
Abstract
The renin-angiotensin system (RAS) plays an important role in the initiation and progression of tissue injuries in the cardiovascular and nervous systems. The detrimental actions of the AT1 receptor (AT1R) in hypertension and vascular injury, myocardial infarction and brain ischemia are well established. In the past twenty years, protective actions of the RAS, not only in the cardiovascular, but also in the nervous system, have been demonstrated. The so-called protective arm of the RAS includes AT2-receptors and Mas receptors (AT2R and MasR) and is characterized by effects different from and often opposing those of the AT1R. These include anti-inflammation, anti-fibrosis, anti-apoptosis and neuroregeneration that can counterbalance pathological processes and enable recovery from disease. The recent development of novel, small-molecule AT2R agonists offers a therapeutic potential in humans with a variety of clinical indications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Research on the AT2 receptor (AT2R) started about 25 years ago with the discovery of two receptors for angiotensin II (Ang II), the AT1R and the AT2R. The AT2R was finally cloned in 1993 by two independent groups [1]. Due to its unusual properties including lack of the classic features of G-protein coupled receptor signaling, the AT2R was described as an “enigmatic” seven transmembrane receptor [2]. Research teams working on AT2R had to cope with several problems including a very selective tissue expression pattern in adult life and unusual, sometimes contradictory, physiological properties. Thus, the AT2R became “a matter of love and hate” [3]. Nowadays, it is well accepted that the AT2R forms part of the “protective arm of RAS” with a great potential in tissue protection and regeneration [4]. The AT2R, while being only sparsely expressed in most healthy tissues, is strongly upregulated following tissue damage [5] such as vascular [6] and neuronal injury [7], myocardial infarction (MI) [8–10] and brain ischemia [11]. The selective stimulation of the AT2R with a recently available small-molecule ligand, compound 21, has not only greatly helped to elucidate the major molecular pathways involved in AT2R-mediated tissue protection, such as anti-inflammation, anti-fibrosis and anti-apoptosis [5], but has also revealed a great potential for pharmacological intervention in the above-mentioned diseases [4]. This review summarizes current knowledge about the beneficial features of AT2R stimulation with restriction to cardiac, vascular and neuronal disease.
AT2R-Mediated Signaling
AT2R stimulation activates at least three different classical signaling pathways: the cGMP/nitric oxide pathway [12, 13], protein phosphatases [14, 15] and phospholipase A2 signaling [16]. Since the tissue-protective properties of the AT2R are characterized mainly by regulation of inflammation, fibrosis and apoptosis [5], this review will focus on signaling pathways involved in these events.
Inflammation
Key molecular mechanisms involved in the AT2R-mediated anti-inflammatory actions are the inhibition of NF-κB activity [17, 18] and the reduction of oxidative stress [19, 20] (Fig. 1).

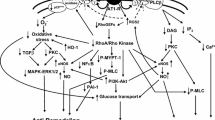
AT2R-mediated molecular pathways involved in tissue injury. The AT2R can be stimulated either by endogenous ligand Ang II or by exogenous agonists (CGP42112 or C21) resulting in activation or inhibition of multiple molecular pathways. This in turn leads to multiple cellular responses (grey boxes) including anti-inflammation, anti-fibrosis, modulation of apoptosis and enhanced neurite outgrowth. The MAPK-pathway and NFκB-pathway play central roles in the AT2R-mediated responses. Some of the molecular pathways are initiated indirectly via cross-talk with other receptors including Fas, TrkA and TrkB receptors by activation of the receptor or upregulation of ligand expression (e.g., FasL or BDNF). All pathways can be inhibited by the AT2R antagonist, PD123319, or by knocking-down the AT2R (not shown on the graph). Please notice that the edges in the pathway have undirected character (i.e., can represent either activation or inhibition). *growth factors are necessary for p85αPI3K activation. Abbreviations: AT 2R angiotensin AT2 receptor; Ang II angiotensin II; C21 compound 21 (non-peptide AT2R agonist); CGP42112 peptide AT2R agonist; PD123319 non-peptide AT2R antagonist; Trk tyrosine kinase receptor; BDNF brain-derived neurotrophic factor; Fas Fas cell surface death receptor; FasL Fas ligand; CYP2C/2J isoform of arachidonic acid-metabolizing cytochrome P450 enzyme; EET 11,12-epoxyeicosatrienoic acid; NFκB nuclear factor NF-kappa-B; TIMP tissue inhibitor of metalloproteinases; MMPs matrix metalloproteinases; PKCα protein kinase C, alpha; p21 RAS Ras family of small GTP binding proteins; ATIP/ATBP AT2 receptor-interacting protein (or AT2 receptor binding protein); SHP-1 protein-tyrosine phosphatase SHP-1; MMS2 methyl methanesulfonate sensitive 2; nNOS neuronal nitric oxide synthase; NO nitric oxide; cGMP cyclic guanosine monophosphate; PKG cGMP-dependent protein kinase; NADPH reduced form of nicotinamide adenine dinucleotide phosphate; ROS reactive oxygen species; MEK mitogen-activated protein kinase kinase; p42/p44 mapk p42/p44 mitogen-activated protein kinase; Bcl-2 apoptosis regulator Bcl-2; p38 mitogen-activated protein kinase p38; JNK JUN N-terminal kinase; PLZF promyelocytic leukemia zinc finger protein; p85αPI3K phosphatidylinositol 3-kinase regulatory subunit alpha
In 2010, Rompe and colleagues demonstrated anti-inflammatory effects of AT2R stimulation via inhibition of cytokine levels in vitro and in vivo, using the orally active, highly selective, non-peptide AT2R agonist, compound 21 (C21). In this study, the authors showed that C21 caused a dose-dependent reduction of TNFα-induced interleukin-6 (Il-6) levels in primary human and murine dermal fibroblasts. Moreover, this study elucidated the anti-inflammatory AT2R-coupled signaling demonstrating that this pathway involves activation of protein phosphatases, CYP-dependent epoxidation of arachidonic acid to EETs, and inhibition of NF-κB activity. With this mechanism, AT2R counteracts not only the pro-inflammatory effects of TNFα but also those mediated by the AT1R, which involve CYP-dependent hydroxylation of arachidonic acid to 20-HETE and induces NF-κB activation [17].
As mentioned above, AT2R-mediated anti-inflammation can be achieved through an inhibition of oxidative stress. In fact, McCarthy et al., found that stimulation of the AT2R caused a reduction of stroke-induced superoxide production. They showed an inverse relationship between superoxide production and AT2R expression and suggested that AT2R reduces oxidative stress related to ischemia [19].
It is well known that Ang II induces oxidative stress via AT1R activation. Pendergrass et al., showed that AT1R-induced oxidative stress involves NADPH oxidase activation to generate reactive oxygen species (ROS) [21]. On the other hand, Dandapat and colleagues hypothesized that the AT2R is anti-inflammatory via reduction of pro-oxidant signals by inhibiting NADPH oxidase expression and ROS generation leading to a downregulation of p38 and p44/42 MAP kinase phosphorylation [22].
It is also known that, during oxidative stress, the production of ROS exceeds the available antioxidant defense systems. As a consequence, increased ROS concentrations reduce the amount of bioactive NO [23]. Moreover, it has been speculated that the signaling cascades activated by NO, including cGMP-dependent protein kinase activation, may be involved in downstream activation of mitogen-activated protein kinases that are required for IL-10 production [24]. In agreement with this hypothesis, Dhande and colleagues have recently demonstrated anti-inflammatory actions of the AT2R via increased interleukin-10 (IL-10) production in an NO-dependent manner [25].
In addition to the above-mentioned effects, increasing evidence demonstrates new protective anti-inflammatory actions of the AT2R via cellular mechanisms [26–29].
Curato and colleagues studied the role of the AT2R in the regulation of the cellular immune response in the context of ischemic heart injury. The authors identified a cardioprotective T cell population, CD8+AT2R+, characterized by upregulated IL-10 and downregulated IL-2 and INF-γ expression compared with CD8+AT2R- T cells, which increased in response to ischemic cardiac injury. The authors demonstrated an immune-regulatory, cardioprotective action of the AT2R involving downregulation of the expression of proinflammatory cytokines and sustained IL-10 production, mediated, at least in part, via CD8+AT2R+ T cells [26].
Another recent study supports the immune regulatory role of the AT2R. Valero-Esquitino et al., evaluated the effects of AT2R stimulation on T cell differentiation in vitro. The authors concluded that AT2R stimulation induces an inhibition of T cells recruitment and modulation of the differentiation of naïve T cells into pro-inflammatory T helper (Th)1 and Th17 subsets while promoting differentiation into anti-inflammatory T regulatory cells [29].
Fibrosis
Several investigators have observed anti-inflammatory actions of the AT2R concomitantly with anti-fibrosis suggesting a possible cross-talk between the two mechanisms [22, 30].
Moreover, different studies demonstrate that the anti-fibrotic activity of the AT2R seems to be due to a regulation of matrix metalloproteinases (MMP) and their inhibitors (TIMP) [22, 31–33] that play a key role in the regulation of the metabolic balance of the extracellular matrix (Fig. 1).
An important mechanism of AT2R-mediated anti-fibrosis appears to be an increased expression and activity of TIMP1 and TIMP2 with consequent inhibition of MMP9 and MMP2; however, the exact signaling pathway is still unknown.
Jing et al., showed in rat vascular smooth muscle cells (VSMCs) expressing the AT2R in a tetracyclin-regulated system, that the AT2R counteracted the effects elicited by AT1R signaling and caused a marked reduction in MMP2 levels [32]. In agreement with this finding, Brassard et al., demonstrated an AT2R-mediated decrease in MMP2, counteracting the AT1R-mediated increase in MMP2 activity and decrease in TIMP2 activity [31].
In a study investigating the role of the AT2R in atherosclerotic plaque, Dandapat et al., found that the AT2R reduced the expression and activity of MMP2 and MMP9, and collagen accumulation. The authors hypothesized that, like the anti-inflammatory pathway, this mechanism involves downregulation of NADPH oxidase and subsequent ROS generation leading to an inhibition of p38 MAPKS and p44/42 MAPKs phosphorylation [22].
In accordance with previous studies, an AT2R-mediated regulation of MMPs has been recently confirmed in a setting of atherosclerosis by Kljajic and colleagues. In this study, stimulation of AT2R, with the peptide AT2R agonist, CGP42112, caused a reduction of MMP2 and MMP9 [34].
Supporting the regulatory role of the AT2R in the TIMP/MMPs axis, Lauer et al., recently reported that in a rat model of infarct-induced heart failure, stimulation of the AT2R with the selective, non-peptide AT2R agonist, C21 induced an activation of TIMP1 and subsequent inhibition of MMP9-mediated proteolysis. Moreover, AT2R stimulation led to a downregulation of TGF-β1 followed by decreased collagen accumulation [33].
Conversely, Rehman et al., found that AT2R stimulation with C21 reduced fibrosis in stroke-prone spontaneously hypertensive rats without any difference in MMP2 and MMP9 expression [28]. A possible explanation for these contrasting results is that, in the latter study, only the expression and not the activity of MMPs and TIMPs was evaluated.
Apoptosis
The role of the AT2R in apoptosis is controversial and seems to differ heavily depending on the experimental conditions (e.g., cell types, presence or absence of grow factors).
The AT2R regulates apoptosis via different pathways. AT2R stimulation can activate tyrosine phosphatases, such as mitogen-activated protein (MAP) kinase-phosphatase-1 (MKP-1) and inactivate MAP kinase extracellular signal-regulated kinase (ERK)1/2, resulting in dephosphorylation of Bcl-2 and upregulation of bax-induced pro-apoptotic effects [15, 35, 36] (Fig. 1).
Recently, we investigated the effect of AT2R stimulation by C21 on neuroprotection and neurite outgrowth and plasticity both in vitro and in vivo in a model of spinal cord injury in mice. This study reported improved neuronal survival with elevated expression of the neurotrophin, brain-derived neurotrophic factor (BDNF), and the neurotrophin receptors, TrkA and TrkB, as well as Bcl-2 that may link AT2R to anti-apoptosis [37].
Another pathway describing the AT2R-mediated regulation of apoptosis includes activation of caspases. Long-term stimulation of AT2R has been reported to cause ceramide generation leading to the activation of stress kinases and caspases and finally to apoptosis [38–40].
In fact, it has been reported in SMC that the AT2R mediates inducible transcriptional regulatory protein GATA-6 expression via activation of mitogen-activated protein kinase kinase (MEK)– ERK1/2 and c-Jun N-terminal kinase (JNK). GATA-6 in turn activates FasL promoter, FasL expression and consequently apoptosis via caspase 8 [41].
Conversely, different studies reported anti-apoptotic properties of the AT2R especially in pathological conditions. For instance, Kaschina et al., demonstrated that direct AT2R stimulation with C21 exerts beneficial effects after MI by anti-apoptotic and anti-inflammatory mechanisms. They showed that, stimulation of the AT2R engendered anti-apoptosis after MI by rescuing p38 MAPK and p44/42 MAPK expression and decreasing Fas-ligand and caspase-3 expression [30] (Fig. 1).
Growth
It has been demonstrated that AT2R stimulation is anti-hypertrophic via two main mechanisms: activation of protein tyrosine phosphatase SHP-1, which blocks growth factor signals [42–44] and interaction with an AT2R binding protein (ATBP = ATIP) [45, 46]. A higher level of complexity has been added to the growth effects of the AT2R after the discovery of the promyelocytic leukemia zinc finger protein (PLZF) as an interacting protein of the AT2R [47], see below (Fig. 1).
Neuronal Regeneration
The AT2R-related pathways in neuronal regeneration have been extensively studied in vitro in PC12W and NG108-15 cell-lines [48, 49]. AT2R-mediated neurite outgrowth seems to be a complex process that involves several pathways necessary for cytoskeleton rearrangements. In contrast to nerve growth factor, stimulation of the AT2R with Ang II leads to upregulation of beta-tubulin and MAP2 but downregulation of MAP1B [50] and neurofilament M [51] as shown in the PC12W cell-line. Signaling pathways involved in neurite outgrowth can be divided into four cascades that can be activated simultaneously or sequentially (Fig. 1). In the NG108-15 cell-line, stimulation of the AT2R leads to a decreased activity of PKCα followed by a decreased p21RAS activity [52]. The second pathway involves phosphorylation of p42/p44mapk mediated by Rap1/B-Raf [53]. However, this pathway may be initiated rather via the phosphorylation of tyrosine kinase receptor TrkA than directly downstream of the AT2R [54]. This is in agreement with our finding, where inhibition of tyrosine kinases with K252a abolished AT2R-mediated neurite outgrowth in primary cortical neurons [37]. However, the upregulation of TrkA, TrkB and BDNF suggest that autocrine and/or paracrine mechanisms may be involved as well. The third pathway is mediated via nitric oxide and cyclic GMP. Activation of nNOS via cGMP induces neurite elongation in NG108-15 cells [55]. Finally, the fourth cascade leading to neuronal differentiation involves ATIP (ATBP) [45, 46] and tyrosine phosphatase SHP-1 leading to transactivation of methyl methanesulfonate sensitive 2 enzyme (MMS2) [56, 57]. Other molecules such as PLZF or PPARγ may be involved in neurite outgrowth as well [56].
AT2R in Cardiovascular Injury
While angiotensin AT1 receptor antagonists (ARBs) are well recognized and commonly used in the treatment of many cardiovascular disorders, the potential cardiovascular protection offered by the AT2R per se is less known. Direct AT2R stimulation has no impact on blood pressure; however, it seems to afford tissue/organ protection via anti-inflammatory and anti-fibrotic actions [58]. As reviewed previously, the expression of the AT2R is increased in different cardiovascular disorders such as left ventricular hypertrophy (LVH), heart failure, cardiac fibrosis and atherosclerosis [5, 59]. Here we review recent studies highlighting the cardiovascular protective actions and their related mechanisms induced by the AT2R.
Cardioprotection
The pathophysiological role of AT1R stimulation in various kinds of heart disease is unanimously accepted. In particular, after myocardial infarction (MI), the AT1R contributes to tissue injury via inflammation and tissue remodeling. The AT2R, on the other hand, is widely recognized to induce actions counteracting those of the AT1R and this seems particularly true during cardiac injury [60].
Kaschina et al., performed the first study looking at the impact of direct AT2R stimulation post MI using the selective AT2R agonist, C21 [30]. In a model of MI following permanent coronary artery ligation in Wistar rats, acute treatment with C21 post-MI for one week improved cardiac function and decreased the infarct scar compared to vehicle treatment [30]. The underlying mechanisms seem to imply an anti-inflammatory as well as an anti-apoptotic action. Moreover, continuous AT2R stimulation can also afford long-term cardioprotection as highlighted recently in the late stage of MI six weeks after the acute event, when heart failure begins to develop [33]. C21 also improved arterial stiffness and reduced cardiac collagen content post-MI compared to the vehicle-treated group. The prevention of the cardiac remodeling by C21 seems to involve, among others, the TIMP1/MMP9 axis to reduce post-MI fibrosis [33].
In a model of MI with permanent ligature of the left anterior descending coronary artery, moderate cardio-selective overexpression of the AT2R also protected cardiac function and attenuated MI-induced cardiac hypertrophy [61]. The upregulation of the RAS components observed after MI in the left ventricle (mRNA) was attenuated in the presence of an overexpressed AT2R. Moreover, the MI-induced increase of collagen I was also attenuated [61]. In addition, other AT2R-mediated cardioprotective mechanisms, implying cardiac stem cells as well as CD8-positive T cells, have been identified [8, 26]. Indeed after MI, an increased AT2R expression in cardiac stem cells has been reported. Treatment of these cells with C21 attenuated the apoptosis of cardiomyocytes [8]. On the other hand, CD8+ AT2R+ T cells did not induce cytotoxicity to cardiomyocytes in opposition to the CD8+ AT2R- T cells [26]. They also exhibited an anti-inflammatory pattern (see above). Moreover, intra-myocardial transplantation of CD8+ AT2R+ T cells after MI reduced the infarct size [26].
In a hypertension-induced model of left ventricular hypertrophy (SHR-SP), administration of C21, alone or in combination with an ARB, led to anti-fibrotic and anti-hypertrophic effects on the heart independently of any blood pressure change [28]. In particular, C21 reduced the interstitial collagen I/III content in the left ventricle and the expression of cardiac genes associated with cardiac hypertrophy. As suggested by the authors, this may be due to antioxidant and anti-inflammatory actions induced by AT2R stimulation [28]. Another recent study investigated the contribution of the AT2R to cardiac remodeling, which is commonly observed with chronic ARB treatment in the aging hypertensive heart [62]. In this study, cardiac fibrosis of aged SHR was reduced by AT1R blockade, and this cardioprotective effect was reversed by blockade of the AT2R, thus, highlighting the anti-fibrotic effects of the AT2R in the heart.
In a model of pulmonary hypertension, a disease that often leads to right ventricular failure due to the increased vasculature resistance in the lung, C21 treatment slowed the progression of the associated pulmonary and cardiac disease [63, 64], as observed by the reduction of right ventricular systolic pressure, right ventricular hypertrophy and dP/dtmax. These protective effects were prevented when the AT2R antagonist, PD123319, was administered concomitantly with C21. Moreover, C21 also attenuated the increase in pro-inflammatory cytokines contributing to the protective mechanisms of the AT2R stimulation in pulmonary hypertension [63, 64].
Cardioprotection afforded by the AT2R, thus, seems to be mainly the result of anti-inflammatory and anti-fibrotic actions. AT2R-mediated anti-inflammation is controlled by mechanisms initiated at two levels: within cardiac as well as within immune cells. This may offer new therapeutic perspectives against cardiac injuries.
In contrast to the results summarized above, the AT2R has also been considered as a mediator of cardiac hypertrophy. Infusion of Ang II in mice induces cardiac hypertrophy. However, in AT2R knock-out mice, cardiac hypertrophy is absent suggesting that the AT2R mediates this effect in response to increased blood pressure [65]. Further investigations suggested that the cardiac hypertrophic response mediated by the AT2R may involve the transcription factor PLZF [47, 66]. PLZF is able to bind the AT2R and recent in vivo studies confirmed its involvement in AT2R-mediated cardiac hypertrophy [67]. Following Ang II infusion, mice developed a major cardiac hypertrophy and fibrosis that was completely absent in PLZF knockout mice or in Ang II-infused wild type mice treated by an AT2R antagonist [67]. However, it should be noted at this point that the actions of PLZF depend on the growth factor environment for activation. This would indicate that the AT2R by itself does not induce cardiac hypertrophy, but that the recruitment of PLZF to the AT2R may, under appropriate conditions, switch the AT2R-mediated cardiac actions from antihypertrophic to hypertrophic ones [66].
Vascular Protection
Using the L-NAME hypertension model that exhibits vascular remodeling with increased stiffness, Paulis et al., investigated the effect of chronic AT2R stimulation with C21 alone or in combination with the ARB olmesartan [68]. L-NAME treatment led to hypertension and hypertension-induced vascular changes characterized by an increased pulse wave velocity (PWV, an index of arterial stiffness), an increased aortic wall thickness, elastic modulus and aortic hydroxyproline content (marker for collagen deposition). C21 alone was able to partly prevent all these vascular injuries without any change in blood pressure. As expected, AT1R blockade with olmesartan completely prevented hypertension as well as the increase in PWV, aortic wall thickness and elastic modulus. Hydroxyproline accumulation in the aorta was slightly reduced by olmesartan, and the combination with C21 further reduced and even restored this parameter. Thus, the C21 + olmesartan combination led to a more pronounced stiffness reduction than each component alone, independently of any effect on blood pressure [68].
Similar mechanical/structural improvement of the vasculature after AT2R stimulation have also been reported in different vascular beds of SHR-SP (aorta, coronary arteries, and mesenteric arteries) [28]. Chronic administration of C21 alone or in combination with an ARB reduced vascular stiffness, collagen and fibronectin content. Moreover, C21 also improved the endothelial function as shown by the improved vasorelaxation to acetylcholine. These vasoprotective actions were observed concomitantly with a reduced superoxide production and monocyte/macrophage infiltration in the aorta [28].
The role of the AT2R has also been investigated in the pathological context of the Marfan syndrome, a disease that predisposes for aortic root aneurysm and aortic rupture. The authors crossed AT2R knock-out mice with a genetic mouse model of Marfan syndrome [69]. They observed a larger aortic root diameter in 2-month-old mice lacking the AT2R compared to mice with Marfan syndrome expressing the AT2R. This effect was maintained in one-year-old mice. In addition, they observed an increased death rate in AT2R KO mice with Marfan syndrome [69]. This highlights the key role of the AT2R in vascular remodeling since its absence accelerates the process of aortic aneurysm. The authors also investigated the contribution of indirect AT2R stimulation by using losartan in Marfan syndrome mice that express or not express the AT2R. They showed that AT2R signaling is needed to observe the protection afforded by losartan in this model [69].
In the pathological context of atherosclerosis, it has been previously suggested that the AT2R may play a role, since its absence increased the extent of the vascular lesions [70, 71]. This notion has been confirmed by a recent study using apoE-deficient transgenic mice overexpressing the AT2R [72]. The authors demonstrated that the AT2R-mediated anti-atherogenic actions were kinin / NO-dependent. Another recent study investigated the effect of direct AT2R stimulation using CGP42112 [34]. ApoE knock-out mice received a high fat diet for 16 weeks and were treated during the last four weeks with CGP42112. In this setting, AT2R stimulation showed vaso-protective and athero-protective effects with increased plaque stability. These effects were observed without any change on blood pressure and were associated with an improvement of the endothelial function and an increased NO bioavailability [34].
Although many studies have highlighted an AT2R-mediated vasoprotection, there are still some controversies. Opposed to the findings described above, a recent study showed that AT2R deficiency has no effect on either aortic aneurysms or atherosclerosis [73]. However the impact of the AT2R during tissue injuries should not be studied indirectly using AT2R blockade and/or AT2R knock-out animals but rather using direct stimulation of the AT2R.
AT2R in Neuronal Injury
This chapter focuses on the AT2R-mediated neuroprotection and neuroregeneration. Neuroprotection can be defined as a process that directly prevents necrotic or apoptotic neuronal cell death (primary neuroprotection) or affords protection of myelin, axons, and neurons by, e.g., anti-inflammation (secondary neuroprotection). Neuroregeneration can be defined as a complex process restoring the interrupted neuronal connectivity and resulting in functional recovery. The underlying mechanisms may involve cell renewal, synaptic plasticity, regrowth of severed axons and sprouting of non-damaged neurons compensating the loss of a neighboring tract [74].
Neuroprotection
Several studies have suggested a protective role of the AT2R in neuronal injury. The expression of AT2R is massively upregulated in neuronal damage as demonstrated in animal models of stroke and of sciatic or optic nerve crush [7, 11, 75]. Genetically modified animals lacking AT2R subjected to cerebral ischemia have larger infarcts as compared to the wild-type animals, mainly due to exacerbated inflammation and generation of ROS [76]. Subsequent studies demonstrated that stimulation of the AT2R exerts neuroprotection either directly, or by the anti-inflammatory activity (secondary neuroprotection).
The neuro-protective potential of AT2R-stimulation was demonstrated in vitro in N-methyl-d-aspartate (NMDA)-mediated cell death [77]. The survival of two differentiated neuronal cell lines, NG108-15 and N1E115, was reduced by treatment with NMDA in a dose-dependent manner. Incubation with 10-7 M Ang II inhibited neuronal apoptosis and suppressed the NMDA-mediated reduction of anti-apoptotic Bcl-2. This effect was inhibited by the co-treatment with PD123319, but not with losartan, suggesting an AT2R-mediated primary neuroprotection [77]. In primary cortical neurons isolated from mouse embryos, AT2R stimulation with CGP42112 significantly reduced cell death during glucose deprivation [78]. This effect was blocked by the co-treatment with PD123319 confirming the AT2R specificity.
Postischemic inflammation significantly contributes to ischemic brain injury. Therefore, anti-inflammatory therapeutic strategies can afford secondary neuroprotection in ischemic stroke [79]. Previously, we demonstrated that AT2R stimulation inhibits inflammatory response in vitro in CNS-derived cells. As shown in primary rat astrocytes exposed to LPS, the elevated expression of IL-6 and TNF-α was significantly reduced by treatment with C21 as compared to vehicle [80]. AT2R stimulation also inhibited microglial activation as shown by reduced nitric oxide release, and abolished cell migration [81].
In animal models of neuronal injury, both primary and secondary neuroprotection may significantly contribute to the beneficial effects mediated by the AT2R. Previously, we had shown that the AT1R blocker telmisartan, but not the ACE inhibitor ramipril, reduced infarct volume, neuronal apoptosis and inflammation in normotensive rats subjected to middle cerebral artery occlusion (MCAO) [82]. This observation may suggest that, in the presence of AT1R blockade, endogenous Ang II stimulates the unopposed AT2R and by this exerts neuroprotective actions.
The AT2R-mediated neuroprotection in cerebral ischemia was extensively studied by another group [19, 78, 83]. In spontaneously hypertensive rats, intracerebroventricular (i.c.v.) administration of CGP42112 for five days prior to endothelin-1 induced stroke reduced infarct volume, locomotor deficits and neuronal apoptosis as measured 72 hours after cerebral ischemia [19]. Importantly, stimulation of the AT2R even six hours after stroke induction was still effective, showing for the first time a neuroprotective effect of delayed AT2R stimulation after cerebral ischemia [83]. In C57Bl6 mice subjected to MCAO, systemic administration of the peptidic AT2R agonist, CGP42112, during the reperfusion reduced infarct and edema volume as well as improved functional outcome [78]. It is unclear, whether CGP42112 can enter brain parenchyma through the damaged blood-brain barrier. However, improved cerebral blood flow during the first ten minutes of reperfusion suggests, that vascular components may contribute to the observed beneficial effects of systemic drug administration [78].
Our group studied the AT2R-mediated neuroprotective mechanisms in MCAO in mice [84]. Systemic administration of C21 reduced post-stroke mortality, neurological deficits and neuronal apoptosis. This was accompanied by a reduced IL-6 expression and elevated BDNF in the infarcted brain areas [84]. These findings suggest that the neurotrophic pathway is involved in the observed neuroprotection by promoting neuronal survival.
Neuroregeneration
Neurite outgrowth has been attributed to the AT2R [85, 86] indicating a neuroregenerative potential [1]. In primary cortical neurons isolated from embryonic rat, Ang II promoted neurite outgrowth as estimated by the measurement of neurite length. Treatment with irbesartan enhanced the Ang II-mediated effect, and PD123319 completely abolished neurite outgrowth [11]. Similar effects were obtained when the AT2R was stimulated directly; either with CGP42112 [85] or with C21 [37, 87].
Lucius et al., demonstrated already in 1998 that AT2R stimulation engendered neurotrophic-like actions in the nervous system of adult animals [75]. Rats subjected to the optic nerve lesion and treated locally with Ang II showed outgrowth of axon bundles within the proximal optic nerve. This effect was AT2R-dependent since it could be inhibited by an AT2R-antagonist [75]. The neuroregenerative potential of AT2R was also demonstrated in a sciatic nerve crush model in rats [88]. Ang II not only increased axonal diameters and promoted re-myelination via AT2R but also accelerated functional recovery as shown by increased toe spread distance (parameter for motor-function) and improved the foot reflex withdrawal reaction (a parameter of sensomotoric function) [88]. Finally, in rats subjected to MCAO, increased AT2R immunoreactivity was associated with intense neurite outgrowth in ischemic striatum [11].
The AT2R may be implied in neuro-regenerative processes beyond the well-described neurite outgrowth. It has been shown that in AT2R-KO mice subjected to MCAO, impaired spatial memory was associated with decreased hippocampal neurogenesis as compared to wild-type animals [89]. This effect was larger in female animals, suggesting gender differences in AT2R-mediated neuroregeneration. In a related manner, the impact of AT2R-stimulation on renal function also differs between sexes in that direct AT2R-stimulation with C21 increased renal blood flow to a greater extent in female rats than in males [90].
We have recently evaluated the impact of the direct AT2R stimulation with C21 on neuroregeneration in an animal model of spinal cord injury (SCI) [37]. In mice subjected to SCI, treatment with C21 elevated the number of regenerating axons cranially and caudally from the lesioned area. The number of regenerating fibers positively correlated with improved locomotor performance indicating functionality of these fibers. Animals treated with C21 showed elevated immunoreactivity for the neutrophic receptor TrkB in the peri-lesional area. Parallel in vitro studies confirmed the importance of the BDNF/TrkB axis in AT2R-mediated neuroregeneration [37].
Apart from neuronal injury, the exogenous delivery of BDNF to the CNS may exert therapeutic effects in a variety of other neurological diseases including Alzheimer’s, Parkinson’s and Huntington’s disease, amyotrophic lateral sclerosis, Rett syndrome and vascular dementia [91, 92]. However, gene delivery in humans remains a challenge. On the other hand, a drug-induced increase of endogenous BDNF expression in the CNS seems to be a rational alternative for gene therapy. Small molecule AT2R agonists, such as C21, via the activation of neutrophic pathways, may provide a therapeutic option in the above-mentioned diseases.
Conclusions
During the twenty-five years following the discovery of the AT2R, our understanding of its protective role in tissue injury has greatly improved, but it is still far from complete. Development of pharmacological tools such as small-molecule ligands and the generation of the AT2R-KO animals have facilitated the elucidation of the major molecular pathways engaged by the AT2R. Results from various experimental disease models revealed the protective and regenerative potential of AT2R stimulation in cardiovascular and neuronal injury. Future research on the AT2R should concentrate on the complexity of molecular AT2R-related pathways and the cross-talk with other receptors and pathways in the context of tissue-protection. In addition to basic science, translational research will have to address the therapeutic potential of AT2R stimulation with available AT2R agonists in humans and define therapeutic cardiovascular indications and non-cardiovascular indications.
References
De Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–72.
Unger T. The angiotensin type 2 receptor: variations on an enigmatic theme. J Hypertens. 1999;17:1775–86.
Steckelings UM, Kaschina E, Unger T. The AT2 receptor–a matter of love and hate. Peptides. 2005;26:1401–9.
Steckelings UM, Unger T. Angiotensin II type 2 receptor agonists–where should they be applied? Expert Opin Investig Drugs. 2012;21:763–6.
Steckelings UM, Rompe F, Kaschina E, Namsolleck P, Grzesiak A, Funke-Kaiser H, et al. The past, present and future of angiotensin II type 2 receptor stimulation. J Renin-Angiotensin-Aldosterone Syst. 2010;11:67–73.
Nakajima M, Hutchinson HG, Fujinaga M, Hayashida W, Morishita R, Zhang L, et al. The angiotensin II type 2 (AT2) receptor antagonizes the growth effects of the AT1 receptor: gain-of-function study using gene transfer. Proc Natl Acad Sci U S A. 1995;92:10663–7.
Gallinat S, Yu M, Dorst A, Unger T, Herdegen T. Sciatic nerve transection evokes lasting up-regulation of angiotensin AT2 and AT1 receptor mRNA in adult rat dorsal root ganglia and sciatic nerves. Brain Res Mol Brain Res. 1998;57:111–22.
Altarche-Xifró W, Curato C, Kaschina E, Grzesiak A, Slavic S, Dong J, et al. Cardiac c-kit + AT2+ cell population is increased in response to ischemic injury and supports cardiomyocyte performance. Stem Cells. 2009;27:2488–97.
Busche S, Gallinat S, Bohle RM, Reinecke A, Seebeck J, Franke F, et al. Expression of angiotensin AT(1) and AT(2) receptors in adult rat cardiomyocytes after myocardial infarction. A single-cell reverse transcriptase-polymerase chain reaction study. Am J Pathol. 2000;157:605–11.
Nio Y, Matsubara H, Murasawa S, Kanasaki M, Inada M. Regulation of gene transcription of angiotensin II receptor subtypes in myocardial infarction. J Clin Invest. 1995;95:46–54.
Li J, Culman J, Hörtnagl H, Zhao Y, Gerova N, Timm M, et al. Angiotensin AT2 receptor protects against cerebral ischemia-induced neuronal injury. FASEB J. 2005;19:617–9.
Gohlke P, Pees C, Unger T. AT2 receptor stimulation increases aortic cyclic GMP in SHRSP by a kinin-dependent mechanism. Hypertension. 1998;31:349–55.
Walters PE, Gaspari TA, Widdop RE. Angiotensin-(1-7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension. 2005;45:960–6.
Fischer TA, Singh K, O’Hara DS, Kaye DM, Kelly RA. Role of AT1 and AT2 receptors in regulation of MAPKs and MKP-1 by ANG II in adult cardiac myocytes. Am J Physiol. 1998;275:H906–916.
Horiuchi M, Hayashida W, Kambe T, Yamada T, Dzau VJ. Angiotensin type 2 receptor dephosphorylates Bcl-2 by activating mitogen-activated protein kinase phosphatase-1 and induces apoptosis. J Biol Chem. 1997;272:19022–6.
Lokuta AJ, Cooper C, Gaa ST, Wang HE, Rogers TB. Angiotensin II stimulates the release of phospholipid-derived second messengers through multiple receptor subtypes in heart cells. J Biol Chem. 1994;269:4832–8.
Rompe F, Artuc M, Hallberg A, Alterman M, Ströder K, Thöne-Reineke C, et al. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor kappaB. Hypertension. 2010;55:924–31.
Wu L, Iwai M, Li Z, Shiuchi T, Min L-J, Cui T-X, et al. Regulation of inhibitory protein-kappaB and monocyte chemoattractant protein-1 by angiotensin II type 2 receptor-activated Src homology protein tyrosine phosphatase-1 in fetal vascular smooth muscle cells. Mol Endocrinol. 2004;18:666–78.
McCarthy CA, Vinh A, Callaway JK, Widdop RE. Angiotensin AT2 receptor stimulation causes neuroprotection in a conscious rat model of stroke. Stroke. 2009;40:1482–9.
Sumners C, Horiuchi M, Widdop RE, McCarthy C, Unger T, Steckelings UM. Protective arms of the renin-angiotensin-system in neurological disease. Clin Exp Pharmacol Physiol. 2013;40:580–8.
Pendergrass KD, Gwathmey TM, Michalek RD, Grayson JM, Chappell MC. The angiotensin II-AT1 receptor stimulates reactive oxygen species within the cell nucleus. Biochem Biophys Res Commun. 2009;384:149–54.
Dandapat A, Hu CP, Chen J, Liu Y, Khan JA, Remeo F, et al. Over-expression of angiotensin II type 2 receptor (agtr2) decreases collagen accumulation in atherosclerotic plaque. Biochem Biophys Res Commun. 2008;366:871–7.
Förstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010;459:923–39.
Li Z, Zhang G, Feil R, Han J, Du X. Sequential activation of p38 and ERK pathways by cGMP-dependent protein kinase leading to activation of the platelet integrin alphaIIb beta3. Blood. 2006;107:965–72.
Dhande I, Ali Q, Hussain T. Proximal tubule angiotensin AT2 receptors mediate an anti-inflammatory response via interleukin-10: role in renoprotection in obese rats. Hypertension. 2013;61:1218–26.
Curato C, Slavic S, Dong J, Skorska A, Altarche-Xifró W, Miteva K, et al. Identification of noncytotoxic and IL-10-producing CD8 + AT2R + T cell population in response to ischemic heart injury. J Immunol. 2010;185:6286–93.
Gelosa P, Pignieri A, Fändriks L, de Gasparo M, Hallberg A, Banfi C, et al. Stimulation of AT2 receptor exerts beneficial effects in stroke-prone rats: focus on renal damage. J Hypertens. 2009;27:2444–51.
Rehman A, Leibowitz A, Yamamoto N, Rautureau Y, Paradis P, Schiffrin EL. Angiotensin type 2 receptor agonist compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension. 2012;59:291–9.
Valero-Esquitino V, Lucht K, Cirere-Salinas D, Stubbe T, Pascual A, Curato C, et al. Direct angiotensin type 2 receptor stimulation attenuates T-lymphocyte infiltration to the central nervous system and modulates T-cell differentiation. 17th Annual Meeting of the European Council for Cardiovascular Research (ECCR). La Colle sur Loup – Nice, France; 2013.
Kaschina E, Grzesiak A, Li J, Foryst-Ludwig A, Timm M, Rompe F, et al. Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation. 2008;118:2523–32.
Brassard P, Amiri F, Schiffrin EL. Combined angiotensin II type 1 and type 2 receptor blockade on vascular remodeling and matrix metalloproteinases in resistance arteries. Hypertension. 2005;46:598–606.
Jing T, Wang H, Srivenugopal KS, He G, Liu J, Miao L, et al. Conditional expression of type 2 angiotensin II receptor in rat vascular smooth muscle cells reveals the interplay of the angiotensin system in matrix metalloproteinase 2 expression and vascular remodeling. Int J Mol Med. 2009;24:103–10.
Lauer D, Slavic S, Sommerfeld M, Thöne-Reineke C, Sharkovska Y, Hallberg A, et al. AT2 receptor agonism regulates TIMP1/MMP9 axis in the heart preventing cardiac fibrosis and improving heart function after experimental myocardial infarction. Hypertension. 2013.
Kljajic ST, Widdop RE, Vinh A, Welungoda I, Bosnyak S, Jones ES, et al. Direct AT2 receptor stimulation is athero-protective and stabilizes plaque in Apolipoprotein E-deficient mice. Int J Cardiol. 2013;169:281–7.
Horiuchi M, Akishita M, Dzau VJ. Molecular and cellular mechanism of angiotensin II-mediated apoptosis. Endocr Res. 1998;24:307–14.
Savoia C, D’Agostino M, Lauri F, Volpe M. Angiotensin type 2 receptor in hypertensive cardiovascular disease. Curr Opin Nephrol Hypertens. 2011;20:125–32.
Namsolleck P, Boato F, Schwengel K, Paulis L, Matho KS, Geurts N, et al. AT2-receptor stimulation enhances axonal plasticity after spinal cord injury by upregulating BDNF expression. Neurobiol Dis. 2013;51:177–91.
Gallinat S, Busche S, Schütze S, Krönke M, Unger T. AT2 receptor stimulation induces generation of ceramides in PC12W cells. FEBS Lett. 1999;443:75–9.
Kacimi R, Gerdes AM. Alterations in G protein and MAP kinase signaling pathways during cardiac remodeling in hypertension and heart failure. Hypertension. 2003;41:968–77.
Wang X, Phillips MI, Mehta JL. LOX-1 and angiotensin receptors, and their interplay. Cardiovasc Drugs Ther. 2011;25:401–17.
Tan NY, Li J-M, Stocker R, Khachigian LM. Angiotensin II-inducible smooth muscle cell apoptosis involves the angiotensin II type 2 receptor, GATA-6 activation, and FasL-Fas engagement. Circ Res. 2009;105:422–30.
Bedecs K, Elbaz N, Sutren M, Masson M, Susini C, Strosberg AD, et al. Angiotensin II type 2 receptors mediate inhibition of mitogen-activated protein kinase cascade and functional activation of SHP-1 tyrosine phosphatase. Biochem J. 1997;325(Pt 2):449–54.
Cui T, Nakagami H, Iwai M, Takeda Y, Shiuchi T, Daviet L, et al. Pivotal role of tyrosine phosphatase SHP-1 in AT2 receptor-mediated apoptosis in rat fetal vascular smooth muscle cell. Cardiovasc Res. 2001;49:863–71.
Horiuchi M, Hayashida W, Akishita M, Tamura K, Daviet L, Lehtonen JY, et al. Stimulation of different subtypes of angiotensin II receptors, AT1 and AT2 receptors, regulates STAT activation by negative crosstalk. Circ Res. 1999;84:876–82.
Nouet S, Amzallag N, Li J-M, Louis S, Seitz I, Cui T-X, et al. Trans-inactivation of receptor tyrosine kinases by novel angiotensin II AT2 receptor-interacting protein, ATIP. J Biol Chem. 2004;279:28989–97.
Wruck CJ, Funke-Kaiser H, Pufe T, Kusserow H, Menk M, Schefe JH, et al. Regulation of transport of the angiotensin AT2 receptor by a novel membrane-associated Golgi protein. Arterioscler Thromb Vasc Biol. 2005;25:57–64.
Senbonmatsu T, Saito T, Landon EJ, Watanabe O, Price Jr E, Roberts RL, et al. A novel angiotensin II type 2 receptor signaling pathway: possible role in cardiac hypertrophy. EMBO J. 2003;22:6471–82.
Guimond M-O, Gallo-Payet N. How does angiotensin AT(2) receptor activation help neuronal differentiation and improve neuronal pathological situations? Front Endocrinol (Lausanne). 2012;3:164.
Guimond M-O, Gallo-Payet N. The angiotensin II type 2 receptor in brain functions: an update. Int J Hypertens. 2012;2012:351758.
Stroth U, Meffert S, Gallinat S, Unger T. Angiotensin II and NGF differentially influence microtubule proteins in PC12W cells: role of the AT2 receptor. Brain Res Mol Brain Res. 1998;53:187–95.
Gallinat S, Csikos T, Meffert S, Herdegen T, Stoll M, Unger T. The angiotensin AT2 receptor down-regulates neurofilament M in PC12W cells. Neurosci Lett. 1997;227:29–32.
Beaudry H, Gendron L, Guimond M-O, Payet MD, Gallo-Payet N. Involvement of protein kinase C alpha (PKC alpha) in the early action of angiotensin II type 2 (AT2) effects on neurite outgrowth in NG108-15 cells: AT2-receptor inhibits PKC alpha and p21ras activity. Endocrinology. 2006;147:4263–72.
Gendron L, Laflamme L, Rivard N, Asselin C, Payet MD, Gallo-Payet N. Signals from the AT2 (angiotensin type 2) receptor of angiotensin II inhibit p21ras and activate MAPK (mitogen-activated protein kinase) to induce morphological neuronal differentiation in NG108-15 cells. Mol Endocrinol. 1999;13:1615–26.
Plouffe B, Guimond M-O, Beaudry H, Gallo-Payet N. Role of tyrosine kinase receptors in angiotensin II AT2 receptor signaling: involvement in neurite outgrowth and in p42/p44mapk activation in NG108-15 cells. Endocrinology. 2006;147:4646–54.
Gendron L, Côté F, Payet MD, Gallo-Payet N. Nitric oxide and cyclic GMP are involved in angiotensin II AT(2) receptor effects on neurite outgrowth in NG108-15 cells. Neuroendocrinology. 2002;75:70–81.
Horiuchi M, Iwanami J, Mogi M. Regulation of angiotensin II receptors beyond the classical pathway. Clin Sci. 2012;123:193–203.
Li J-M, Mogi M, Tsukuda K, Tomochika H, Iwanami J, Min L-J, et al. Angiotensin II-induced neural differentiation via angiotensin II type 2 (AT2) receptor-MMS2 cascade involving interaction between AT2 receptor-interacting protein and Src homology 2 domain-containing protein-tyrosine phosphatase 1. Mol Endocrinol. 2007;21:499–511.
Foulquier S, Steckelings UM, Unger T. Impact of the AT(2) receptor agonist C21 on blood pressure and beyond. Curr Hypertens Rep. 2012;14:403–9.
Jones ES, Vinh A, McCarthy CA, Gaspari TA, Widdop RE. AT2 receptors: functional relevance in cardiovascular disease. Pharmacol Ther. 2008;120:292–316.
Foulquier S, Steckelings UM, Unger T. Perspective: a tale of two receptors. Nature. 2013;493:S9.
Qi Y, Li H, Shenoy V, Li Q, Wong F, Zhang L, et al. Moderate cardiac-selective overexpression of angiotensin II type 2 receptor protects cardiac functions from ischaemic injury. Exp Physiol. 2012;97:89–101.
Jones ES, Black MJ, Widdop RE. Influence of angiotensin II subtype 2 receptor (AT(2)R) antagonist, PD123319, on cardiovascular remodelling of aged spontaneously hypertensive rats during chronic angiotensin II subtype 1 receptor (AT(1)R) blockade. Int J Hypertens. 2012;2012:543062.
Bruce E, Shenoy V, Francis J, Steckelings U, Unger T, Sumners C, et al. AT2 receptor agonist, compound 21, attenuates pulmonary hypertension and associated cardiac pathophysiology via the vasoprotective ACE2/Ang-(1-7)/Mas axis. High Blood Pressure Research (HBPR) 2012 Scientific Sessions. Washington, USA; 2012.
Bruce E, Shenoy V, Francis J, Steckelings U, Unger T, Sumners C, et al. Stimulation of angiotensin type 2 receptor as a potential therapy for pulmonary hypertension. High Blood Pressure Research (HBPR) 2012 Scientific Sessions. Washington, USA; 2012.
Senbonmatsu T, Ichihara S, Price Jr E, Gaffney FA, Inagami T. Evidence for angiotensin II type 2 receptor-mediated cardiac myocyte enlargement during in vivo pressure overload. J Clin Invest. 2000;106:R25–29.
Funke-Kaiser H, Reinemund J, Steckelings UM, Unger T. Adapter proteins and promoter regulation of the angiotensin AT2 receptor–implications for cardiac pathophysiology. J Renin-Angiotensin-Aldosterone Syst. 2010;11:7–17.
Wang N, Frank GD, Ding R, Tan Z, Rachakonda A, Pandolfi PP, et al. Promyelocytic leukemia zinc finger protein activates GATA4 transcription and mediates cardiac hypertrophic signaling from angiotensin II receptor 2. PLoS ONE. 2012;7:e35632.
Paulis L, Becker STR, Lucht K, Schwengel K, Slavic S, Kaschina E, et al. Direct angiotensin II type 2 receptor stimulation in Nω-nitro-L-arginine-methyl ester-induced hypertension: the effect on pulse wave velocity and aortic remodeling. Hypertension. 2012;59:485–92.
Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, et al. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361–5.
Iwai M, Chen R, Li Z, Shiuchi T, Suzuki J, Ide A, et al. Deletion of angiotensin II type 2 receptor exaggerated atherosclerosis in apolipoprotein E-null mice. Circulation. 2005;112:1636–43.
Sales VL, Sukhova GK, Lopez-Ilasaca MA, Libby P, Dzau VJ, Pratt RE. Angiotensin type 2 receptor is expressed in murine atherosclerotic lesions and modulates lesion evolution. Circulation. 2005;112:3328–36.
Takata H, Yamada H, Kawahito H, Kishida S, Irie D, Kato T, et al. Vascular angiotensin II type 2 receptor attenuates atherosclerosis via a kinin/NO-dependent mechanism. J Renin-Angiotensin-Aldosterone Syst. 2013.
Daugherty A, Rateri DL, Howatt DA, Charnigo R, Cassis LA. PD123319 augments angiotensin II-induced abdominal aortic aneurysms through an AT2 receptor-independent mechanism. PLoS ONE. 2013;8:e61849.
Martino G, Pluchino S, Bonfanti L, Schwartz M. Brain regeneration in physiology and pathology: the immune signature driving therapeutic plasticity of neural stem cells. Physiol Rev. 2011;91:1281–304.
Lucius R, Gallinat S, Rosenstiel P, Herdegen T, Sievers J, Unger T. The angiotensin II type 2 (AT2) receptor promotes axonal regeneration in the optic nerve of adult rats. J Exp Med. 1998;188:661–70.
Iwai M, Liu H-W, Chen R, Ide A, Okamoto S, Hata R, et al. Possible inhibition of focal cerebral ischemia by angiotensin II type 2 receptor stimulation. Circulation. 2004;110:843–8.
Schelman WR, Andres R, Ferguson P, Orr B, Kang E, Weyhenmeyer JA. Angiotensin II attenuates NMDA receptor-mediated neuronal cell death and prevents the associated reduction in Bcl-2 expression. Brain Res Mol Brain Res. 2004;128:20–9.
Lee S, Brait VH, Arumugam TV, Evans MA, Kim HA, Widdop RE, et al. Neuroprotective effect of an angiotensin receptor type 2 agonist following cerebral ischemia in vitro and in vivo. Exp Transl Stroke Med. 2012;4:16.
Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–7.
Steckelings UM, Larhed M, Hallberg A, Widdop RE, Jones ES, Wallinder C, et al. Non-peptide AT2-receptor agonists. Curr Opin Pharmacol. 2011;11:187–92.
Namsolleck P, Valero-Esquitino V, Lucht K, Unger T, Steckelings U. Inhibition of microglia activation and migration by direct AT2-receptor stimulation. 24th Scientific Meeting of the International Society of Hypertension. Sydney, Australia; 2012.
Thoene-Reineke C, Rumschüssel K, Schmerbach K, Krikov M, Wengenmayer C, Godes M, et al. Prevention and intervention studies with telmisartan, ramipril and their combination in different rat stroke models. PLoS ONE. 2011;6:e23646.
McCarthy CA, Vinh A, Broughton BRS, Sobey CG, Callaway JK, Widdop RE. Angiotensin II type 2 receptor stimulation initiated after stroke causes neuroprotection in conscious rats. Hypertension. 2012;60:1531–7.
Schwengel K, Thoene-Reineke C, Lucht K, Namsolleck P, Horiuchi M, Iwai M, et al. Direct AT2-receptor stimulation improves survival and neurological outcome after experimental stroke (MCAO) in mice. 34. Wissenschaftlicher Kongress “Hypertonie 2010” der Deutschen Hochdruckliga e. V. DHL® – Deutsche Hypertonie Gesellschaft. Berlin, Germany; 2010.
Laflamme L, Gasparo M, Gallo JM, Payet MD, Gallo-Payet N. Angiotensin II induction of neurite outgrowth by AT2 receptors in NG108-15 cells. Effect counteracted by the AT1 receptors. J Biol Chem. 1996;271:22729–35.
Meffert S, Stoll M, Steckelings UM, Bottari SP, Unger T. The angiotensin II AT2 receptor inhibits proliferation and promotes differentiation in PC12W cells. Mol Cell Endocrinol. 1996;122:59–67.
Wan Y, Wallinder C, Plouffe B, Beaudry H, Mahalingam AK, Wu X, et al. Design, synthesis, and biological evaluation of the first selective nonpeptide AT2 receptor agonist. J Med Chem. 2004;47:5995–6008.
Reinecke K, Lucius R, Reinecke A, Rickert U, Herdegen T, Unger T. Angiotensin II accelerates functional recovery in the rat sciatic nerve in vivo: role of the AT2 receptor and the transcription factor NF-kappaB. FASEB J. 2003;17:2094–6.
Sakata A, Mogi M, Iwanami J, Tsukuda K, Min L-J, Fujita T, et al. Sex-different effect of angiotensin II type 2 receptor on ischemic brain injury and cognitive function. Brain Res. 2009;1300:14–23.
Hilliard LM, Jones ES, Steckelings UM, Unger T, Widdop RE, Denton KM. Sex-specific influence of angiotensin type 2 receptor stimulation on renal function: a novel therapeutic target for hypertension. Hypertension. 2012;59:409–14.
Balaratnasingam S, Janca A. Brain derived neurotrophic factor: a novel neurotrophin involved in psychiatric and neurological disorders. Pharmacol Ther. 2012;134:116–24.
Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10:209–19.
Compliance with Ethics Guidelines
Conflict of Interest
Pawel Namsolleck, Chiara Recarti, Sébastien Foulquier, and Thomas Unger declare that they have no conflict of interest.
Ulrike Muscha Steckelings has received modest research support (short term fellowship; free supply with drug) from Vicore Pharma.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Pawel Namsolleck and Chiara Recarti contributed equally to this work.
This article is part of the Topical Collection on Mediators, Mechanisms, and Pathways in Tissue Injury
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Namsolleck, P., Recarti, C., Foulquier, S. et al. AT2 Receptor and Tissue Injury: Therapeutic Implications. Curr Hypertens Rep 16, 416 (2014). https://doi.org/10.1007/s11906-013-0416-6
Published:
DOI: https://doi.org/10.1007/s11906-013-0416-6