] and 32 (51%) patients had progressive disease; 14 were not evaluable for response due to the absence of an on-treatment tumor assessment (Fig. 4). No objective tumor responses were observed; thus, the disease control rate was 27%. Median duration of disease control was 99 days (95% confidence interval 87–141 days), and four patients achieved disease control for ≥ 150 days [pancreatic adenocarcinoma (n = 2), platinum-resistant ovarian carcinoma (n = 1), and soft tissue sarcoma (n = 1)].

Confirmed best overall tumor response and treatment duration by tumor type (expansion cohort; 200 mg QD). AE adverse event, QD once daily
Fifty of the 63 patients were evaluable for assessment of tumor shrinkage. Median best percentage change from baseline in the sum of longest diameters of target lesions was 8.97% (range − 25.7 to 168.6%).
3.2.7 Evaluation of E-Cadherin as a Potential Predictive Biomarker
Of the 57 patients for whom E-cadherin expression could be determined, 17 (30%) had low expression [soft tissue sarcoma (n = 13), esophageal carcinoma (n = 2), pancreatic adenocarcinoma (n = 1), and ovarian cancer (n = 1)]. Approximately one-quarter of patients in both the low- and high-expression E-cadherin groups achieved SD, as confirmed best overall tumor response (H-score ≤ 100: 4/17 patients, 23.5%; and H-score > 100: 11/40 patients, 27.5%). Median duration of disease control was also similar in both groups (low expression 103 days; high expression 95 days).
4 Discussion
This phase I trial demonstrated that continuous monotherapy with BI 853520 is feasible in heavily pretreated patients with advanced or metastatic nonhematologic malignancies; the MTD is determined at 200 mg QD.
The tolerability profile for BI 853520 is acceptable and is similar to that observed with other FAK inhibitors [e.g. VS-6063 (defactinib) and GSK2256098] [21, 22, 24]. The most common drug-related AEs included gastrointestinal events (nausea, diarrhea, vomiting), proteinuria, and fatigue, and were predominantly mild to moderate in severity.
No grade 4 or 5 drug-related AEs were reported; drug-related grade 3 AEs occurred in 35% of patients. Thirteen (21%) patients experienced grade 3 proteinuria, which was unexpected as preclinical toxicology studies did not identify any kidney abnormalities [31]. After the first occurrence of grade 3 proteinuria in a patient treated at the 300-mg dose, frequent monitoring of urine protein levels was implemented to facilitate early detection. Importantly, proteinuria was generally improved to grade 1 upon treatment interruption and subsequent dose reduction, suggesting a dose-dependent effect. To date, no predisposing or risk factors for the development of BI 853520-associated proteinuria have been identified, and no clear correlation was identified with prior nephrotoxic therapy. In kidney biopsies from two patients with grade 3 proteinuria, dysjunction of podocytes from the glomerular basement membrane and podocyte effacement was observed. FAK has been localized to the cytoplasm and nuclei of glomerular podocytes, and studies suggest that proteinuria may be related to activation of FAK in the glomerulus [32, 33]. However, the underlying mechanism of proteinuria following FAK inhibition remains unclear and requires further investigation. Proteinuria has also been reported as an AE following treatment with GSK2256098 [24] and PF-00562271 [21].
PK analysis demonstrated that BI 853520 was rapidly absorbed and exhibited at least biphasic disposition kinetics. A trend was observed toward an overproportional increase in mean exposure within the entire dose range investigated; however, interpatient variability of PK parameters was moderate to high. While this variability cannot yet be fully explained, it was within the expected range for an orally administered anticancer drug in the first dose-finding study. The PK data demonstrate oral bioavailability of BI 853520 and support a QD dosing schedule.
In five of eight cases, including three patients with soft tissue sarcoma, the ratio of active pFAK/total FAK was substantially reduced at the end of the first cycle, thus providing clinical evidence supporting target engagement with BI 853520 in patients treated with the 200 mg QD dose.
Disease control was observed in 27% of patients with a median duration of 99 days, and four patients achieved disease control for ≥ 150 days. No objective tumor responses were observed. In order to identify patients most likely to derive benefit from FAK inhibitors, we undertook an exploratory analysis of E-cadherin expression as a potential predictive biomarker [34]. There were no clear differences in disease control rate with respect to E-cadherin levels. However, it should be noted that the majority of patients with E-cadherin-negative tumors had soft tissue sarcomas, which are of mesenchymal origin and would not be expected to express E-cadherin at all. Based on this study, the potential predictive value of E-cadherin could not be determined, and further studies are warranted to identify other potential biomarkers to guide patient selection for FAK inhibitor treatment.
Given the cytostatic activity of BI 853520, the combination with other compounds such as immunotherapy [13, 19] should be considered for further development. In vitro and clinical studies suggest that combining FAK inhibitors with other biologic or chemotherapeutic agents such as MEK inhibitors, vascular endothelial growth factor (VEGF) inhibitors, immune checkpoint inhibitors, gemcitabine, paclitaxel, or cisplatin may overcome resistance, reduce metastasis and/or improve antitumor efficacy [13, 35,36,37,38,39]. A separate phase I study of BI 853520 has been conducted in 21 Japanese and Taiwanese patients with advanced or metastatic solid tumors [40]. In this study, a similar safety and PK profile was observed, with an equivalent MTD of 200 mg QD. The most common drug-related AEs were proteinuria and gastrointestinal events. One patient with gastric cancer who received BI 853520 100 mg QD achieved a confirmed partial response, with disease control seen in 29% of patients.
5 Conclusion
These data demonstrate that BI 853520 has a manageable and acceptable safety profile, favorable PK, and preliminary antitumor activity, including, for some patients, SD lasting ≥ 150 days, and PD modulation at the MTD of 200 mg QD in patients with selected advanced nonhematologic malignancies. As single-agent activity is modest, future development should focus on BI 853520 in combination with other agents.
References
Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28(1–2):35–49.
Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14(9):598–610.
Tai YL, Chen LC, Shen TL. Emerging roles of focal adhesion kinase in cancer. Biomed Res Int. 2015;2015:690690.
Lee BY, Timpson P, Horvath LG, Daly RJ. FAK signaling in human cancer as a target for therapeutics. Pharmacol Ther. 2015;146:132–49.
Kornberg LJ. Focal adhesion kinase and its potential involvement in tumor invasion and metastasis. Head Neck. 1998;20(8):745–52.
Oktay MH, Oktay K, Hamele-Bena D, Buyuk A, Koss LG. Focal adhesion kinase as a marker of malignant phenotype in breast and cervical carcinomas. Hum Pathol. 2003;34(3):240–5.
Owens LV, Xu L, Craven RJ, Dent GA, Weiner TM, Kornberg L, et al. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995;55(13):2752–5.
Lark AL, Livasy CA, Calvo B, Caskey L, Moore DT, Yang X, et al. Overexpression of focal adhesion kinase in primary colorectal carcinomas and colorectal liver metastases: immunohistochemistry and real-time PCR analyses. Clin Cancer Res. 2003;9(1):215–22.
Judson PL, He X, Cance WG, Van LL. Overexpression of focal adhesion kinase, a protein tyrosine kinase, in ovarian carcinoma. Cancer. 1999;86(8):1551–6.
Tremblay L, Hauck W, Aprikian AG, Begin LR, Chapdelaine A, Chevalier S. Focal adhesion kinase (pp125FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int J Cancer. 1996;68(2):164–71.
Weiner TM, Liu ET, Craven RJ, Cance WG. Expression of focal adhesion kinase gene and invasive cancer. Lancet. 1993;342(8878):1024–5.
Zhang J, Hochwald SN. The role of FAK in tumor metabolism and therapy. Pharmacol Ther. 2014;142(2):154–63.
Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–60.
Serrels A, Lund T, Serrels B, Byron A, McPherson RC, von Kriegsheim A, et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell. 2015;163(1):160–73.
Bagi CM, Roberts GW, Andresen CJ. Dual focal adhesion kinase/Pyk2 inhibitor has positive effects on bone tumors: implications for bone metastases. Cancer. 2008;112(10):2313–21.
Halder J, Kamat AA, Landen CN Jr, Han LY, Lutgendorf SK, Lin YG, et al. Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy. Clin Cancer Res. 2006;12(16):4916–24.
Parsons JT, Slack-Davis J, Tilghman R, Roberts WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin Cancer Res. 2008;14(3):627–32.
Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68(6):1935–44.
Roy-Luzarraga M, Hodivala-Dilke K. Molecular pathways: endothelial cell FAK-A target for cancer treatment. Clin Cancer Res. 2016;22(15):3718–24.
Shi Q, Hjelmeland AB, Keir ST, Song L, Wickman S, Jackson D, et al. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol Carcinog. 2007;46(6):488–96.
Infante JR, Camidge DR, Mileshkin LR, Chen EX, Hicks RJ, Rischin D, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J Clin Oncol. 2012;30(13):1527–33.
Jones SF, Siu LL, Bendell JC, Cleary JM, Razak AR, Infante JR, et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2015;33(5):1100–7.
Shimizu T, Fukuoka K, Takeda M, Iwasa T, Yoshida T, Horobin J, et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;77(5):997–1003.
Soria JC, Gan HK, Blagden SP, Plummer R, Arkenau HT, Ranson M, et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann Oncol. 2016;27(12):2268–74.
Hirt UA, Braunger J, Schleicher M, Weyer-Czemilofsky U, Garin-Chesa P, Bister B, et al. BI 853520, a potent and highly selective inhibitor of protein tyrosine kinase 2 (focal adhesion kinase), shows efficacy in multiple xenograft models of human cancer [abstract no. A249]. Mol Cancer Ther. 2011;10(11 Suppl):A249.
Hirt UA, Waizenegger IC, Schweifer N, Haslinger C, Gerlach D, Braunger J, et al. Efficacy of the highly selective focal adhesion kinase inhibitor BI 853520 in adenocarcinoma xenograft models is linked to a mesenchymal tumor phenotype. Oncogenesis. 2018;7(2):21.
Auger KR, Smitheman KN, Korenchuk S, McHugh C, Kruger R, Van Aller GS, et al. The focal adhesion kinase inhibitor GSK2256098: a potent and selective inhibitor for the treatment of cancer [abstract no. 387]. Eur J Cancer. 2012;48:118.
Tiede S, Meyer-Schaller N, Kalathur RKR, Ivanek R, Fagiani E, Schmassmann P, et al. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis. 2018;7(9):73.
National Institute of Health. National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03). 2009. https://www.eortc.be/services/doc/ctc/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed 2 Aug 2017.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47.
Boehringer Ingelheim. Data on file. c01566501–04.
Ma H, Togawa A, Soda K, Zhang J, Lee S, Ma M, et al. Inhibition of podocyte FAK protects against proteinuria and foot process effacement. J Am Soc Nephrol. 2010;21(7):1145–56.
Yuan X, Wang W, Wang J, Yin X, Zhai X, Wang L, et al. Down-regulation of integrin beta1 and focal adhesion kinase in renal glomeruli under various hemodynamic conditions. PLoS One. 2014;9(4):e94212.
Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28(1–2):15–33.
Arkenau H-T, Gazzah A, Plummer R, Blagden SP, Mak G, Soria J-C, et al. A phase Ib dose-escalation study of GSK2256098 (FAKi) plus trametinib (MEKi) in patients with selected advanced solid tumors [abstract no. 2593]. J Clin Oncol. 2015;33(15 Suppl):2593.
Moen I, Gebre M, Alonso-Camino V, Chen D, Epstein D, McDonald DM. Anti-metastatic action of FAK inhibitor OXA-11 in combination with VEGFR-2 signaling blockade in pancreatic neuroendocrine tumors. Clin Exp Metastasis. 2015;32(8):799–817.
Patel MR, Infante JR, Moore KN, Keegan M, Poli A, Padval M, et al. Phase 1/1b study of the FAK inhibitor defactinib (VS-6063) in combination with weekly paclitaxel for advanced ovarian cancer [abstract no. 5521]. J Clin Oncol. 2014;32(15 Suppl):5521.
Wang DD, Chen Y, Chen ZB, Yan FJ, Dai XY, Ying MD, et al. CT-707, a novel FAK inhibitor, synergizes with cabozantinib to suppress hepatocellular carcinoma by blocking cabozantinib-induced FAK activation. Mol Cancer Ther. 2016;15(12):2916–25.
Wang-Gillam A, Lockhart AC, Tan BR, Suresh R, Lim K-H, Ratner L, et al. Phase I study of defactinib combined with pembrolizumab and gemcitabine in patients with advanced cancer [abstract no. 2561]. J Clin Oncol. 2018;36(15 Suppl):2561.
Doi T, Yang JC-H, Shitara K, Naito Y, Cheng A-L, Sarashina A, et al. Phase I study of the focal adhesion kinase inhibitor, BI 853520, in Japanese and Taiwanese patients with advanced or metastatic solid tumors. Target Oncol. 2019. https://doi.org/10.1007/s11523-019-00620-0.
Acknowledgements
The authors thank all patients and their families, and investigators and staff at all clinical sites, for their valuable contributions to this study. The authors acknowledge Emma Landers, PhD, of GeoMed, an Ashfield company, part of UDG Healthcare plc, for writing support during the development of this manuscript, which was funded by Boehringer Ingelheim.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This trial was sponsored by Boehringer Ingelheim. This work was supported by Boehringer Ingelheim, Ingelheim am Rhein, Germany. BI 853520 is an asset of Boehringer Ingelheim. This article was published open access under a Springer Compact agreement with Dutch universities and Academy institutes.
Conflict of Interest
Hal W. Hirte reports receiving honoraria from AstraZeneca, Roche and Merck; Albiruni R. Abdul Razak reports paid expert testimony for, and grants from, Boehringer Ingelheim; and Filip Y.F.L. De Vos reports paid expert testimony for Bristol Myers Squibb, and grants from Novartis. Remy B. Verheijen is an employee of AstraZeneca; David Schnell and Linda C. Pronk are employees of Boehringer Ingelheim; and Monique Jansen was an employee of Boehringer Ingelheim at the time of study conduct and manuscript preparation. Lillian L. Siu reports institutional clinical trial funding for this study provided by Boehringer Ingelheim. Maja J.A. de Jonge, Neeltje Steeghs, Martijn P. Lolkema, Sebastien J. Hotte, and Diane A.J. van der Biessen declare that they have no conflicts of interest.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
de Jonge, M.J.A., Steeghs, N., Lolkema, M.P. et al. Phase I Study of BI 853520, an Inhibitor of Focal Adhesion Kinase, in Patients with Advanced or Metastatic Nonhematologic Malignancies. Targ Oncol 14, 43–55 (2019). https://doi.org/10.1007/s11523-018-00617-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-018-00617-1