
Abstract
Introduction
The lack of large panels of validated antibodies, tissue handling variability, and intratumoral heterogeneity potentially hamper comprehensive study of the functional proteome in non-microdissected solid tumors. The purpose of this study was to address these concerns and to demonstrate clinical utility for the functional analysis of proteins in non-microdissected breast tumors using reverse phase protein arrays (RPPA).
Methods
Herein, 82 antibodies that recognize kinase and steroid signaling proteins and effectors were validated for RPPA. Intraslide and interslide coefficients of variability were <15%. Multiple sites in non-microdissected breast tumors were analyzed using RPPA after intervals of up to 24 h on the benchtop at room temperature following surgical resection.
Results
Twenty-one of 82 total and phosphoproteins demonstrated time-dependent instability at room temperature with most variability occurring at later time points between 6 and 24 h. However, the 82-protein functional proteomic “fingerprint” was robust in most tumors even when maintained at room temperature for 24 h before freezing. In repeat samples from each tumor, intratumoral protein levels were markedly less variable than intertumoral levels. Indeed, an independent analysis of prognostic biomarkers in tissue from multiple tumor sites accurately and reproducibly predicted patient outcomes. Significant correlations were observed between RPPA and immunohistochemistry. However, RPPA demonstrated a superior dynamic range. Classification of 128 breast cancers using RPPA identified six subgroups with markedly different patient outcomes that demonstrated a significant correlation with breast cancer subtypes identified by transcriptional profiling.
Conclusion
Thus, the robustness of RPPA and stability of the functional proteomic “fingerprint” facilitate the study of the functional proteome in non-microdissected breast tumors.
Similar content being viewed by others



Introduction
Much progress has been made in genomic breast cancer classification [1–10]. However, as mRNA levels may not translate precisely into protein function due to posttranslational modifications and other factors, mRNA profiling may not be able to fully characterize the functional proteome. Proteins are the ultimate effectors of cellular outcomes. Thus, the lack of a validated, practical, moderate- to high-throughput, quantitative functional proteomics platform applicable to patient tumors remains a key barrier to the identification of solid tumor biomarkers.
Traditional protein assays including enzyme-linked immunosorbent assay, immunoblotting, and immunohistochemistry (IHC) can assess only small numbers of proteins and are expensive, semiquantitative, and require large amounts of material. Although mass spectroscopy is promising, it is not currently sufficiently robust or cost-effective for clinical implementation.
By providing high-throughput, low-cost, objective analysis of multiple proteins in small amounts of sample, reverse phase protein lysate arrays (RPPA) offer an emerging approach to comprehensive quantitative profiling of the levels and function of multiple proteins in tumors and have the potential to map protein levels and function in intracellular pathways in a comprehensive, convenient, and sensitive manner [11–23].
Although RPPA has been extensively validated for in vitro analyses [11–23], several obstacles remain to be addressed prior to its routine application to non-microdissected human breast tumors. These potential obstacles include:
-
1.
Antibody validation: The validation of a large panel of antibodies is required since RPPA is essentially a high-throughput “dot-blot” and therefore is unable to distinguish between specific and off-target antibody–protein interactions.
-
2.
Variability in tissue handling prior to freezing: Variability in tissue handling may result in unpredictable changes in the levels and posttranslational modification (e.g., phosphorylation) of proteins.
-
3.
Intratumoral heterogeneity: RPPA does not provide information concerning spatial organization. Intratumoral heterogeneity in protein expression and activation thus poses a potential challenge.
These problems could clearly impair the integrity of data derived from the study of the functional proteome in human breast tumors using RPPA. Thus, the goals of this study were:
-
a.
to address these obstacles to the successful application of RPPA to the study of non-microdissected human breast tumors
-
b.
to investigate reproducibility and the correlation of RPPA with standard IHC in human breast tumors
-
c.
to evaluate the potential clinical utility of this approach for the analysis of the breast cancer functional proteome.
Methods
Antibodies and Reagents
Eighty-two antibodies, chosen because of the importance of the detected proteins to breast carcinogenesis [24–44], were used (Table 1). The AKT inhibitor perifosine was obtained from Keryx Pharmaceuticals (New York, NY). The phosphatidylinositol-3 kinase inhibitor LY294002 was obtained from Calbiochem (San Diego, CA). Rapamycin was obtained from Cell Signaling, Inc. (Danvers, MA). Epidermal growth factor (EGF) was purchased from R&D Systems, Inc. (Minneapolis, MN).
Cell Lines and Tumor Samples
The MDAMB231, MDAMB468, MCF7, T47D, ZR75-1, OVCAR3, and SKOV3 cell lines were obtained from the American Type Culture Collection (Manassas, VA). Protein lysates of 52 breast cancer cell lines were prepared as previously described [29]. The human tumor sets used herein were obtained using Institutional Review Board-approved protocols and are as follows:
-
1.
Set A (128 tumors): For comparison of RPPA with transcriptional profiling (e.g., for protein–mRNA correlations), 128 stored primary breast tumors were obtained from patients treated in the Danish DBCG82 b and c studies [45] (Table 2).
Table 2 Clinical details of human breast tumors utilized in this study -
2.
Set B (ten tumors): For the studies of intratumoral heterogeneity and total and phosphoprotein stability, a prospective study was undertaken to collect primary breast tissue at breast surgery in ten patients with breast cancer under an Institutional Review Board (IRB)-approved protocol. Each tumor was sectioned with assistance from a breast pathologist and immediately snap frozen (three pieces) or left at room temperature in closed eppendorf tubes without any added buffer for 0.5/1/2/4/6/24 h (1 piece/time point) prior to freezing (−85°C). Protein was extracted from each piece of tumor without thawing.
-
3.
Set C (95 tumors): Ninety-five stored primary breast tumors were obtained from the breast tumor frozen tissue bank at M. D. Anderson Cancer Center under an IRB-approved protocol (Table 2). Protein was extracted from these 95 tumors, including from two independent sections (“biologic replicates”) derived from 49 of the 95 tumors.
Note that Table 2 does not show the clinical data for Set B since the clinical data for this set were not utilized in this study.
MDAMB231 and MDAMB435 breast cancer xenografts were assessed for total and phosphoprotein stability using the same approach as with human tumor set B above. After animal sacrifice, the xenograft tumors were sectioned and immediately snap frozen or left at room temperature in closed eppendorf tubes without any added buffer for 0.5/1/2/4/6 h (1 piece/time point) prior to freezing (−85°C). As with the human tumors, protein was extracted from each piece of tumor without thawing.
Lysate Preparation and Array Spotting
Breast cancer cell lines were cultured in their optimal medium (recommended by the American Type Culture Collection) with 5% fetal bovine serum in 6-well plates. For experiments involving cell line treatment or stimulation, the cells were starved overnight and treated with inhibitor with or without EGF stimulation (20 ng/ml for 10 min) where indicated. Cells were then washed twice with PBS and lysed in ice-cold lysis buffer (1% Triton X-100, 50 mm HEPES, pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM Na pyrophosphate, 1 mM Na3VO4, 10% glycerol) supplemented with proteinase inhibitors (Roche Applied Science, Indianapolis, IN). Cellular protein concentration was determined by bicinchoninic acid reaction (Pierce, Rockford, IL). Frozen tumor tissue (≤10 mg) was homogenized after macrodissection without microdissection in lysis buffer at 40 mg/ml by PowerGen polytron homogenizer (Fisher Scientific, Hampton, NH) and concentration of the protein lysates corrected to 1.33 mg/ml. After centrifugation, post-nuclear detergent lysates (three parts) were boiled with a solution (one part) of 4XSDS (90%)/B mercaptoethanol (10%). Five serial 2-fold dilutions were performed in lysis buffer containing 1% SDS (dilution buffer). The diluted lysates were spotted on nitrocellulose-coated FAST slides (Whatman, Schleicher & Schuell BioScience, Inc., Keene, NH) by a robotic GeneTAC (Genomic Solutions, Inc., Ann Arbor, MI) G3 arrayer or an Aushon Biosystems (Burlington, MA) 2,470 arrayer.
Antibody Probing and Signal Detection of RPPA
The DAKO (Carpinteria, CA) catalyzed signal amplification system was used for antibody blotting. Each slide was incubated with a primary antibody (Table 1) in the appropriate dilution. The signal was captured by biotin-conjugated secondary antibody and amplified by tyramide deposition. The analyte was detected by avidin-conjugated peroxidase reactive to its substrate chromogen diaminobenzidine. Subsequently, the slides were individually scanned, analyzed, and quantitated using MicroVigene software (VigeneTech Inc., North Billerica, MA). This software provides automated spot identification, background correction, and individual spot intensity determination (expressed in logarithmic units).
Immunoblotting
Lysates were prepared as described above. Proteins were resolved in SDS-PAGE and transferred to PVDF membranes. The membranes were blocked by 5% BSA and hybridized with different primary antibodies as indicated. Signals were captured by horse radish peroxidase-conjugated secondary antibody and visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ). The abundance of immunoreactive protein was quantified using a computing densitometer (NIH Imaging) and presented as arbitrary units of density.
Transcriptional Profiling
Expression data for Set A (Table 2) were generated at the Norwegian Radium Hospital using the Applied Biosystems Human Genome Survey Microarray version 2.0 consisting of whole genome arrays spotted with 32,878 probes covering 29,098 genes. Signal was detected by chemiluminescence in a single channel system. Details can be found at the website: http://www3.appliedbiosystems.com/cms/groups/mcb_marketing/documents/generaldocuments/cms_040420.pdf
Statistical Analysis
R and NCSS (Kaysville, Utah) software were used. The spot signal intensity data from MicroVigene are processed by the R package SuperCurve (version 1.01) [18], available at “http://bioinformatics.mdanderson.org/OOMPA.” A fitted curve (called “supercurve”) is plotted with the signal intensities on the y-axis and the relative log2 concentration of each protein on the x-axis using the non-parametric, monotone increasing B-spline model (Fig. 1) [18]. The protein concentrations are derived from supercurve for each sample lysate on the slide by curve fitting and then normalized by median polish. Each total and phosphoprotein measurement is subsequently corrected for loading using the average expression of all measured proteins. For the study of total and phosphoprotein stability, the expression of each protein in the three immediately frozen replicate sections of ten primary breast tumors was averaged, measurements at six later time points (0.5/1/2/4/6/24 h) were treated as separate observations, and the effects of time to freezing on total and phosphoprotein expression were tested using an analysis of variance (ANOVA) model. The effects of intratumoral and intertumoral variability on protein expression were tested by applying ANOVA models to RPPA data derived from the three immediately frozen replicate sections of ten breast tumors. To estimate disease-free survival (DFS), the time to any breast cancer relapse or any death (whichever came first) since diagnosis was computed. DFS time was censored at last follow-up if neither relapse nor death occurred. To estimate distant metastasis-free survival, the time to distant breast cancer metastasis since diagnosis was computed. Distant metastasis-free survival time was censored at last follow-up or death if no distant metastasis was detected. To estimate overall survival (OS), the time to death from any cause since diagnosis was computed. OS time was censored at last follow-up if death had not occurred. Survival probabilities were estimated using Kaplan–Meier’s product limit method.


a Akt and b Aktp473 antibody validation for reverse phase protein array (RPPA). MDAMB468 (red), ZR75-1 (black) and T47D (blue) cells were left untreated followed by no stimulation (control) or by stimulation with epidermal growth factor (EGF) or were treated with LY294002 (phosphatidylinositol-3-kinase (PI3K) inhibitor), perifosine (Akt inhibitor), rapamycin (mTOR inhibitor), or ultraviolet (UV) irradiation and then stimulated with epidermal growth factor (EGF) in the case of treatment with the three inhibitors. Lysates were then probed with antibody to total Akt (a) or to phosphorylated Akt at serine 473 (Aktp473, b) by RPPA in triplicate (panels A–C) and by western blotting (panel D) and the derived signals for total Akt and for Aktp473 were quantified and correlated (panel E in a and b). For RPPA, each lysate was arrayed in five serial 2-fold dilutions on nitrocellulose slides (with increasing dilution from left to right on each slide for each lysate as shown in panel B). A control spot (a mixed cell line lysate) was placed at the end of each sample lysate’s five serial 2-fold dilution series to give six spots. Four samples are arrayed in this fashion in each grid of 24 spots on the nitrocellulose slides shown. The correlation coefficients between signals derived using RPPA and western blotting for Akt and Aktp473 were 0.897 and 0.93, respectively (panel E in a and b). These correlation coefficients were based on 18 data points as shown and indicate valid antibodies for RPPA. Panel A in a and b demonstrates the process of curve fitting for RPPA that is applied by the R package SuperCurve (version 1.01)18. In the upper left of panel A, estimated protein concentration (x-axis) is plotted against signal intensity (y-axis). In the upper right of panel A, residuals from model fitting (y-axis) are plotted against estimated protein concentration (x-axis). Ideally, the residuals should be symmetrical about the horizontal 0 line and should not increase with increasing concentration. In the lower left of panel A is an image plot of squared residuals from model fitting. This plot shows that the squared residuals are largely homogeneous. In the lower right of panel A, the intensity differences of adjacent dilution steps are plotted (y-axis) against the averaged intensities of adjacent dilution steps (x-axis). If this curve is flat and close to the horizontal line, the dilutions were unsuccessful and the data are not reliable
Results
-
A.
Obstacles to the Successful Application of RPPA to the Study of Non-microdissected Breast Tumors:
-
Obstacle 1:
Antibody Validation
Antibody validation for RPPA is critical to ensure that the detected signal is representative of the protein of interest. We chose 82 antibodies that recognize kinase and steroid signaling events and their effectors (Table 1) because of the importance of these proteins to breast carcinogenesis [24–44]. The relative protein levels derived from RPPA [18] were correlated with the density of the appropriately sized band on immunoblots of the corresponding protein lysates. An arbitrary correlation coefficient (R) of ≥0.7 is required for each antibody (Fig. 1). Antibodies that interact with multiple “off-target” western blot bands or a dominant non-specific band are not suitable for RPPA, and an alternative antibody is sought. For phospho-specific (p) antibodies, cell lines are manipulated in a fashion (e.g., with inhibitors and growth factors) that will alter the phosphorylation site to ensure that observed signal changes are correlated between immunoblotting and RPPA (Fig. 1). For proteins whose expression does not demonstrate a sufficient dynamic range to facilitate antibody validation, siRNA is used to manipulate the signal to allow evaluation of RPPA–immunoblotting correlations. Further, protein and mRNA levels are compared (Table 3); when levels are concordant, as they are with 41% of assayed targets in human breast tumors in Set A (at p ≤ 0.05), this provides additional confidence in the validity of the RPPA analysis (these correlations must be interpreted in the context of the other data above for antibody validation since a poor protein–mRNA correlation does not necessarily indicate that an antibody is not valid). Using these approaches, we continue to expand the antibody list with particular emphasis on proteins implicated in breast carcinogenesis. A web site will be made available with publication of this manuscript with demonstration of the utility of all antibodies in Table 1 in the format shown in Fig. 1 (http://10.106.178.152:8080/AntibodyDatabase/index.html).
-
Obstacle 2:
Variability in Tissue Handling Prior to Freezing
A major challenge to the study of patient tumors is the potential that protein levels and particularly posttranslational modifications will change between the time of tissue collection and analysis. To evaluate total and phosphoprotein stability, ten human breast tumors (Set B) were obtained at surgery, processed, and analyzed by RPPA (see the “Methods” section). Strikingly, the levels of 61/82 proteins including several phosphoproteins were stable (defined using an ANOVA p ≤ 0.05) up to 24 h after tumor collection before freezing (Figs. 2 and 3 and Table 4). Indeed, only 13 of the assessed proteins actually showed a 40% or greater percentage change from baseline with increasing time to freezing (Table 4). Thus, most of proteins were very stable in the samples over the analyzed time course. Of all proteins, only phosphorylated acetyl coenzyme A carboxylase at serine 79 showed marked loss with an estimated half-life of 2.7 h. The remainder of the proteins did not reach an estimated half-life by 24 h at room temperature prior to freezing. Indeed, the RPPA data demonstrated less variability over time than western blotting (Fig. 3). This could be due to RPPA being a “dot-blot” approach which is less susceptible to proteolysis than immunoblotting. Thus, although human breast tumors should be frozen as soon as possible after excision to preserve the ability to assess signaling events, many total and phosphoprotein levels do not change markedly over time, potentially allowing analysis of stable proteins in samples that have not been rapidly frozen. Importantly, this was also confirmed in MDAMB231 and MDAMB435 breast cancer xenografts. For example, no significant changes (at p ≤ 0.05) were observed in phosphorylation of AKT (Ser473), glycogen synthase kinase 3 (Ser21/9), mammalian target of rapamycin (Ser2448), p70S6K (Thr389), or JNK (Thr183/Tyr185) after xenograft tissue was left at room temperature for up to 6 h from the time of animal sacrifice before freezing. In contrast, as in human tumor tissue (Table 4), phosphorylation of MAPK (Thr202/Tyr204) and p38 (T180/182) did decrease over time prior to freezing. Importantly, in neither the human tumor nor the xenograft experiments did we observe early increases in the majority of phosphorylation events when tumor tissue was left at room temperature for 30 min prior to freezing.
-
Obstacle 3:
Intratumoral Heterogeneity
The effects of intratumoral and intertumoral variability on protein and phosphoprotein expression were assessed by applying ANOVA models to RPPA data derived from Set B. Of 82 proteins in three time 0 breast tumor replicates, 80 demonstrated significant (at p ≤ 0.05) variability across the ten tumors, while the expression of only eight total and phosphoproteins demonstrated significant intratumoral variability (Table 5). Clearly, intratumoral total and phosphoprotein levels are much less variable than intertumoral levels. Therefore, RPPA has the potential to provide accurate and reproducible analysis of protein expression and function across patient samples despite potential challenges with intratumoral heterogeneity.
To determine the impact of intratumoral heterogeneity on the robustness and reproducibility of functional proteomic biomarkers, we firstly determined the correlation coefficients between protein expression levels in protein lysates derived from each of two separate sections (“biologic replicates”) obtained from 49 primary hormone receptor-positive breast tumors in Set C (Table 6). These correlation coefficients were not as high as those associated with replicate protein lysates derived from the same tumor sections (“technical replicates”) likely due in part to the modest degree of intratumoral heterogeneity described above. However, 72% of the correlation coefficients between “biologic replicates” were statistically significant (at p < 0.001).
Next, the total and phosphoproteins associated with differential DFS times were determined using either of the two 49 “biologic replicates” in Set C. High expression of p53 and cyclin B1, which both showed minimal intratumoral variability, were significantly associated with short DFS times regardless of which biological replicate was used to classify the patient (Fig. 4), while, low levels of phospho-MAPK (Thr202/Tyr204) were significantly associated with short DFS in both biopsy sets (not shown). In both biopsies, low levels of estrogen (ERα) and progesterone receptors (PR) and low phosphorylation of stat3 at Ser727 were associated with a trend (p = 0.05–0.1) to shorter DFS times.
An integrated analysis of multiple proteins may facilitate more accurate prediction of clinical end points than analysis of individual proteins. Thus, we next determined if the expression and activation levels of multiple proteins yield a stable functional proteomic “fingerprint” despite intratumoral heterogeneity and variability in tumor handling prior to freezing. Using the ten breast tumors obtained at surgery, on unsupervised clustering, the 82-protein functional proteomic “fingerprint” was faithfully preserved across three snap frozen (time 0) sections derived from nine of the ten tumors (Fig. 5a). Further, the unique “fingerprint” was maintained in most tumors with increasing time to tumor freezing up to 24 h after resection (Fig. 5b). In two cohorts of separate sections (“biologic replicates”) derived from each of the 49 breast tumors in Set C, the functional proteomic signatures associated with each corresponding pair of sections was significantly correlated (at p ≤ 0.05) in 43 tumors (Fig. 6). Overall, in terms of intratumoral heterogeneity, the data suggest that the quantification of total and phosphoproteins by RPPA in primary breast tumors is reproducible in snap frozen tissue without microdissection. Although the expression of 21/82 total and phosphoproteins was affected by time to tumor freezing as shown above, the functional proteomic “fingerprint” is reproducible in most tumors even after a delay of 24 h before freezing.
Table 3 Eighty-two protein–mRNA correlation coefficients (rho) and corresponding p values Fig. 2 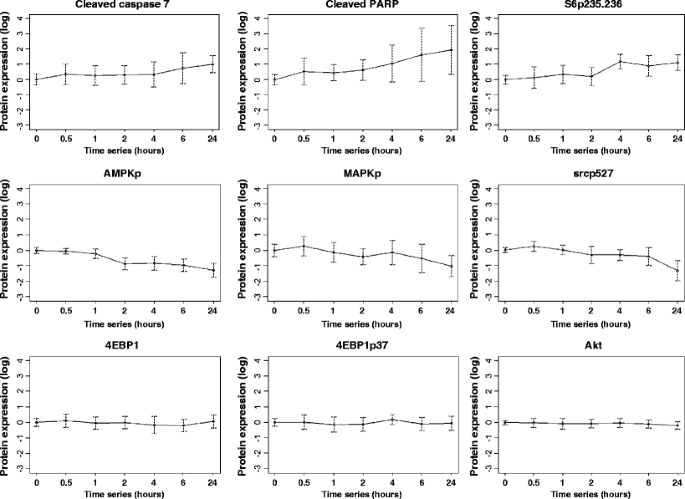
Changes in proteins with increasing time to breast tumor freezing. Ten human breast tumors were collected immediately at surgery and frozen after increasing time intervals up until 24 h. Of the nine total and phosphoproteins shown as examples, three showed a progressive increase with increasing time to breast tumor freezing (cleaved caspase 7, cleaved PARP, and phosphorylation of S6 at serines 235/236 (S6p235–236)), three showed a progressive deterioration with increasing time to breast tumor freezing (phosphorylation of AMP-activated protein kinase (AMPKp), MAPK (MAPKp), and src (srcp527)) and three did not change with increasing time to breast tumor freezing up to 24 h (4EBP1 expression and phosphorylation (4EBP1p37) and Akt expression). The mean expression of each total and phosphoprotein across the ten tumors relative to the mean expression level at time 0 was expressed in log2 units on the y-axis of each plot (with 95% confidence intervals (CI) also shown) and the series of times until breast tumor tissue freezing is shown on the x-axis of each plot (0, 0.5, 1, 2, 4, 6, and 24 h)
Fig. 3 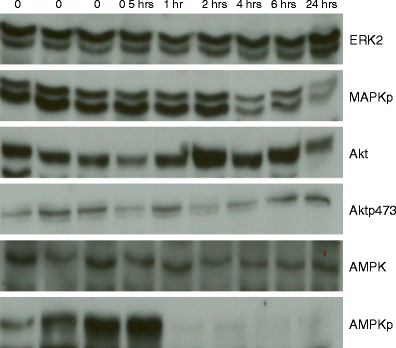
Changes in total and phosphoproteins with increasing time to breast tumor freezing. Six western blots demonstrate stability of mitogen-activated protein kinase (ERK2), Akt, and AMP-activated protein kinase (AMPK) expression and of Akt phosphorylation (Aktp473) with increasing time to tumor freezing. In contrast, consistent with RPPA data, a progressive deterioration was seen with increasing time to breast tumor freezing in the phosphorylation of mitogen-activated protein kinase (MAPKp) and in the phosphorylation of AMPK (AMPKp). The time before tumor freezing is shown along the top of the figure
Table 4 Time-dependent variability in total and phospho (p) protein expression with increasing time to breast tumor freezing Table 5 Inter- versus intratumoral heterogeneity Table 6 Reproducibility associated with biologic replicates in reverse phase protein arrays (RPPA) Fig. 4 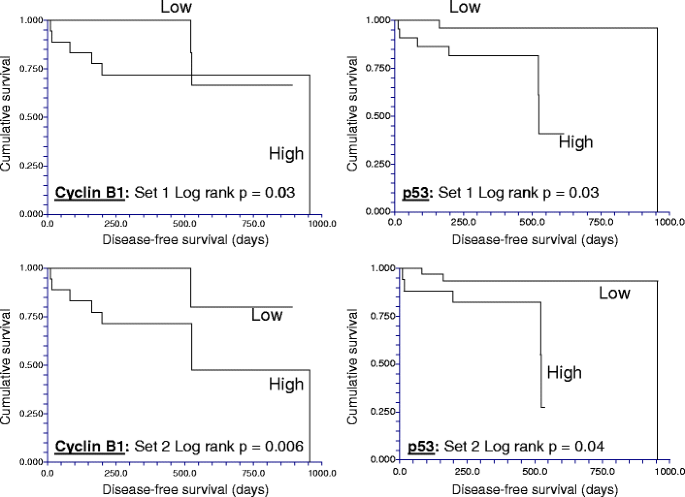
The reproducibility of clinically important breast cancer protein biomarkers detected by reverse phase protein array (RPPA) despite intratumoral heterogeneity. In two cohorts of separate sections derived from each of 49 non-microdissected hormone receptor-positive breast cancers, high expression of cyclin B1 and of p53 proteins as determined using RPPA (>log mean centered cutoff of 0) was associated with short disease-free survival times
Fig. 5 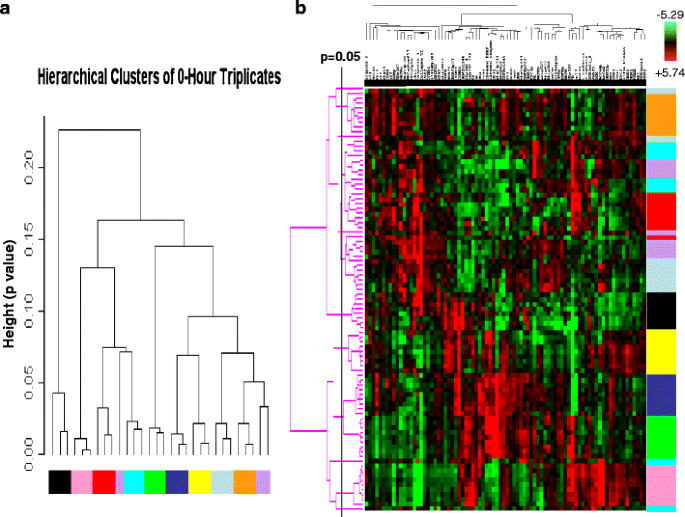
Stability of the primary human breast tumor functional proteomic “fingerprint” despite variability resulting from intratumoral heterogeneity and tissue handling/time to tumor freezing. The overall total and phosphoprotein expression pattern or “signature” was determined by unsupervised hierarchical clustering of data derived from reverse phase protein array (RPPA) analysis of ten primary human breast tumors using the antibodies shown in Table 1. This “signature” was faithfully preserved in the majority of cases a across three separate immediately (snap) frozen (time 0) sections derived from each tumor (FT01–10) and b across nine separate sections frozen at increasing time delays after surgical resection up to 24 h. Note that all sections derived from the same tumor are designated with the same color and that sections derived from different tumors are designated with different colors in the figure. In b, the p = 0.05 bar indicates the position to the right of which dendrogram branches that emerge from the same node represent samples that have statistically similar functional proteomic “fingerprints” (at p ≤ 0.05)
Fig. 6 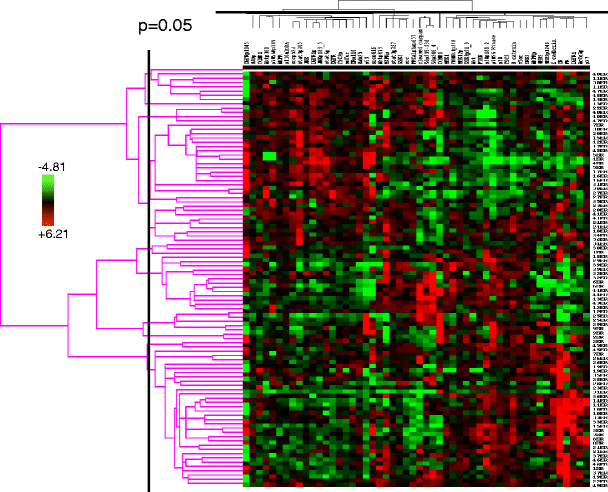
Stability of a human breast tumor functional proteomic “fingerprint” despite individual protein variability resulting from intratumoral heterogeneity. This figure shows unsupervised clustering of total and phosphoprotein quantification data obtained by applying reverse phase protein arrays (RPPA) to protein lysates derived from two independent sections obtained from each of 49 human hormone receptor-positive breast cancers. In only six of the 49 cases did the tumor functional proteomic “fingerprints” in each of the two corresponding tumor sections not significantly correlate with each other (at p ≤ 0.05)
-
Obstacle 1:





-
B.
Reproducibility and the Correlation of RPPA with IHC in Human Breast Tumors:
Reproducibility
Intra- and interslide reproducibility was excellent (see Figs. 7 and 8 for representative examples) for validated antibodies. Antibodies with coefficients of variation (CVs) that are not consistently <15% are discarded and alternate antibodies are sought.
Fig. 7 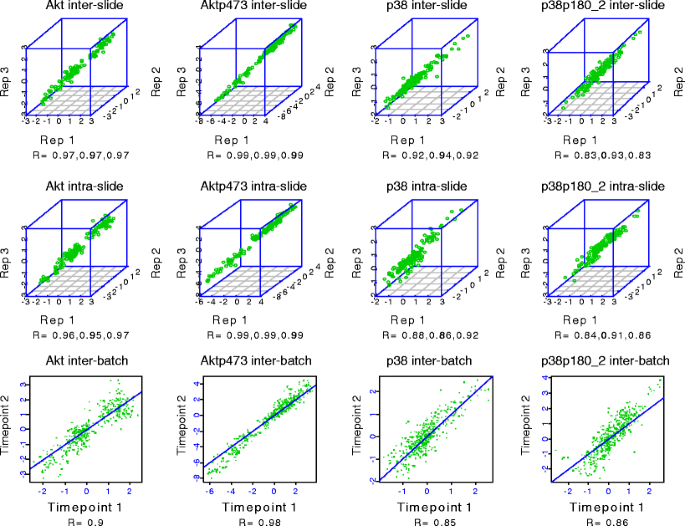
Reverse phase protein array (RPPA) reproducibility for four antibodies. Five serial 2-fold dilutions were made from 48 protein lysates (experimental outline shown in Fig. 8) and the serial dilutions were spotted in triplicate on three sets of nitrocellulose-coated slides at two time points separated by 1 month (“technical replicates”) followed by probing of each slide set with four antibodies to determine intraslide, interslide, and interbatch reproducibility, respectively. The individual correlation coefficients (R) for pairs of replicates for intraslide, interslide, and interbatch reproducibility are shown under each correlation plot for each antibody
Fig. 8 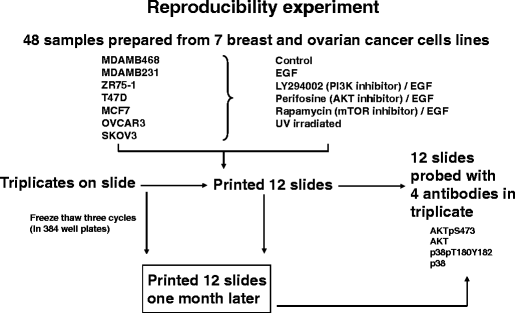
Experimental outline for the reverse phase protein array (RPPA) reproducibility experiment in Fig. 7. Five serial 2-fold dilutions were made from 48 protein lysates, and the serial dilutions were spotted in triplicate on three sets of nitrocellulose-coated slides at two time points separated by 1 month, followed by probing of each slide set with four antibodies to determine intraslide, interslide, and interbatch reproducibility for the total and phosphoproteins detected by these four antibodies
Correlations Between RPPA and IHC
In 95 breast tumors (Set C (Table 2)), the levels of ERα and PR proteins, respectively, determined by RPPA were significantly higher in tumors that are categorized by IHC and fluorescent in situ hybridization as hormone receptor-positive compared with levels in triple receptor-negative (p = 0.00004 and p < 0.001, respectively) and HER2-amplified breast cancers (p = 0.01 and p = <0.001). There were significant positive correlations between ERα and PR levels determined by RPPA and the percentage positivity of these proteins as assessed using IHC (p = 0.002 and p = 0.0006, respectively). Among 64 hormone receptor-positive tumors in Set C, RPPA detected a 866-fold difference in ERα between the tumor with the highest versus the lowest level of ERα. The maximum fold change for PR was 142. This dynamic range may allow RPPA to identify clinically relevant biomarkers that may not be predictive using IHC or that may require a larger sample set to detect using IHC.


-
C.
Potential Clinical Utility of RPPA for the Analysis of the Breast Cancer Functional Proteome: Breast Cancer Classification by Functional Proteomics
Based on the validation approaches described above, protein quantification by RPPA in single sections derived from human breast tumors has the potential to provide sufficient information to faithfully represent the tumor proteome, particularly if the tissue is frozen expeditiously. In 128 tumors (Set A (Table 2)), a highly significant correlation was found on cross tabulation (p < 0.000001) between six breast tumor clusters defined by RPPA (details shown in Fig. 9a (the six groups are described in some detail in the legend of this figure)) and the subtypes defined by transcriptional profiling [1] (Table 7).
Fig. 9 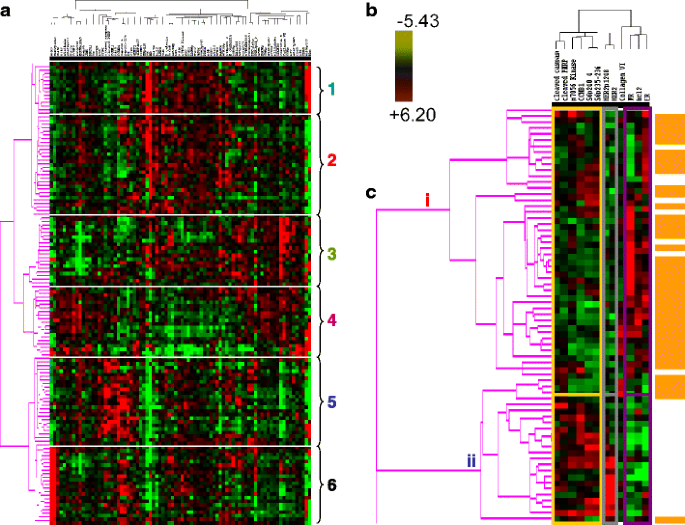
A functional proteomic classification of breast cancer. a Unsupervised hierarchical clustering of 128 breast tumors with data derived from reverse phase protein array (RPPA) using 82 antibodies (Table 1). Six groups were identified as follows: groups 1 and 2—high expression of estrogen receptor alpha (ER) ± progesterone receptor (PR); groups 3 and 4—high expression of stromal markers, including collagen VI and caveolin; group 5—high expression of proliferation markers, including cyclin B1 (CCNB1), with very low expression of ER; and group 6—high HER2 expression and phosphorylation at tyrosine 1248 (HER2p1248). b A log2 scale for the data used to generate the heat maps in a and c. c Hierarchical clustering analysis using 12 markers to distinguish luminal A from luminal B breast cancers in Set A (see Table 2). Luminal A tumors are designated by a brown color to the right of the heat map. The 12 markers can be subdivided into three functional groups—a proliferation group (cleaved caspase 7, cleaved PARP, CCNB1, p70S6 Kinase, and phosphorylation of ribosomal S6 protein at serines 235–236 (S6p235–236) and 240–244 (S6p240_4)), a receptor tyrosine kinase (RTK) group (HER2/HER2p1248), and a functional ER alpha (“ERness”) group (ER, PR, and bcl2). The order of these 12 markers from left to right at the top of panel c are: cleaved caspase 7, cleaved PARP, p70S6 Kinase, CCNBI, S6p240_4, S6p235–236, HER2p1248, HER2, Collagen VI, PR, bcl 2, ER
Table 7 Significant correlation between subtypes of breast cancer identified by reverse phase protein array (RPPA) and by transcriptional profiling The proteomic differences between luminal A and luminal B breast cancers are not well understood [1]. We hypothesized [24–44] that a metric assessing ERα function (ERα/PR/Bcl2), HER2 levels and activity (HER2/HERp1248), apoptosis (cleaved caspase 7/cleaved PARP/Bcl2), protein synthesis (p70S6K/S6 phosphorylation), cell cycle progression (cyclin B1), and stroma (collagen VI) would accurately distinguish luminal A from luminal B cancers (Fig. 9c). The expression levels of these markers from RPPA were weighted equally but in opposing directions for their association with either the luminal A (positive weighting) or luminal B (negative weighting) subtype and summed to create a classifier. This analysis yielded a log mean centered “luminalness” score cutoff of −0.907, with 81% sensitivity, 90% specificity, 85% positive predictive value, and 88% negative predictive value for distinguishing luminal A from luminal B breast cancers in Set A. Figure 10 demonstrates the survival curves associated with the functional proteomic breast cancer classification systems illustrated in Fig. 9a and c. Just as luminal A tumors as defined by transcriptional profiling did significantly better than luminal B tumors in terms of distant metastasis-free survival (at p < 0.05 (not shown)), the “luminalness” score defined by RPPA identified two groups of breast tumors with significantly different distant metastasis-free survival times (Fig. 10c). The receiver operator curves for these analyses are shown in Fig. 11. The ability of RPPA to assay total protein levels as well as functional correlates (phosphorylation/cleavage) likely contributes to the ability of RPPA to accurately distinguish luminal A from luminal B breast cancers (Fig. 9c) and to predict outcomes using a limited number of markers.
Fig. 10 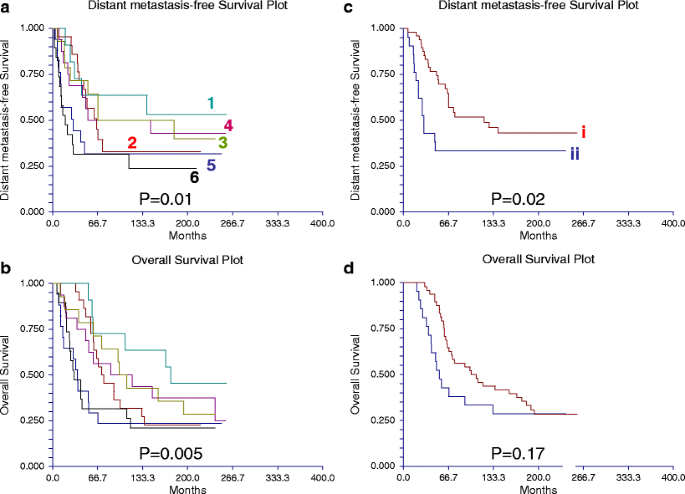
Survival curves for subgroups identified by a functional proteomic classification of breast cancer. a Distant metastasis-free and b overall survival curves for six subgroups (Fig. 9a) identified by unsupervised hierarchical clustering of 128 breast tumors (Set A) with data derived from reverse phase protein array (RPPA) using 82 antibodies. The same color scheme is used to illustrate the six breast cancer subgroups in Figs. 9a and 10a and b. c Distant metastasis-free and d overall survival curves for two subgroups identified by hierarchical clustering analysis of 69 luminal breast tumors using 12 markers (Fig. 9c). The same color scheme is used to illustrate the two breast cancer subgroups in Fig. 9c and 10c and d. Subgroup i represents “luminal a” breast tumors as defined using functional proteomics and subgroup ii represents “luminal b” breast tumors as defined using functional proteomics
Fig. 11 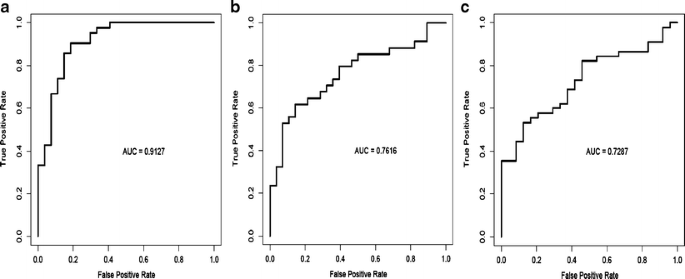
Receiver operator (ROC) curves. These panels show ROC curves associated with prediction of a luminal A vs. luminal B breast cancers, b distant metastasis-free and c overall survival using the reverse phase protein array (RPPA) signature shown in Fig. 9c



Conclusion
Much progress has been made in genomic classification of breast cancer, with these results already impacting patient care [1–10]. However, proteins are the ultimate effectors of cellular outcomes, and functional proteomic data represent an under-evaluated information resource for the identification of useful biomarkers in solid tumors. RPPA represents an emerging functional proteomic assay that has the potential to provide a cost- and material-effective, high-throughput, comprehensive, sensitive, and quantitative approach to molecular classification and pathophysiology studies [11–23]. RPPA has been demonstrated to have utility in the analysis of functional proteomic events in vitro [11–23] and allows exploration of the intricacy of cellular signaling in a manner that cannot be accomplished by immunoblotting or IHC.
Although the application of RPPA to analysis of the functional proteome in cell lines and xenografts has proven relatively straightforward, the application of this technology to the study of non-microdissected human tumors presents a number of potential obstacles. These include the need to validate a large panel of antibodies, variability in tissue handling prior to freezing, and intratumoral heterogeneity. In this study, 82 antibodies that recognize kinase and steroid signaling proteins and their effectors were validated for RPPA. Further, our study demonstrates that both increasing time to tissue freezing and intratumoral heterogeneity result in variability in protein levels in breast tumors. However, the reproducibility and robustness of RPPA, the faithfulness with which total and phosphoproteins and the functional proteomic “fingerprint” are preserved in different sections derived from snap frozen primary breast tumors, and the stability of this “fingerprint” with increasing time to freezing all facilitate the application of RPPA to the study of individual and multiple protein biomarkers in non-microdissected breast tumor specimens.
A previous study demonstrated the half-life of Aktp473 as measured by western blotting in human HT-29 human colon tumor xenografts at room temperature to be 20 min, whereas total Akt was lost with a half-life of 180 min [46]. Indeed, we also noted that the half-life for proteins on western blotting was less than that for RPPA likely due to RPPA being a dot blot approach that is less sensitive to the protein degradation. During the preparation of this manuscript, a study of protein stability in patient samples derived from a number of human tissues and tumor types was published [47]. This manuscript demonstrated transient increases in phosphorylation of a number of proteins over the first 30 min to 1-h post-collection with a return to the original levels at later time points. The data showing a post-collection increase in a number of phosphorylation events followed by a return to the baseline steady state levels are intriguing and will require exploration of potential mechanisms. In our study, we specifically assessed a larger set of only breast cancers than in the study by Espina and colleagues [47], as well as human breast xenograft tissues, to determine the relative effect of protein stability as related to the dynamic range of each marker across patient samples. Our goal was to determine the effects of tissue handling on the ability to characterize the functional proteome in human breast tumors. For the majority of markers characterized in breast tumors, we did not detect early increases in phosphoprotein or other protein levels in either human tumor or xenograft tissues that would alter our ability to classify tumors based on protein levels. Both the human breast tumor and xenograft tissues in our study were left at room temperature in closed eppendorf tubes with no added buffer until the point of freezing.
The primary purpose of this study was to address and overcome obstacles to the successful application of RPPA to the study of the breast cancer functional proteome. Subsequent to our addressing a key set of potential obstacles and our demonstration of the reproducibility of RPPA and of significant (at p < 0.05) correlations between results derived using RPPA and IHC studies, we attempted to evaluate the potential clinical utility of RPPA for the analysis of the breast cancer functional proteome. We selected 82 antibodies (Table 1) that recognize multiple kinase and steroid signaling events and their downstream effectors implicated in breast carcinogenesis [24–44]. Utilizing these markers, RPPA classifies breast cancer into six groups by assaying functional correlates (e.g., phosphorylation, cleavage) in addition to total protein levels (Fig. 9a). The ability to assess both total levels and functional correlates likely confers upon RPPA the ability to accurately distinguish luminal A from luminal B breast cancers using a limited number of markers (Fig. 9c). Further, the classification of breast cancer by RPPA demonstrates a significant correlation on cross tabulation with the well-established classification of breast cancer by transcriptional profiling (Table 7). Thus, the information content captured by RPPA reflects the underlying characteristics of breast tumors, including the likely cell of origin, and potentially patient outcomes and tumor responsiveness to therapy.
As with the current study, the studies of other groups also support the feasibility and potential utility of comprehensive signal pathway activation profiling using RPPA for molecular analysis of human cancers [19–23, 48, 49]. Many of these studies utilized microdissected human tissue and human tumor material. Our study also validates RPPA as a robust tool for the study of the functional proteome in non-microdissected human breast cancers, and this is clearly important for a number of reasons. In terms of potential clinical utility, RPPA could potentially be used to develop signatures that may be useful in terms of prediction of therapy responsiveness in specific subsets of women with breast cancer. Such signatures could conceivably resemble the mRNA-based test, Oncotype Dx® [44]. In addition to potential utility in the identification of prognostic and predictive biomarkers in breast cancer, RPPA has potential utility for the identification of baseline and pharmacodynamic biomarkers that predict benefit from novel therapies targeting signaling pathways. Indeed, we have already established a preclinical precedent for the latter approach [13] and are currently testing this model in an ongoing clinical trial of the Akt inhibitor perifosine in the treatment of women with advanced ovarian cancer.
Several questions remain to be answered. RPPA has advantages over IHC and immunoblotting, including throughput, cost, sensitivity, amount of material required, objective quantification, and a superior dynamic range. However, since IHC provides information concerning spatial organization and RPPA does not, the integrated use of the two technologies may provide a complementary approach to the study of functional proteomics in breast and other solid tumors. Since the routine storage of frozen tumor tissue is a relatively recent approach in most institutions, it will be important to determine which proteins are sufficiently robust to allow RPPA to be applied to the study of the functional proteome in formalin-fixed paraffin-embedded tumor tissue. Further, as the functional proteome is composed of many more proteins than are shown in Table 1, validation of additional high quality affinity reagents could greatly extend the utility of the technology. Ultimately, the true test of RPPA will lie in its ability to determine robust functional proteomic biomarkers that can impact clinical practice.
Abbreviations
- AcCoA:
-
acetyl coenzyme A carboxylase
- AcCoAp:
-
phosphorylated acetyl coenzyme A carboxylase at serine 79
- AMP:
-
adenosine monophosphate
- AMPK:
-
AMP-activated protein kinase
- AMPKp:
-
phosphorylated AMP-activated protein kinase at serine 172
- ANOVA:
-
analysis of variance
- BCA:
-
bicinchoninic acid
- CCNB1:
-
cyclin B1
- CCND1:
-
cyclin D1
- CCNE1:
-
cyclin E1
- CMF:
-
cyclophosphamide, methotrexate, and 5-fluorouracil chemotherapy
- CV:
-
coefficients of variation
- DAB:
-
diaminobenzidine
- DFS:
-
disease-free survival
- EGF:
-
epidermal growth factor
- EGFR:
-
epidermal growth factor receptor
- ERα:
-
estrogen receptor alpha
- FC:
-
fold change
- FFPE:
-
formalin-fixed paraffin-embedded
- FISH:
-
fluorescent in situ hybridization
- FT:
-
frozen tumor
- GSK3:
-
glycogen synthase kinase 3
- HER2:
-
human epidermal receptor 2
- HR:
-
hormone receptor
- IRB:
-
Institutional Review Board
- IHC:
-
immunohistochemistry
- Log2:
-
log to the base 2
- MDACC:
-
The University of Texas M. D. Anderson Cancer Center
- mRNA:
-
messenger ribonucleic acid
- mTor:
-
mammalian target of rapamycin
- PI3K:
-
phosphatidylinositol-3 kinase
- PR:
-
progesterone receptor
- RPPA:
-
reverse phase protein lysate array
- S:
-
serine
- Stat3:
-
signal transducer and activator of transcription
- T:
-
threonine
- Y:
-
tyrosine
References
Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–74.
van de Vijver MJ, He YD, van’t Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009.
Ayers M, Symmans WF, Stec J, et al. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J Clin Oncol. 2004;22:2284–93.
Sjöblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74.
Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–47.
Bullinger L, Döhner K, Bair E, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350:1605–16.
Jazaeri AA, Yee CJ, Sotiriou C, Brantley KR, Boyd J, Liu ET. Gene expression profiles of BRCA1-linked, BRCA2-linked, and sporadic ovarian cancers. J Natl Cancer Inst. 2002;94:990–1000.
Pedersen N, Mortensen S, Sørensen SB, et al. Transcriptional gene expression profiling of small cell lung cancer cells. Cancer Res. 2003;63:1943–53.
Yu YP, Landsittel D, Jing L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–9.
Sirotnak FM, She Y, Khokhar NZ, Hayes P, Gerald W, Scher HI. Microarray analysis of prostate cancer progression to reduced androgen dependence: studies in unique models contrasts early and late molecular events. Mol Carcinog. 2004;41:150–63.
Tibes R, Qiu Y, Lu Y, et al. Reverse phase protein array (RPPA): validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–21.
Sheehan KM, Calvert VS, Kay EW, et al. Use of reverse phase protein microarrays and reference standard development for molecular network analysis of metastatic ovarian carcinoma. Mol Cell Proteomics. 2005;4:346–55.
Hennessy BT, Lu Y, Poradosu E, et al. Pharmacodynamic markers of perifosine efficacy. Clin Cancer Res. 2007;13:7421–31.
Cheng KW, Lu Y, Mills GB. Assay of Rab25 function in ovarian and breast cancers. Methods Enzymol. 2005;403:202–15.
Charboneau L, Tory H, Chen T, et al. Utility of reverse phase protein arrays: applications to signalling pathways and human body arrays. Brief Funct Genomic Proteomic. 2002;1:305–15.
Iwamaru A, Kondo Y, Iwado E, et al. Silencing mammalian target of rapamycin signaling by small interfering RNA enhances rapamycin-induced autophagy in malignant glioma cells. Oncogene. 2007;26:1840–51.
Wulfkuhle JD, Edmiston KH, Liotta LA, Petricoin 3rd EF. Technology insight: pharmacoproteomics for cancer—promises of patient-tailored medicine using protein microarrays. Nat Clin Pract Oncol. 2006;3:256–68.
Hu J, He X, Baggerly KA, Coombes KR, Hennessy BT, Mills GB. Non-parametric quantification of protein lysate arrays. Bioinformatics. 2007;23:1986–94.
Paweletz CP, Charboneau L, Bichsel VE, et al. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene. 2001;20:1981–9.
Grubb RL, Deng J, Pinto PA, et al. Pathway biomarker profiling of localized and metastatic human prostate cancer reveal metastatic and prognostic signatures. J Proteome Res. 2009;8:3044–54.
Sheehan KM, Gulmann C, Eichler GS, et al. Signal pathway profiling of epithelial and stromal compartments of colonic carcinoma reveal epithelial-mesenchymal transition. Oncogene. 2008;27:323–31.
Ornstein DK, Gillespie JW, Paweletz CP, et al. Proteomic analysis of laser capture microdissected human prostate cancer and in vitro prostate cell lines. Electrophoresis. 2000;21:2235–42.
Emmert-Buck MR, Gillespie JW, Paweletz CP, et al. An approach to proteomic analysis of human tumors. Mol Carcinog. 2000;27:158–65.
Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27.
Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65:2554–9.
Monni O, Barlund M, Mousses S, et al. Comprehensive copy number and gene expression profiling of the 17q23 amplicon in human breast cancer. Proc Natl Acad Sci USA. 2001;98:5711–6.
Bellacosa A, de Feo D, Godwin AK, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280–5.
Espina V, Woodhouse EC, Wulfkuhle J, Asmussen HD, Petricoin 3rd EF, Liotta LA. Protein microarray detection strategies: focus on direct detection technologies. J Immunol Methods. 2004;290:121–33.
Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27.
Stoica GE, Franke TF, Moroni M, et al. Effect of estradiol on estrogen receptor-alpha gene expression and activity can be modulated by the ErbB2/PI 3-K/Akt pathway. Oncogene. 2003;22:7998–8011.
Bachman KE, Argani P, Samuels Y, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3:772–5.
Shou J, Massarweh S, Osborne CK, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–35.
Knuefermann C, Lu Y, Liu B, et al. HER2/PI-3 K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003;22:3205–12.
Liang K, Jin W, Knuefermann C, et al. Targeting the phosphatidylinositol 3-kinase/Akt pathway for enhancing breast cancer cells to radiotherapy. Mol Cancer Ther. 2003;2:353–60.
Brown RE. HER-2/neu-positive breast carcinoma: molecular concomitants by proteomic analysis and their therapeutic implications. Ann Clin Lab Sci. 2002;32:12–21.
Ueda Y, Wang S, Dumont N, Yi JY, Koh Y, Arteaga CL. Overexpression of HER2 (erbB2) in human breast epithelial cells unmasks transforming growth factor beta-induced cell motility. J Biol Chem. 2004;279:24505–13.
Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–10.
Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci USA. 2005;102:18443–8.
Ellis MJ, Coop A, Singh B, et al. Letrozole is more effective neoadjuvant endocrine therapy than tamoxifen for ErbB-1- and/or ErbB-2-positive, estrogen receptor-positive primary breast cancer: evidence from a phase III randomized trial. J Clin Oncol. 2001;19:3808–16.
Smith IE, Dowsett M, Ebbs SR, et al. Neoadjuvant treatment of postmenopausal breast cancer with anastrozole, tamoxifen, or both in combination: the immediate preoperative anastrozole, tamoxifen, or combined with tamoxifen (IMPACT) multicenter double-blind randomized trial. J Clin Oncol. 2005;23:5108–16.
Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11:155–68.
Jirström K, Stendahl M, Rydén L, et al. Adverse effect of adjuvant tamoxifen in premenopausal breast cancer with cyclin D1 gene amplification. Cancer Res. 2005;65:8009–16.
Yamashita H, Toyama T, Nishio M, et al. p53 protein accumulation predicts resistance to endocrine therapy and decreased post-relapse survival in metastatic breast cancer. Breast Cancer Res. 2006;8:R48.
Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–26.
Danish Breast Cancer Cooperative Group, Nielsen HM, Overgaard M, Grau C, Jensen AR, Overgaard J. Study of failure pattern among high-risk breast cancer patients with or without postmastectomy radiotherapy in addition to adjuvant systemic therapy: long-term results from the Danish Breast Cancer Cooperative Group DBCG 82 b and c randomized studies. J Clin Oncol. 2006;24:2268–75.
Baker AF, Dragovich T, Ihle NT, Williams R, Fenoglio-Preiser C, Powis G. Stability of phosphoprotein as a biological marker of tumor signaling. Clin Cancer Res. 2005;11:4338–40.
Espina V, Edmiston KH, Heiby M, et al. A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Mol Cell Proteomics. 2008;7:1998–2018.
Wulfkuhle JD, Speer R, Pierobon M, et al. Multiplexed cell signaling analysis of human breast cancer applications for personalized therapy. J Proteome Res. 2008;7:1508–17.
Petricoin 3rd EF, Bichsel VE, Calvert VS, et al. Mapping molecular networks using proteomics: a vision for patient-tailored combination therapy. J Clin Oncol. 2005;23:3614–21.
Acknowledgments
This work was supported in part by the Kleberg Center for Molecular Markers at M. D. Anderson Cancer Center, by the Janice & Robert McNair Foundation, a career development award (CDA) from The ASCO (American Society Clinical Oncology) Cancer Foundation (TACF), and the M. D. Anderson Cancer Center Physician Scientist Program to BTH, by NCI PO1CA099031 (to G.B.M.), by The Susan G. Komen Foundation Biomarkers Identification and Validation Award FAS0703849 (to B. T. H., A. M. G., G. B. M.), and by the Research Council of Norway grant 175240/S10 (to A.L.B.-D.).
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Drs. Hennessy and Lu contributed equally to this work.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hennessy, B.T., Lu, Y., Gonzalez-Angulo, A.M. et al. A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin Proteom 6, 129–151 (2010). https://doi.org/10.1007/s12014-010-9055-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12014-010-9055-y