
Abstract
The internal transcribed spacer (ITS) region of the ribosomal DNA (rDNA) has been established (and is generally accepted) as a primary “universal” genetic barcode for fungi for many years, but the actual value for taxonomy has been heavily disputed among mycologists. Recently, twelve draft genome sequences, mainly derived from type species of the family Hypoxylaceae (Xylariales, Ascomycota) and the ex-epitype strain of Xylaria hypoxylon have become available during the course of a large phylogenomic study that was primarily aimed at establishing a correlation between the existing multi-gene-based genealogy with a genome-based phylogeny and the discovery of novel biosynthetic gene clusters encoding for secondary metabolites. The genome sequences were obtained using combinations of Illumina and Oxford nanopore technologies or PacBio sequencing, respectively, and resulted in high-quality sequences with an average N50 of 3.2 Mbp. While the main results will be published concurrently in a separate paper, the current case study was dedicated to the detection of ITS nrDNA copies in the genomes, in an attempt to explain certain incongruities and apparent mismatches between phenotypes and genotypes that had been observed during previous polyphasic studies. The results revealed that all of the studied strains had at least three copies of rDNA in their genomes, with Hypoxylon fragiforme having at least 19 copies of the ITS region, followed by Xylaria hypoxylon with at least 13 copies. Several of the genomes contained 2–3 copies that were nearly identical, but in some cases drastic differences, below 97% identity were observed. In one case, ascribable to the presence of a pseudogene, the deviations of the ITS sequences from the same genome resulted in only ca. 90% of overall homology. These results are discussed in the scope of the current trends to use ITS data for species recognition and segregation of fungi. We propose that additional genomes should be checked for such ITS polymorphisms to reassess the validity of this non-coding part of the fungal DNA for molecular identification.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The establishment of DNA barcoding methodology for identification of fungal organisms based on pure cultures, as well as in environmental samples, has been a great challenge in the past decades (Nilsson et al. 2008). Starting from the early times when sequencing costs were very high and the application of PCR-based methods to large contingents was still prohibitive, the internal transcribed spacer (ITS) of the ribosomal DNA (rDNA) has become the favorite DNA region because of the high success rate with sequencing and its almost universal applicability. The groundbreaking work by White et al. (1990) has since then received thousands of citations, and many interesting correlations between morphological traits and molecular phylogeny have been revealed from polyphasic taxonomic approaches across the fungal kingdom. The ITS has therefore been ultimately selected as primary barcode for fungi (Schoch et al. 2012), and it serves this purpose very well. In particular the numerous recently published sequences of type and authentic species (Vu et al. 2019), as well as respective efforts by the curators of public domain databases to increase the quality of sequence data such as the RefSeq projects (e.g., https://www.ncbi.nlm.nih.gov/bioproject/177353; see also Schoch et al. 2014), have certainly been very helpful for taxonomists as well as for the various user communities of taxonomic data. However, some of the aforementioned DNA sequences of type strains are obviously erroneous (e.g., because BLAST searches sometimes clearly show that they have affinities to different orders or even classes than the taxon from which they were supposed to be derived). Therefore, a lot of work remains to be done to validate even the DNA sequences of type material.
Regardless of the great value of the ITS region for barcoding, its utility for species discrimination has often been disputed. While some mycologists have even promoted the use of similarity rates of the ITS and LSU to define species boundaries (see, e.g., Kurtzman et al. 2015 for yeast-like fungi), others have found that secondary barcodes are necessary for both, the construction of stable phylogenies and for establishment of molecular identification systems. In some families and genera of the Ascomycota, it was necessary to use up to eight different DNA loci to enable a clear-cut resolution of the taxa involved (Cai et al. 2009; Cannon et al. 2012; Houbraken et al. 2014; Phillips et al. 2013), even though finally one or two secondary barcodes may be found to be sufficient to discriminate all species.
However, this can evidently only be established after the respective monographs have been completed! Presently, we are far away from reaching such a situation in the mycological community. One reason for this could be that too many projects deal with tentative molecular ecology or other environmental studies and too little funding goes to polyphasic taxonomic work.
The situation is worsened by the fact that no sequence data at all are available for the majority of species that were initially described from morphological characters in many taxonomic groups of fungi, and in particular in those groups that cannot be cultured easily because of their symbiotic or biotrophic relationships with animals and plants (see discussions by Thines et al. 2018 and Zamora et al. 2018).
On the other hand, some mycologists have recently proposed that new taxa should be recognized on the basis of novelty of the ITS sequences alone, even including data from environmental samples in the absence of a physical type (Hawksworth et al. 2016; Lücking et al. 2018). In one case, new taxa based on DNA sequence comparisons have been invalidly erected on purpose (de Beer et al. 2016), to emphasize the need for a sequence-based classification of fungi of which no physical specimen exists.
The biggest problems associated with such “innovations” are (i) the lack of suitability of the ITS region (which is predominant among the environmental sequence data) for resolution of species complexes in many taxa and (ii) the low quality of the next generation sequence (NGS) data (which often have high error rates, are based on very short reads, and are also prone to chimera formation; cf. Hongsanan et al. 2018 for an extensive discussion of this topic).
Fortunately, the chaos that would have arisen from such measures was avoided as the vast majority of the mycological community voted against it (May et al. 2018). Nevertheless, there remains a great need to classify the unseen fungi that can only be detected from their DNA in environmental samples, by developing more concise and accurate methods for species identification (Wu et al. 2019). This will give the mycological community some time to cope with the current challenges and finally develop a workable strategy to find a way to concisely identify the “dark taxa.”
A problem that has been rarely addressed during the numerous studies that were carried out in the past decade is the polymorphism of rDNA (Hibbett et al. 2011). It has firstly been reported by O'Donnell and Cigelnik (1997) that such polymorphisms occur in the important plant pathogenic and mycotoxigenic genus Fusarium. Further studies focused on intragenomic ITS variation reported sequence divergence of around 0.1–2%, usually not exceeding 3% (Kovács et al. 2011; Smith et al. 2007; Simon and Weiss 2008; Vydryakova et al. 2012). Perhaps the most worrisome source of error in sequence-based taxon description is intragenomic heterogeneity exceeding 3% of divergence. Simon and Weiss (2008) reported 3.6% ITS variability in Davidiella tassiana, and even higher levels (10–15%) were found in Amanita, Cordyceps, and Laetiporus (Lindner and Banik 2011, Hughes et al. 2018, Li et al. 2013). Such extremely deviating variants were sometimes considered non-functional pseudogenes.
Nevertheless, many authors have ignored this fact and have instead promoted ITS as sole means for species identification in fungi. This situation has even led non-specialists to a false belief that they can classify environmental isolates, such as secondary metabolite–producing fungal endophytes based on ITS data alone, such as the Xylariales (see examples cited by Helaly et al. 2018) and this has resulted in numerous erroneous publications. The natural product community has recently been made aware of the problem (Raja et al. 2017), but still there are dozens of papers published each year with incorrectly assigned taxonomy of biotechnologically interesting new organisms. Studies such as the one by U’Ren et al. (2016) have been very helpful because they characterized various fungal endophytes by using a multi-locus phylogeny. However, the authors did not use many type strains for comparison; and not even authentic, ascospore-derived cultures from the geographic region from where the endophytic isolates originated were studied for comparison. The species concepts based on morphological traits of the teleomorphs were traditionally broad. On the other hand, many species of Xylariales that were erected based on specimens from North America have never been recollected and subjected to DNA sequencing. Mycologists have in the past also frequently used European species names for North American fungi that were later shown to be misapplied (see, e.g., Fournier et al. 2012 for Xylaria; Stadler et al. 2014a, b for Daldinia). Therefore, the species identification of these endophytes remains tentative.
We have recently generated high-quality genome sequence data of twelve representative strains of the Xylariales, most of which were chosen from the type and authentic strains representing the major phylogenetic clades resulting from multi-locus phylogenies (Daranagama et al. 2018; Wendt et al. 2018). PacBio sequencing or a combination of Illumina sequencing with the Oxford nanopore long-read method gave us the ability to read through highly repetitive sequences such as those found in rDNA. The major aims of this study were the construction of a phylogenomic tree and its comparison with the classical four locus genealogy (Wendt et al. 2018), and the creation of a data matrix that would allow us to perform genome mining and discovery of unique and/or silent biosynthetic gene clusters. However, we found by coincidence that multiple copies of the ITS were present in the genomes and wish to report and discuss these findings separately in the present paper. This is the first study mapping the structure of the ITS array using the continuous long reads that span multiple rDNA cistrons and can reliably identify multiple ITS copy numbers as well as sequence polymorphisms.
Materials and methods
A list of the strains whose genome sequences were used for the current study is provided in Table 1. In cases where substantial polymorphisms were observed, a BLAST search for the most common ITS paralog was carried out in order to find the most similar sequences in the public domain.
Nanopore library preparation and MinION® sequencing
A MinION sequencing library with genomic DNA from the different fungi strains was prepared using the Nanopore Rapid DNA Sequencing kit (SQK-RAD04, Oxford Nanopore Technologies, Oxford, UK) according to the manufacturer’s instructions. Sequencing was performed on an Oxford Nanopore MinION Mk1b sequencer using a R9.5 flow cell, which was prepared according to the manufacturer’s instructions.
Illumina library preparation and MiSeq sequencing
Whole-genome-shotgun PCR-free libraries were constructed from 5 μg of gDNA with the Nextera XT DNA Sample Preparation Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol. The libraries were quality controlled by analysis on an Agilent 2000 Bioanalyzer equipped with Agilent High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA, USA) for fragment sizes of 500–1000 bp. Sequencing was performed on the MiSeq platform (Illumina; 2 × 300 bp paired-end sequencing, v3 chemistry). Adapters and low quality reads were removed by an in house software pipeline prior to polishing as recently described (Wibberg et al. 2016).
Base calling, reads processing, and assembly
MinKNOW (v1.13.1, Oxford Nanopore Technologies) was used to control the run using the 48-h sequencing run protocol; base calling was performed offline using albacore (v2.3.1, https://github.com/Albacore/albacore). The assembly was performed using canu v1.6 and v1.7 (Koren et al. 2017), resulting in a single, circular contig. This contig was then polished with Illumina short read data using Pilon (Walker et al. 2014), applying eight iterative cycles. BWA-MEM (Li 2013) was used for read mapping in the first four iterations and Bowtie2 v2.3.2 (Langmead and Salzberg 2012) in the second set of four iterations.
PacBio library preparation and sequencing
For Hypoxylon fragiforme SMRTbell™ template library was prepared according to the instructions from Pacific Biosciences (Menlo Park, CA, USA), following the Procedure & Checklist – Greater Than 10 kb Template Preparation. Briefly, for preparation of 15 kb libraries 8 μg genomic DNA was sheared using g-tubes™ from Covaris, Woburn, MA, USA, according to the manufacturer’s instructions. DNA was end-repaired and ligated overnight to hairpin adapters applying components from the DNA/Polymerase Binding Kit P6 from Pacific Biosciences. Reactions were carried out according to the manufacturer’s instructions. BluePippin™ Size-Selection to greater than 7 kb was performed according to the manufacturer’s instructions (Sage Science, Beverly, MA, USA). Conditions for annealing of sequencing primers and binding of polymerase to purified SMRTbell™ template were assessed with the Calculator in RS Remote (Pacific Biosciences). One SMRT cell was sequenced on the PacBio RSII (Pacific Biosciences) taking one 240-min movie. Two further SMRT cells were sequenced on the Sequel System (Pacific Biosciences) taking one 600-min movie for each SMRT cell. Sequencing on the Sequel System resulted in 727,768 (sub)reads with a mean length of 7823 bp. Data was assembled within SMRTLink 5.1.0.26412 using HGAP4 protocol and a target genome size of 35 Mbp. The genome assembly resulted in 36 contigs summing up to a final genome size of 38.1 Mbp (3.6 Mbp N50 contig length, 127 × genome coverage).
Identification of rDNA sequence copies
Genome sequences were used to create individual BLAST databases in Geneious 9.1.8 (https://www.geneious.com). Previously published ITS sequences from the respective strains were used as template for homology search to locate rDNA regions. Subsequently, the hits within each genome were aligned using the internal ClustalW methodology with standard settings.
Molecular phylogenetic inference
Extracted ITS sequences were aligned using the MAFFT v. 7.017 plugin in Geneious 7.1.9 (Katoh et al. 2002). The alignment was subjected to the ATGC PhyML Online Server (http://www.atgc- montpellier.fr/phyml/) selecting options for the PhyML 3.0 algorithm with subtree-pruning and regrafting (SPR, Guindon et al. 2010) and preliminary selection of the best nucleotide substitution model by the Bayesian information criterion (BIC) using the in-built Smart model Selection (SMS, Lefort et al. 2017) feature with 1000 bootstrap replicates.
Testing for the presence of a pseudogene in the genome of Hypomontagnella monticulosa
Putative pseudogenes were identified by a combination of mutation and secondary structure analyses of 5.8S rDNA (Li et al. 2013). Secondary structures of the 5.8S region at 25 °C were predicted using Mfold version 2.3 on a web server (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form2.3; cf. Zuker 2003) using a universal model as a guide. Descriptive statistics (nucleotide frequency, length) was done on the extracted ITS sequences in MEGA X Version 10.1 (Kumar et al. 2018)
Results and discussion
Reports of genome assemblies produced from Nanopore or PacBio reads have increased, and the improvement to contiguity in such genome assemblies is seen as a benefit due to the long reads (Shin et al. 2019). Therefore, we mainly applied a combination of Nanopore and Illumina reads to assemble high-quality fungal draft genomes. Assemblies with our Nanopore reads were sufficient to result in almost complete fungal genomes. Genome polishing with high-quality Illumina reads effectively improved the quality of the genome assembly produced using Nanopore reads (~ 99.9998% accuracy) resulting in highly accurate rDNA sequences. In other studies it was shown that even very long, near identical repeats were resolved by Nanopore reads (Schmid et al. 2018). Nevertheless, long read techniques have limitations in sequencing the pericentromeric, centromeric, and teleomeric regions, and acrocentric short arms of chromosomes, which contain satellite DNA and large numbers of tandem repeats (Jain et al. 2018). In such regions, ribosomal DNA operon tandem repeats can also be detected. If these operons occur in high consecutive copy numbers without sequence variations (including NTS regions), the respective genomic regions cannot be fully resolved with the read lengths achieved in this study (averaging 10 kb for ONT and 8 kb PacBio herein, which equals the size of a full rDNA operon). The given number of rDNA copies in the following paragraphs therefore represents only the minimum set of tandem repeats that can be covered and identified with the currently available state-of-the-art genome sequencing methods. As the following paragraphs exclusively focus on the ITS and 5.8S regions, we would like to briefly mention that the 28S rDNA in all nanopore sequenced strains showed a much higher degree of intragenomic variation in comparison (ranging from 6–25 bp in J. multiformis to 8–275 bp in X. hypoxylon). These polymorphisms contributed strongly to the accuracy of the assemblies and thus allowed for clear separation of rDNA copies.
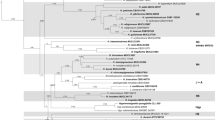
The results of the genome mining study are summarized in Table 1, illustrated in Figs. 1 and 2, and will be briefly commented in the following paragraphs.

Inferred maximum-likelihood (ML) tree (lnL = − 6064.64571) using the Kimura (1980) nucleotide substitution model as selected by smart-model selection (SMS) with an estimated gamma shape parameter of 0.597 and fixed proportion of invariable sites (0.0) derived from extracted ITS-like regions using PhyML 3.0. Bootstrap values (BS) are shown at corresponding branches following the 50% majority-consensus rule. Sequences found to be identical within each clade are highlighted by a blue color pattern. Branch length is indicated as nucleotide substitutions per site

Infragenomic polymorphism between the ITS regions of the identified and aligned nrDNA copies of different Xylariales species (aHypoxylon lienhwacheense MFLUCC 14-1231, bHypomontagnella monticulosa MUCL 54604, cPyrenopolyporus hunteri MUCL 49339, dXylaria hypoxylon CBS 122620). The shown region spans the partial 18S rDNA, ITS1, 5.8S rDNA, ITS2, and partial 28S rDNA. The fragment sizes are indicated by numbers. Identical regions are shown in black, and variable areas are highlighted in dark or light gray. Gaps are depicted in white
Most of the sequences were generated by Oxford Nanopore/Illumina, and only the genome of Hypoxylon fragiforme was sequenced using PacBio. This genome, derived from the ex-epitype strain of H. fragiforme, contained at least 19 copies of the ITS. While fourteen of these copies were identical, five deviated by 1–4 base pairs (similarity of 99.7–99.8% to the major genotype observed).
A BLAST comparison of the ITS sequences deposited in GenBank with our sequences derived from the genomes revealed that the closest 30 sequences with 100–96% identity were also derived from vouchers of H. fragiforme, and the closest match of an ITS sequence derived from a different species (H. howeanum 94.4%; GenBank Acc no JQ009323) originated from the study by Hsieh et al. (2005). Among the ITS sequences in GenBank that were reported to be derived from “H. fragiforme,” the sequence of an endophyte associated with Viscum album (Acc. no FN435671; cf. Peršoh et al. 2010) showed the highest divergence from that of the ex-epitype strain. While the identity of this voucher with H. fragiforme remains dubious, all sequences that were obtained from ascospore-derived cultures showed a higher homology to the sequences derived from the ex-type strain. In conclusion, ITS seems to be workable for identification of H. fragiforme, even though some intragenomic and intraspecific variability remains.
Another interesting case is the genome sequence of H. lienhwacheense, where the four ITS copies obtained showed a divergence of 15–23 nucleotides, equivalent to 97.7–98.5% identity (Fig. 2a). All of the observed ITS copies were different from one another. A similar observation was made for the five identified ITS copies in P. hunteri, which all slightly varied. The identity between the sequences ranged from 99.8 to 96.5% equaling 2–40 bp mismatches (Fig. 2c).
Little variability was noted in the sequences of Annulohypoxylon truncatum, Daldinia concentrica, and Jackrogersella multiformis, which contained three ITS copies that were almost identical to one another and showed differences in only 1–3 bp. Regarding Daldinia concentrica, Stadler et al. (2014b) have shown that this species is a representative of a complex that contains several other taxa that can be discriminated by morphological studies of the anamorphic states, as well as from a comparison of ascospore dimensions and ultrastructural studies on the ascospore ornamentation. Furthermore, species of the D. concentrica group also deviate in the apparent host specificity of their stromata. However, they all share similar secondary metabolite profiles and ITS sequences and cannot be segregated by using the universal primary barcode.
In the case of J. multiformis strong ITS sequence variation has been detected in a different strain by Lindner et al. (2013), as Annulohypoxylon multiforme) using 454 amplicon pyrosequencing. They identified a high number of haplotypes (71) with at least one of them being strongly diverged. Even though pyrosequencing suffers from PCR and sequencing errors resulting in overestimation of polymorphism, these data suggested that intraspecific variations in rDNA copy numbers and their sequences likely exist in the Hypoxylaceae. Unfortunately, the origin and authenticity of their strain cannot be evaluated and thus the results of this comparison have to be treated with caution.
Four highly homologous copies of the ITS were observed in the genomes of “Entonaema liquescens” (a strain of dubious identity, since the genomic data did not reveal the presence of the azaphilone gene cluster encoding for the characteristic stromatal metabolite of this species; see concurrent paper by Wibberg et al. 2020), as well as from Hypoxylon pulicicidum and H. rickii. These species can be determined with certainty at least on the basis of the currently available nucleotide sequence collection in GenBank, since no other taxon has been reported to show a similar ITS sequence.
Two genomes of species referable to Hypomontagnella monticulosa were available for comparison. One of those was derived from an isolate obtained from ascospores of a terrestrial specimen collected from wood in a rainforest in French Guiana (the location where the type of the fungus was originally collected), while the second sequence was generated from a sponge-derived isolate originating from the South Pacific (Leman-Loubière et al. 2017). Comparison of the genomes revealed remarkable differences, in accordance with the ecology of the two strains. While details will be reported concurrently, the two isolates deviated from one another in the number of ITS copies, and also with respect to the intragenomic diversity of the ITS paralogs (Table 2). The marine strain CLL-205 had 5 ITS copies, of which two were identical and three showed minimal deviations of 1–2 nucleotides. The genome of the ascospore-derived strain, however, contained four ITS copies (Fig. 1), two of which were identical and the third showed slight deviations of 2 base pairs. The fourth ITS copy, however, deviated by 64 base pairs and only showed ca. 87% identity to the other paralogs. A BLAST search revealed only 90% identity to the next homologous sequences, which were derived from H. monticulosa. However, if such differences had been observed for different specimens, these data might have prompted those mycologists who believe in the classification of fungi on mere homology of rDNA sequences to erect a new species, if not even two different genera for the aberrant ITS paralog. Such a striking divergence suggested the possible presence of a pseudogene. In comparison to shallow divergent paralogues, which are still controlled by selection, rRNA in pseudogenes mutates without functional constraints, exhibits higher variability in conserved regions, differs in length, and possesses a number of methylation-induced substitutions, which in turn lead to a reduced GC content due to the methylation-induced substitutions C→T and G→A and reduced stability of the secondary structure, which is normally ensured by GC-rich helices (Feliner and Rossell 2007; Kolařík and Vohník 2018). The strongly deviating H. monticulosa ITS variant fullfils completely all those criteria and can be considered a pseudogene. In particular, it is extremely AT rich, with a 5.8S of the less stable (i.e., with high free energy) secondary structure and deviating length (Table 2). The 5.8S rDNA sequence is highly conserved among the studied strains (2 substitutions only). The deviating ITS copy differs by 10 substitutions and 16 indels. The substitution pattern of the positions unique to the deviating ITS copy was strongly biased towards C→T (6 cases), whereas other substitution types occurred in 1–2 cases. The same pattern became obvious when both ITS variants of H. monticulosa MUCL 54604 were compared (C→T, 12 cases; T→C, 3 cases; others, 1–2 cases).
Thus far, rDNA pseudogenes were detected or postulated to be present in only a few studies (Rooney and Ward 2005; Lindner and Banik 2011; Li et al. 2013, 2017). The only two studies that systematically screened whole genome sequence data for the presence of pseudogenes and reported that pseudogene rDNA copies seem to be more prevalent than functional ones were those by Rooney and Ward (2005) and Li et al. (2017). Similar data have occasionally been reported, e.g., for Melanconiella chrysodiscosporina (Voglmayr et al. 2012), where classical PCR and Sanger sequencing revealed aberrant ITS and LSU sequences of, while rbp2 and tef1 were in agreement with others in the same genus. However, these aberrant sequences had not been studied in-depth as in our case of H. monticulosa.
In our study, however, the pseudogenic rDNA represents 1.5% of all detected variants. Due to the highly repetitive nature of the rDNA genes, traditional bioinformatic approaches attempt to assemble contiguous sequences from short reads, collapse the arrays into a single contiguous sequence, and only retain polymorphisms. This results in the overestimation of the divergent sequence variants (Li et al. 2017). Notably, almost all of the published ITS sequences are basically a consensus of the amplified ITS copies and this does not necessarily reflect the true situation.
The sequencing methods used in our study can overcome these limits, even though we have only based our findings on a relatively small number of datasets. We thus present the first reliable data about the frequency of rDNA pseudogenes in fungi, which appears to be much higher than previously known. We have also found evidence by checking other parts of the rDNA cistron (and in particular the LSU), for such incongruities and the potential presence of pseudogenes. As the matrix of LSU data from Hypoxylaceae species in the public domain is by far not as exhaustive as the data matrix for the ITS, we chose not to integrate these into the current study. However, we would like to encourage other mycologists to carefully analyze the other domains of the rDNA cistron before drawing premature taxonomic conclusions if they intend to use those data for species discrimination.
Finally, the ex-epitype strain of Xylaria hypoxylon (the only species included in the current study that is not a member of the Hypoxylaceae) was found to contain at least 13 ITS copies, six of which were identical, while the remainder differed by 1–19 nucleotides from the main type, equivalent to 96.9–99.8% identity (Fig. 2d). The most similar sequences in GenBank were several that had already been assigned to X. hypoxylon s. str. by Peršoh et al. (2009) and Fournier et al. (2012), and the most similar sequence to those (with an identity of 97.5%) was derived from the type of X. vasconica (MF755269). The latter species is closely related to X. hypoxylon, but was shown by Fournier et al. (2012) to differ in various morphological characters. The most aberrant sequence obtained from the genome of X. hypoxylon showed even stronger deviation from the most frequent ITS genotype observed in the genome of the type strain of X. hypoxylon than to X. vasconica. Therefore, it does not seem possible to discriminate the two taxa using ITS.
We have also noted various slight deviations between the published ITS sequences of our strains and the data we obtained from the genomes. This may be due to PCR sequencing errors, the fact that PCR equally targets all rDNA repeats resulting in a “consensus” sequence across all ITS copies (if amplicons have not been selected by cloning), as well as by mutations that occurred in the genomes since the time the sequences were first generated. On the other hand, these discrepancies were not high and mostly concerned single base pair substitutions.
From the results of this case study, we draw the following conclusions.
-
a)
Oxford nanopore or PacBio sequencing is not able to resolve the total rDNA operon copy numbers due to insufficient read lengths, which are unable to cover highly identical rDNA tandem repeat areas. However, at least Illumina polished ONT assemblies allow reliable identification of polymorphism in rDNA copies unraveling the degree of intragenomic sequence diversity of ITS regions and thus the applicability of such a marker locus for subsequent analysis (i.e., barcoding, phylogenetics, species diversity assessment, etc.) in a given taxonomic group.
-
b)
What was previously often referred to as “interspecific variability” when such polymorphisms were observed from studies of various vouchers of a given fungal species, may actually be due to intragenomic variability of the ITS paralogs located in the same genome.
-
c)
Even though the ITS region seems to be workable for species identification in many cases, it cannot be taken for granted that it works with all the species in a given genus. A concise identification of a certain species based on ITS data alone, or its recognition as a new taxon, in particular from environmental data, is problematic. It may work after all the known next relatives have been studied (cf. de Beer et al. 2016; Pažoutová et al. 2013). However, it may actually be necessary to sequence the genomes of the fungi in order to obtain high-quality data and then look for polymorphisms of the ITS (or alternatively any other DNA locus that is favored for species discrimination).
-
d)
We have been probing the published “full genomes” or related species in the public domain, but were not able to use these data in a similar manner, also because the genomes sequenced using Illumina had many gaps and we could only detect fragments of the ITS in most of them. We propose that more high-quality sequence data should be generated in the future, since the “shotgun genomes” also have great disadvantages concerning other potential application relating to the exploration of functional biodiversity. For instance, secondary metabolite biosynthesis gene clusters are also easier to detect in the genome sequences that were generated using techniques that result in longer reads such as those applied in the present study.
The number of rDNA copies in fungi has been subject to various investigations for more than 50 years, which mainly addressed the question by indirect estimation approaches such as DNA-RNA hybridization, qPCR, or gel electrophoresis of rDNA arrays (Ganley and Kobayashi 2007; Herrera et al. 2009; Raidl et al. 2005; Schweizer et al. 1969). These studies only involved a single or very few species for which rDNA copy numbers between 45 and 140 were calculated. The most recent investigation on this topic involved a broad selection of taxa and revealed a much higher variation in rDNA copy numbers ranging from at least 11 in the ascomycete Oidiodendron maius to almost 1500 in the early diverging fungus Basidiobolus meristosporus (Lofgren et al. 2019). The dataset is based on 91 publically available (short-read) genome-sequenced fungal species from various taxonomic groups, and copy numbers were estimated by the normalized read depths of multi-copy genomic regions. This study also included an endophytic isolate of the Hypoxylaceae tentatively identified as Daldinia eschscholtzii based on ITS sequence data (Wu et al. 2007), which presumably possesses 45 rDNA copies. Despite the use of indirect data, these estimated values strongly indicate that the rDNA copy numbers identified in our study are likely to be much smaller than the actual occurring copy numbers in the respective strains demonstrating that state-of-the-art long read sequencing technologies are still unable to resolve long (> 30 kb) highly repetitive DNA regions. Nevertheless, we have provided the first direct sequencing-based evidence for multiple rDNA copy numbers in fungi.
The observed phenomenon of intragenomic polymorphic ITS regions in fungi has been previously reported for various fungal species, but was mainly evidenced by PCR and sequencing of the corresponding cloned amplicons, making identification of single nucleotide polymorphisms prone to errors. Several such studies were carried out on yeast-like fungi, where sequence divergence has traditionally been discussed much more intensively as a means of species recognition because relatively few phenotypic characters are available for discrimination of morphotypes. Alper et al. (2011) reported polymorphisms in the dimorphic yeast Geotrichum candidum and identified 60 polymorphic sites from a comparison of multiple strains, which were mostly derived from dairy. They attributed most of these polymorphisms to base pair substitutions and found one indel in the ITS1. After the authors detected dual peaks in the electropherograms during sequencing of the rDNA, they cloned the ITS regions of four strains and found 32 deviating ITS paralogs among the 68 clones they obtained from four strains. The authors concluded that the utility of the ITS region for fungal community profiling was dubious, but unfortunately, many other colleagues do not seem to realize the potential pitfalls.
There are even some critical issues regarding species identification of human pathogens, where the ITS and LSU do not work. For instance, recently Passer et al. (2019) have reported such issues during their study on pathogenic and saprotrophic species of the basidiomycete yeast genus Cryptococcus. Even the pathogenic multi-drug-resistant yeast, Candida auris (Hyde et al. 2018), can unfortunately not be easily identified by ITS or LSU sequencing. Heeger et al. (2018) have used long read metabarcoding and came across polymorphisms in several species of the yeast Metschnikovia, which prevented their accurate identification. The estimate of species numbers will be too high in such cases where extreme polymorphism is observed. It can be expected that similar studies may reveal that this problem is common in the fungal kingdom. At least it should not be claimed that polymorphic rDNA sequences that are obtained from the same specimen are due to the existence of “cryptic species” without concurrent studies of the genomes of the respective organisms.
The above examples and the findings reported in the present study should give reason to study this phenomenon more thoroughly in other groups of filamentous fungi. In any case, suitable techniques are now available to obtain high-quality genome sequences and this can certainly help to tackle this challenge and ultimately allow for establishment of a more realistic way to estimate species diversity in fungi than by profiling of the ITS region. We expect that high-quality genome sequence data will increasingly become popular in fungal phylogeny, and hope the results of the current study will be helpful for other researchers to interpret their results.
References
Alper I, Frenette M, Labrie S (2011) Ribosomal DNA polymorphisms in the yeast Geotrichum candidum. Fungal Biol 115:1259–1269
Bills GF, González-Menéndez V, Martín J, Platas G, Fournier J, Peršoh D, Stadler M (2012) Hypoxylon pulicicidum sp. nov. (Ascomycota, Xylariales), a pantropical insecticide-producing endophyte. PLoS One 7:e46687
Cai L, Hyde KD, Taylor PWJ, Weir BS, Waller J, Abang MM, Zhang JZ, Yang YL, Phoulivong S, Liu ZY, Prihastuti H, Shivas RG, McKenzie EHC, Johnston PR (2009) A polyphasic approach for studying Colletotrichum. Fungal Divers 39:183–204
Cannon PF, Damm U, Johnston PR, Weir BS (2012) Colletotrichum–current status and future directions. Stud Mycol 73:181–213
Daranagama DA, Hyde KD, Sir EB et al (2018) Towards a natural classification and backbone tree for Graphostromataceae, Hypoxylaceae, Lopadostomataceae and Xylariaceae. Fungal Divers 88:1–165
de Beer ZW, Marincowitz S, Duong TA, Kim JJ, Rodrigues A, Wingfield MJ (2016) Hawksworthiomyces gen. nov. (Ophiostomatales), illustrates the urgency for a decision on how to name novel taxa known only from environmental nucleic acid sequences (ENAS). Fungal Biol 120:1323–1340
Feliner GN, Rossell JA (2007) Better the devil you know? Guidelines for insightful utilization of nrDNA ITS in species-level evolutionary studies in plants. Mol Phylogenet Evol 44:911
Fournier J, Flessa F, Peršoh D, Stadler M (2012) Three new Xylaria species from southwestern Europe. Mycol Prog 10:33–52
Ganley AR, Kobayashi T (2007) Highly efficient concerted evolution in the ribosomal DNA repeats: total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res 17(2):184–191
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59(3):307–321
Hawksworth DL, Hibbett DS, Kirk PM, Lücking R (2016) Proposals to permit DNA sequence data to serve as types of names of fungi. Taxon 65(4):899–900
Heeger F, Bourne EC, Baschien C, Yurkov A, Bunk B, Spröer C, Overmann J, Mazzoni CJ, Monaghan MT (2018) Long-read DNA metabarcoding of ribosomal RNA in the analysis of fungi from aquatic environments. Mol Ecol Resour 18:1500–1514
Helaly SE, Thongbai B, Stadler M (2018) Diversity of biologically active secondary metabolites from endophytic and saprotrophic fungi of the ascomycete order Xylariales. Nat Prod Rep 35:992–1014
Herrera ML, Vallor AC, Gelfond JA, Patterson TF, Wickes BL (2009) Strain-dependent variation in 18S ribosomal DNA copy numbers in Aspergillus fumigatus. J Clin Microbiol 47:1325–1332
Hibbett DS, Ohman A, Glotzer D, Nuhn M, Kirk P, Nilsson RH (2011) Progress in molecular and morphological taxon discovery in fungi and options for formal classification of environmental sequences. Fungal Biol Rev 25:38–47
Hongsanan S, Xie N, Liu JK, Dissanayake A, Ekanayaka AH, Raspé O, Jayawardena RS, Hyde KD, Jeewon R, Purahong W, Stadler M, Peršoh D (2018) Can we use environmental DNA as holotypes? Fungal Divers 92:1–30
Houbraken J, de Vries RP, Samson RA (2014) Modern taxonomy of biotechnologically important Aspergillus and Penicillium species. Adv Appl Microbiol 86:199–249
Hsieh HM, Ju YM, Rogers JD (2005) Molecular phylogeny of Hypoxylon and closely related genera. Mycologia 97:844–865
Hughes KW, Tulloss RH, Petersen RH (2018) Intragenomic nuclear RNA variation in a cryptic Amanita taxon. Mycologia 110:93–103
Hyde KD; Al-Hatmi A, Andersen B; Boekhout T; Buzina W; Dawson Jr TL; Eastwood DC; Gareth Jones EB; de Hoog S, Kang Y; Longcore JE; McKenzie EHC; Meis J, Gadais JP, Rathnayaka AR; Forget FR, Stadler M, Theelen B, Thongbai B, Tsui CKM (2018). The world’s ten most feared fungi. Fungal Divers 93:161–194
Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, Malla S (2018) Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36(4):338
Katoh K, Misawa K, Kuma KI, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16(2):111–120
Kolařík M, Vohník M (2018) When the ribosomal DNA does not tell the truth: the case of the taxonomic position of Kurtia argillacea, an ericoid mycorrhizal fungus residing among Hymenochaetales. Fungal Biol 122:1–18
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM (2017) Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27(5):722–236
Kovács GM, Jankovics T, Kiss L (2011) Variation in the nrDNA ITS sequences of some powdery mildew species: do routine molecular identification procedures hide valuable information? Eur J Plant Pathol 131:135
Kuhnert E, Fournier J, Peršoh D, Luangsa-ard JJD, Stadler M (2014) New Hypoxylon species from Martinique and new evidence on the molecular phylogeny of Hypoxylon based on ITS rDNA and β-tubulin data. Fungal Divers 64:181–203
Kuhnert E, Sir EB, Lambert C et al (2017) Phylogenetic and chemotaxonomic resolution of the genus Annulohypoxylon (Xylariaceae) including four new species. Fungal Divers 85:1–43
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Kurtzman CP, Mateo RQ, Kolecka A, Theelen B, Robert V, Boekhout T (2015) Advances in yeast systematics and phylogeny and their use as predictors of biotechnologically important metabolic pathways. FEMS Yeast Res 15:6
Lambert C, Wendt L, Hladki AI et al (2019) Hypomontagnella (Hypoxylaceae): a new genus segregated from Hypoxylon by a polyphasic taxonomic approach. Mycol Prog 18:187–201
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Lefort V, Longueville JE, Gascuel O (2017) SMS: smart model selection in PhyML. Mol Biol Evol 34(9):2422–2424
Leman-Loubière C, Le Goff G, Retailleau P, Debitus C, Ouazzani J (2017) Sporothriolide-related compounds from the fungus Hypoxylon monticulosum CLL-205 isolated from a Sphaerocladina sponge from the Tahiti coast. J Nat Prod 80:2850–2854
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv Prepr arXiv
Li Y, Jiao L, Yao YJ (2013) Non-concerted ITS evolution in fungi, as revealed from the important medicinal fungus Ophiocordyceps sinensis. Mol Phylogenet Evol 68:373–379
Li Y, Yang RH, Jiang L, Hu XD, Wu ZJ, Yao YJ (2017) rRNA pseudogenes in filamentous ascomycetes as revealed by genome data. G3 7:2695–2703
Lindner DL, Banik MT (2011) Intragenomic variation in the ITS rDNA region obscures phylogenetic relationships and inflates estimates of operational taxonomic units in genus Laetiporus. Mycologia 103:731–740
Lindner DL, Carlsen T, Henrik Nilsson R, Davey M, Schumacher T, Kauserud H (2013) Employing 454 amplicon pyrosequencing to reveal intragenomic divergence in the internal transcribed spacer rDNA region in fungi. Ecol Evol 3(6):1751–1764
Lofgren LA, Uehling JK, Branco S, Bruns TD, Martin F, Kennedy PG (2019) Genome-based estimates of fungal rDNA copy number variation across phylogenetic scales and ecological lifestyles. Mol Ecol 28(4):721–730
Lücking R, Kirk PM, Hawksworth DL (2018) Sequence-based nomenclature: a reply to Thines et al. and Zamora et al. and provisions for an amended proposal “from the floor” to allow DNA sequences as types of names. IMA Fungus 9(1):185
May TW, Redhead SA, Lombard L, Rossman AY (2018) XI International Mycological Congress: report of Congress action on nomenclature proposals relating to fungi. IMA Fungus 9(2):xxii
Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson K-H (2008) Intraspecific ITS variability in the kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol Bioinformatics Online 4:193
O'Donnell K, Cigelnik E (1997) Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous. Mol Phylogenet Evol 7:103–116
Passer AR, Coelho MA, Billmyre RB, Nowrousian M, Mittelbach M, Yurkov AM, Averette AF, Cuomo CA, Sun S, Heitman J (2019) Genetic and genomic analyses reveal boundaries between species closely related to Cryptococcus pathogens. mBio 10(3):e00764–e00719
Pažoutová S, Follert S, Bitzer J et al (2013) A new endophytic insect-associated Daldinia species, recognised from a comparison of secondary metabolite profiles and molecular phylogeny. Fungal Divers 60:107–123
Peršoh D, Melcher M, Graf K, Fournier J, Stadler M, Rambold G (2009) Molecular and morphological evidence for the delimitation of Xylaria hypoxylon. Mycologia 101:256–268
Peršoh D, Melcher M, Flessa F, Rambold G (2010) First fungal community analyses of endophytic ascomycetes associated with Viscum album ssp. austriacum and its host Pinus sylvestris. Fungal Biol 114:585–596
Phillips AJL, Alves A, Abdollahzadeh J, Slippers B, Wingfield MJ, Groenewald JZ, Crous PW (2013) The Botryosphaeriaceae: genera and species known from culture. Stud Mycol 76:51–167
Raidl S, Bonfigli R, Agerer R (2005) Calibration of quantitative real-time Taqman PCR by correlation with hyphal biomass and ITS copies in mycelia of Piloderma croceum. Plant Biol 7:713–717
Raja HA, Miller AN, Pearce CJ, Oberlies NH (2017) Fungal identification using molecular tools: a primer for the natural products research community. J Nat Prod 80:756–770
Rogers JD (1982) Entonaema liquescens: description of the anamorph and thoughts on its systematic position. Mycotaxon 15:500–506
Rooney AP, Ward TJ (2005) Evolution of a large ribosomal RNA multigene family in filamentous fungi: birth and death of a concerted evolution paradigm. PNAS 102:5084–5089
Schmid M, Frei D, Patrignani A, Schlapbach R, Frey JE, Remus-Emsermann MN, Ahrens CH (2018) Pushing the limits of de novo genome assembly for complex prokaryotic genomes harboring very long, near identical repeats. Nucleic Acids Res 46(17):8953–8965. https://doi.org/10.1093/nar/gky726
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W et al (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. PNAS 109:6241–6246
Schoch CL, Robbertse B, Robert V, Vu D, Cardinali G, Irinyi L, Meyer W, Nilsson RH, Hughes K, Miller AN, Kirk PM (2014) Finding needles in haystacks: linking scientific names, reference specimens and molecular data for Fungi. Database. https://doi.org/10.1093/database/bau061
Schweizer E, MacKechnie C, Halvorson H (1969) The redundancy of ribosomal and transfer RNA genes in Saccharomyces cerevisiae. J Mol Biol 40(2):261–w77
Shin SC, Kim H, Lee JH, Kim HW, Park J, Choi BS, Lee SC, Kim JH, Lee H, Kim S (2019) Nanopore sequencing reads improve assembly and gene annotation of the Parochlus steinenii genome. Sci Rep 9(1):5095
Simon UK, Weiss M (2008) Intragenomic variation of fungal ribosomal genes is higher than previously thought. Mol Biol Evol 25:2251–2254
Sir EB, Kuhnert E, Lambert C, Hladki AI, Romero AI, Stadler M (2016) New species and reports of Hypoxylon from Argentina recognized by a polyphasic approach. Mycol Prog 15:42
Sir EB, Becker K, Lambert C et al (2019) Observations on Texas hypoxylons, including two new Hypoxylon species and widespread environmental isolates of the H. croceum complex identified by a polyphasic approach. Mycologia 11(5):832–856
Smith ME, Douhan GW, Rizzo DM (2007) Intra-specific and intra-sporocarp ITS variation of ectomycorrhizal fungi as assessed by rDNA sequencing of sporocarps and pooled ectomycorrhizal roots from a Quercus woodland. Mycorrhiza 18:5–22
Stadler M, Hawksworth DL, Fournier J (2014a) The application of the name Xylaria hypoxylon, based on Clavaria hypoxylon of Linnaeus. IMA Fungus 5:57–66
Stadler M, Læssøe T, Fournier J, Decock C, Schmieschek B, Tichy HV, Peršoh D (2014b) A polyphasic taxonomy of Daldinia (Xylariaceae). Stud Mycol 77:1–143
Thines M, Crous PW, Aime MC, Aoki T, Cai L, Hyde KD, Miller AN, Zhang N, Stadler M (2018) Ten reasons why a sequence-based nomenclature is not useful for fungi anytime soon. IMA Fungus 9:177–183
Triebel D, Peršoh D, Wollweber H, Stadler M (2005) Phylogenetic relationships among Daldinia, Entonaema, and Hypoxylon as inferred from ITS nrDNA analyses of Xylariales. Nova Hedwigia 80:25–43
U’Ren JM, Miadlikowska J, Zimmerman NB et al (2016) Contributions of North American endophytes to the phylogeny, ecology, and taxonomy of Xylariaceae (Sordariomycetes, Ascomycota). Mol Phylogenet Evol 98:210–232
Voglmayr H, Rossman AY, Castlebury LA, Jaklitsch WM (2012) Multigene phylogeny and taxonomy of the genus Melanconiella (Diaporthales). Fungal Divers 57:1–44
Vu D, Groenewald M, de Vries M, Gehrmann T, Stielow B, Eberhardt U, Al-Hatmi A, Groenewald JZ, Cardinali G, Houbraken J, Boekhout T (2019) Large-scale generation and analysis of filamentous fungal DNA barcodes boosts coverage for kingdom Fungi and reveals thresholds for fungal species and higher taxon delimitation. Stud Mycol 92:135–154
Vydryakova GA, Van DT, Shoukouhi P, Psurtseva NV, Bissett J (2012) Intergenomic and intragenomic ITS sequence heterogeneity in Neonothopanus nambi (Agaricales) from Vietnam. Mycology 3:89–99
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9(11):e112963
Wendt L, Sir EB, Kuhnert E et al (2018) Resurrection and emendation of the Hypoxylaceae, recognised from a multigene phylogeny of the Xylariales. Mycol Prog 17:115–154
White TJ, Bruns T, Lee S, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic, New York, pp 315–322
Wibberg D, Andersson L, Tzelepis G, Rupp O, Blom J, Jelonek L, Pühler A, Fogelqvist J, Varrelmann M, Schlüter A, Dixelius C (2016) Genome analysis of the sugar beet pathogen Rhizoctonia solani AG2-2IIIB revealed high numbers in secreted proteins and cell wall degrading enzymes. BMC Genomics 17(1):245
Wibberg D, Stadler M, Bunk B, Spröer C, Lambert C, Kalinowski J, Cox RJ, Kuhnert E (2020) High quality genome sequences of twelve Hypoxylaceae (Ascomycota) strengthen the phylogenetic family backbone and enable the discovery of new taxa. Submitted to Fungal Diversity
Wu W, Davis RW, Tran-Gyamfi MB, Kuo A, LaButti K, Mihaltcheva S, Hundley H, Chovatia M, Lindquist E, Barry K, Grigoriev IV (2007) Characterization of four endophytic fungi as potential consolidated bioprocessing hosts for conversion of lignocellulose into advanced biofuels. Appl Microbiol Biotechnol 101(6):2603–2618
Wu B, Hussain M, Zhang W, Stadler M, Liu X, Xiang M (2019) Current insights into fungal species diversity and perspective on naming the environmental DNA sequences of fungi. Mycology 10:126–140
Zamora JC, Svensson M, Kirschner R, Olariaga I, Ryman S et al (2018) Considerations and consequences of allowing DNA sequence data as types of fungal taxa. IMA Fungus 9:167–175
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415
Acknowledgments
This work benefitted from the sharing of expertise within the DFG priority program “Taxon-Omics: New Approaches for Discovering and Naming Biodiversity” (SPP 1991) funded by the Deutsche Forschungsgemeinschaft. The authors would like to thank Barbara Robbertse, Hermann Voglmayr and Andrey Yurkov for valuable comments and for pointing our important literature. We are grateful to Cathrin Spröer and Boyke Bunk (DSMZ) for providing the genome data of Hypoxylon fragiforme.
Funding
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Section Editor: Zhu-Liang Yang
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stadler, M., Lambert, C., Wibberg, D. et al. Intragenomic polymorphisms in the ITS region of high-quality genomes of the Hypoxylaceae (Xylariales, Ascomycota). Mycol Progress 19, 235–245 (2020). https://doi.org/10.1007/s11557-019-01552-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11557-019-01552-9