
Abstract
As prominent immune cells in the central nervous system, microglia constantly monitor the environment and provide neuronal protection, which are important functions for maintaining brain homeostasis. In the diseased brain, microglia are crucial mediators of neuroinflammation that regulates a broad spectrum of cellular responses. In this review, we summarize current knowledge on the multifunctional contributions of microglia to homeostasis and their involvement in neurodegeneration. We further provide a comprehensive overview of therapeutic interventions targeting microglia in neurodegenerative diseases. Notably, we propose microglial depletion and subsequent repopulation as promising replacement therapy. Although microglial replacement therapy is still in its infancy, it will likely be a trend in the development of treatments for neurodegenerative diseases due to its versatility and selectivity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Microglia were first described by the Spanish neuroscientist Pío del Río-Hortega in 1919 [1] and were gradually found to be maverick immune cells in the central nervous system (CNS) that are both specialized and diverse. In mice, microglia originate from a pool of primitive macrophages in the yolk sac on embryonic (E) day 8.5 [2]. During development, microglia survive through colony-stimulating factor 1 receptor (CSF1R) signaling [3], and further differentiation depends on the activity of the transcription factors PU.1 and interferon regulatory factor 8 [4]. In the adult brain, microglia have a ramified morphology and express TMEM119, CD11b, and P2ry12/P2ry13, which participate in the immune response [5]. In the normal brain, microglia are highly dynamic and perpetually scan the CNS [6], influencing fundamental processes such as protein aggregation, soluble factor secretion, phagocytosis, and neural circuit refinement [7]. In the context of CNS injury, microglia undergo complex, multistage activation and respond through signaling molecules such as cytokines and chemokines for subsequent tissue repair [8].
The recent introduction of single-cell RNA sequencing and single-nucleus RNA sequencing analyses has greatly advanced our knowledge of microglial responses in neurodegenerative diseases [9,10,11], leading to the identification of special microglial subsets associated with neurodegeneration in both mouse models and human specimens. For example, using transcriptional single-cell sorting, Keren-Shaul et al. identified disease-associated microglia (DAMs) in proximity to amyloid-β (Aβ) plaques involved in the immune response in AD mouse brains [12]. Krasemann et al. reported that the microglial neurodegenerative phenotype is characterized by genetic upregulation of ApoE signaling and suppression of TGFβ signaling, and this phenotype partially overlaps with that of the DAMs found in AD mouse models [13]. Another study reported that activated response microglia are composed of specialized subgroups overexpressing MHC (major histocompatibility complex) type II genes and show strongly upregulated expression of AD risk genes in the AD mouse model [14]. By using single-cell RNA sequencing, Mathys et al. identified multiple disease stage-specific cell states, including IFN-I and IFN-II co-regulated DAMs in a mouse model of severe neurodegeneration with AD-like phenotypes [15]. Microglial features determined from snRNA-seq data from brain specimens of humans with neurodegenerative diseases show considerable heterogeneity and appear to be different from those in mouse models. For example, Friedman et al. analyzed RNA profiles in the brains of AD patients and showed that they were not replicated in animal models or individuals with early-onset and late-onset AD [16]. They also identified multiple dimensions of CNS myeloid cell activation that occur differentially in response to specific disease conditions. Beyond a detailed description of different microglial subsets, it will be essential to investigate additional biological characteristics of microglia.
It has long been assumed that microglia are long-lived cells that persist throughout the entire lifespan of mice or humans under physiological conditions. Lawson et al. [17] used [3H] thymidine incorporation and autoradiography and reported that 0.05% of the microglia proliferated within 1 hour. Askew et al. [18] analyzed the proliferation of resident microglia by using BrdU incorporation and found that 0.69% of the total microglial cells proliferated after a single pulse of BrdU (the estimated value in humans is ~2%). Therefore, at the individual cell level, microglia replicate frequently. Regarding pathological conditions, Tay et al. [19] established a new multicolor fluorescence fate mapping system and found that CNS pathology shifts from random self-renewal of the microglial network toward a rapid expansion of selected microglial clones. Füger et al. [20] imaged individually-labeled microglia in APP/PS1 mice and found increased microglial turnover in the presence of amyloid lesions.
The maintenance and local expansion of microglia are solely dependent on the self-turnover of CNS-resident cells under normal conditions. When microglia are depleted by inhibiting CSF1R signaling, newborn microglia rapidly replenish the population in the whole brain in a short time after drug withdrawal [21]. Importantly, the repopulated microglia are solely derived from residual microglia [22]. In the context of disease, numerous cells of the myeloid lineage appear in the nervous parenchyma in pathological settings, such as in AD models and experimental autoimmune encephalitis (EAE) models. Myeloid cells enter the nervous parenchyma, and some of them appear to transform into cells resembling microglia [23].
In this review, we highlight new evidence demonstrating the unique and diverse properties of microglia in the healthy and diseased brain, including in the context of Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (MS). In addition, we elaborate on the theoretical basis of microglial replacement and other microglia-based therapies for translational clinical applications.
The Multifaceted Functions of Microglia in the CNS
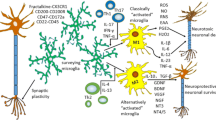
The biological functions of microglia in the CNS are multifaceted. They exert both neuroprotective and neurotoxic effects (Fig. 1). In a healthy brain, microglia are responsible for mediating inflammation, regulating a broad spectrum of cellular responses, and eliminating microbes, dead cells, and protein aggregates [24]. Microglia were originally known for their immune functions. They are the first responders to neuroinflammation and rapidly adapt their phenotypes and functions to fit the brain milieu [1]. Microglia secrete cytokines and chemokines that contribute to various aspects of immune responses in the CNS. During infection, chronic activation of microglia leads to sustained production of proinflammatory cytokines that cause damage to the surrounding neurons. This notion has been confirmed in various brain disorders, such as AD, PD, and MS. For example, in AD patients, the neurotoxic factors produced by activated microglia cause nitration of tyrosine 10 of the Aβ peptide, which promotes the aggregation of senile plaques [25]. In PD patients, inflammatory mediators such as IL-1β and IL-6, and epidermal growth factor released by activated microglia, can lead to the death of dopaminergic neurons [26]. PET studies have indicated that motor disability is correlated with the abundance of activated microglia in MS patients [27].

Microglia play multifaceted roles in the CNS. Upper: homeostatic microglia promote neurogenesis by releasing neurotrophic factors, whereas inflammation-associated microglia lead to neuronal cell death. Right: microglia are crucial for synaptic pruning and circuit refinement. Synaptic over-pruning induces synaptic dysfunction, which is found in neurodegenerative diseases. Lower: phagocytosis is essential for CNS homeostasis. Microglia can remove apoptotic cells and prevent the release of toxic intracellular contents. However, hyperactive microglia engulf newborn neurons, leading to a decrease in the number of mature neurons. Left: microglia–neuron/oligodendrocyte/astrocyte crosstalk promotes neurogenesis and axon formation. Microglial hyperactivation disrupts the blood-brain barrier and recruits peripheral cells into the brain during infection.
In addition to immune function, as resident macrophages in the brain, microglia vigilantly perform surveillance to ensure parenchymal homeostasis. Once they migrate into the brain parenchyma, microglia actively communicate with other cells. Early after birth, microglia release neurotrophic factors such as IGF-1 to promote neuronal survival and axon fiber formation [28]. Brain-derived neurotrophic factors released by activated microglia promote neurogenesis in the CNS [29]. At the same time, microglia are also well poised to induce programmed cell death, which has been demonstrated to be CD11b-dependent [30]. Afterward, microglia clean up the resulting cellular debris by phagocytosis. Microglial phagocytosis is mediated by signaling via triggering receptors expressed on myeloid cell-2 (TREM2) [31]. In addition, microglia participate in maintaining synaptic homeostasis via neuronal pruning and synaptogenesis [32, 33]. Oligodendrocytes, another important group of cells that are pivotal for myelin production around axons, are deeply involved in microglial maturation [34, 35]. In addition, microglia establish a delicate balance with astrocytes and are responsible for the maintenance of neuronal functions and brain homeostasis [36, 37].
Microglia are active sensors and versatile effector cells not only in healthy brains but also in diseased brains. In the following section, we emphasize the role of microglia in neurodegenerative diseases of the CNS, including AD, PD, and MS (Fig. 2).

Microglia are active sensors and versatile effector cells in the brain under pathological conditions. Microglia constantly screen the microenvironment in the CNS and transform into a reactive state under certain pathological conditions. However, the tilt toward harmful or beneficial outcomes of microglial activation varies between neurological diseases. (1) Microglia in AD. AD is typified by the accumulation of extracellular Aβ peptides and intracellular deposits of hyperphosphorylated tau. Microglia are committed to an inflammatory response and can engulf Aβ deposits and impair cells. Microglial treatments for AD include targeting the microglial inflammatory response, TREM2 activation, lipid metabolism, and lysosomal function. (2) Microglia in PD. PD is primarily characterized by the death of dopaminergic neurons that project from the SNpc. Reactive microglia are involved in dopaminergic cell death as proinflammatory mediators. Reactive microglia also block the delivery of α-syn to the nigrostriatal tract. Microglia-based treatments for PD include the promotion of α-synuclein transfer and clearance, anti-inflammatory treatments, and HSCT therapy. (3) Microglia in MS. MS is a chronic inflammatory disease that leads to focal plaques of primary demyelination and diffuse neurodegeneration. Active demyelination is usually associated with microglial activation. Microglial treatments for MS include the regulation of microglial inflammatory signaling, the depletion of microglia to reduce demyelination, and the targeting of microglial phagocytosis. AD, Alzheimer's disease; PD, Parkinson’s disease; MS, multiple sclerosis; SNpc, substantia nigra pars compacta; α-syn, alpha-synuclein; HSCT, hematopoietic stem cell transplantation.
Microglia are a “Double-edged Sword” in the Progression of AD
AD is the major cause of dementia and is mainly characterized by progressive neuronal loss followed by cognitive impairment. The current understanding is that AD is closely associated with genetic, aging-related, and environmental factors [38,39,40]. The main pathological features of AD include the deposition of insoluble Aβ peptides, as well as the aggregation of hyperphosphorylated tau protein, which lead to the formation of neurofibrillary tangles in the brain [38,39,40].
Reactive gliosis and the inflammatory response are hallmarks of AD. Microglia-mediated inflammation is a 'double-edged sword', performing both detrimental and beneficial functions. For example, activated microglia in AD mice show upregulation of inflammatory markers such as CD36, CD14, CD11c, MHC-II, and iNOS, which might disturb neuronal functions and lead to cognitive decline [41]. Clinical imaging studies have reported negative correlations between microglial activation measured by [11C]PK11195 PET and the structural integrity and functional activity of the brain in AD patients [42], whereas some inducers of inflammation, such as lipopolysaccharide (LPS), activate microglia to promote the degradation of Aβ [43]. Microglia interact with Aβ and amyloid-beta precursor protein (APP) through specific pattern-recognition receptors, including CD14, CD36, and Toll-like receptors, which are strongly expressed on the microglial surface [44]. This interaction increases microglial phagocytosis, resulting in the clearance of Aβ from the brain [45, 46]. As an example, the phagocytic index and total Aβ load are higher in IL-1α (+) and IL-1Ra (+) microglia and microglia producing TNF-α and IL-1β are associated with a lower Aβ load and phagocytic index in AD mice [47]. Therefore, strategies need to be developed to modulate the activation of microglia by inhibiting the release of inflammatory factors. Preclinical trials have shown that genetic and pharmacological blockade of TNF effectively alleviates amyloid pathology and tau phosphorylation [48]. Intraperitoneal administration of an antibody blocking IL-1 receptors has also been shown to decrease the activity of several tau kinases in the brains of 3×Tg-AD mice [49]. However, further clinical trials are needed to assess the safety and efficacy of this antibody in humans.
Differences exist between microglia in humans and mouse AD models. At the site of neurodegeneration, plaques are surrounded by activated microglia named DAMs [12]. Genome-wide studies have shown that some genes, such as APOE4, TREM2, and CD33, have unique expression patterns in DAMs [13, 50, 51]. However, no DAM signature has been found in human AD via single-nucleus RNA sequencing (snRNA-seq), and only a few microglial genes, including APOE and SPP1, have been identified [52]. In another study of patients, the differentially expressed microglial genes included EEF1A1, GLULL, KIAA1217, LDLRAD3, and SPP1, which differ from the characteristic genes of DAMs [53]. DAMs have been reported in other conditions, including aging, ALS, and frontotemporal dementia. The scRNA-seq profiles of dissected human brain specimens from MS patients have revealed various microglial populations expressing DAM genes [54]. Given the heterogeneity of brain pathology, certain DAM genes may be involved in different diseases. Research into the treatment of AD should consider the feasibility of implementing this approach in the clinic.
Overall, microglia, as a “double-edged sword” in the progression of AD pathology, have both beneficial and detrimental effects. A better understanding of the role of microglia in the progression of AD pathology is needed, as microglia could be a target and a tool for AD treatment.
Microglia: The Culprit in PD
PD is the second most common neurodegenerative disorder and is characterized by motor and non-motor symptoms. The widely recognized pathology of PD involves the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the presence of cytoplasmic protein aggregates known as Lewy bodies in the remaining dopaminergic neurons [55]. In the early stages of PD, α-synuclein undergoes aggregation and fibrillization, which can lead to neurotoxicity [56, 57]. In late-onset PD, a large genome-wide association study identified a single nucleotide polymorphism in the human leukocyte antigen (HLA-DRA) gene. HLA-DR is a genetic risk factor for PD patients and is expressed by antigen-presenting cells such as microglia [58]. Another study showed that HLA-DR-positive microglia (“reactive” microglia) are also found in the substantia nigra of PD patients [59].
Although molecular profiling of microglia in animal models of PD has yet to be attempted, activated microglia are known to be prevalent in mouse models and PD patients. There is emerging evidence that microglial dysfunction contributes to PD pathogenesis and progression [60], as exhibited by a weakened inflammatory response, reduced phagocytosis, and decreased interactions with neurons. In a PD mouse model, neuroinflammation and associated “reactive” microglia precede the onset of astrogliosis and dopaminergic cell death [61]. Many studies have described reactive microglia in postmortem brain samples from PD patients. Jyothi et al. reported that the microglial count showed a biphasic increase in the vicinity of the few remaining nigral dopamine neurons and that microglia displayed a morphology characteristic of activated cells in PD patients [62]. A PET study using [11C] (R)-PK11195 in early PD, an in vivo marker of microglial activation, reported that reactive microglia are noted in brain regions such as the pons and basal ganglia [63]. These activated microglia then produce a wide range of inflammatory mediators that lead to the continuous death of dopamine neurons. Consistent with this, in PD model mice established by 1-methyl-4-phenyl-1,2,3,6tetrahydropyridine (MPTP), brain microglial activation via NLRP3 inflammasomes plays a role in neurotoxicity [64].
Cell-to-cell propagation of α-synuclein aggregates is thought to contribute to the pathogenesis of PD. IL-4-activated microglia effectively reduce the extracellular α-syn and decrease α-syn transfer from neuron to neuron, whereas LPS-treated microglia are less prone to carry out phagocytosis [65]. Pro-inflammatory cytokines can both be neurotoxic and attenuate microglial phagocytosis. LPS, a pro-inflammatory stimulus, attenuates microglial phagocytosis and leads to the increased presence of α-syn within microglia and grafted dopaminergic neurons in a PD mouse model [65]. Croisier et al. found that CD68 immunoreactivity is negatively correlated with disease duration, suggesting that elevated phagocytic activity is associated with sustained tissue destruction in PD patients [66]. Therefore, regulating the activation of microglia into a neuroprotective function is a potential therapeutic target.
PD is a chronic disease, making it likely that prevention and treatment will require long-term therapy. Thus, mediating microglia to play a neuroprotection role is a promising field for research.
Microglia: The Damaging Element in MS
MS is a demyelinating disease of the CNS, including the brain and spinal cord. In MS, demyelination occurs when the immune system inappropriately attacks and destroys myelin, which breaks down communication between neurons, ultimately leading to a variety of sensory, motor, and cognitive problems [67]. MS is a complex and heterogeneous disease with different lesion patterns and mechanisms of tissue injury [68]. MS is also an inflammatory disease of the CNS in which microglia play an important role.
The role of microglia in MS is complex and controversial. Microglia are considered to be damaging elements in MS. Focal lesions of active demyelination and neurodegeneration are characterized by inflammation and microgliosis. A study in an EAE mouse model indicated that microglia are the first cells to take up myelin antigens and subsequently recruit leukocytes to the CNS through MHC class II molecules, thus initiating immune infiltration and the demyelination cascade in the early stage [69]. Microglia also release proinflammatory cytokines such as IL-18 and IL-6 and chemokines to aggravate MS [70]. In MS patients, TMEM119-positive microglia are less abundant in active MS, with restoration associated with disease inactivity [71]. In chronic slowly expanding MS lesions, a ring of activated microglia is also present at the site of lesion expansion [72]. CSF1R signaling (specifically in microglia) is activated in MS and might drive deleterious neuroinflammation, particularly during disease progression [73, 74]. Disruption of remyelination is another pathogenic factor in MS. TREM2 activation on microglia promotes myelin debris clearance and remyelination [75], and TREM2 genetic deficiency might be a risk factor for MS. Thus, microglia are recognized as key players in MS pathology.
Perspectives and Reflections on Microglial Replacement Therapy
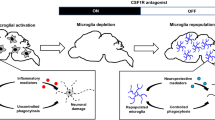
As shown above, we demonstrated the role of microglia in the pathology of neurodegenerative disease and the potential therapeutic role of microglia in disease treatment. Some researchers have reported that microglial repopulation can largely resolve the proinflammatory response and promote functional recovery [76]; however, the functional restoration of repopulated microglia depends on exogenous environmental and endogenous genetic changes. Here, we propose that microglia replacement by allogenic cells is an effective method to treat microglia-associated neurodegenerative disorders (Fig. 3).

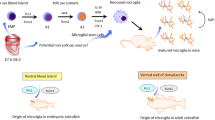
Microglial replacement therapy is a promising strategy for treating neurological disorders. A microglia-free niche is a necessary precondition for microglial replacement. The depletion of microglia is an effective model to create the niche, such as in CX3CR1-CreER::DTA mice. Pharmacological microglial depletion includes the administration of a CSF1R inhibitor or DT in the brain in CX3CR1-CreER::iDTR mice. There are three highly efficient and clinically feasible strategies for microglial replacement in the whole CNS or brain regions of interest: microglial replacement by bone marrow transplantation (Mr BMT), microglial replacement by peripheral blood (Mr PB), and microglial replacement by microglial transplantation (Mr MT). Mr PB clinically boosts donor cell availability. Mr BMT can correct gene deficiency, thus better treating gene-related diseases. Mr MT can achieve region-specific microglial replacement. These three approaches provide options for personalized treatment. DT, diphtheria toxin; DTR, diphtheria toxin receptor; DTA, diphtheria toxin activator. The figure was created with BioRender.com.
The Origin of Microglia
Microglial cells originate from cells produced at ~E7.5 in the yolk sac, at the time of the first wave of hematopoiesis [77, 78]. The second wave starts at E10.5; hematopoietic stem cells (HSCs) are produced in the aorta–gonad–mesonephros region and settle in the fetal liver and other hematopoietic organs of the embryo, where they mature and differentiate into definitive erythrocytes and all myeloid cells, including monocytes. HSCs from the fetal liver finally colonize the bone marrow, the only hematopoietic organ in adults [77]. Due to the specificity of microglial origin, non-autologous cell replacement of microglia may be an effective treatment for microglia-associated neurodegenerative diseases.
A Microglia-free Niche is a Necessary Precondition for Microglial Re-population
The macrophage niche theory is a concept that first postulates that each macrophage cell occupies its territory [79]. When the niche is available, monocytes can efficiently differentiate into macrophages, and the same process occurs for microglia. The depletion or death of microglia may provide a space that triggers the proliferation of neighboring macrophages to repopulate the space. Microglial niches become temporarily available via irradiation-mediated damage or depletion [79]. The irradiation method opens the blood-brain barrier, and this helps a drug to enter the brain through blood circulation and avoids further damage to the CNS. Early approaches depleted microglia by generating mice transgenic for CD11b–thymidine kinase of the herpes simplex virus (HSVTK). In the presence of ganciclovir, HSVTK is activated and induces apoptosis in CD11b+ mitotic cells [74]. While CD11b+ cells are necessary for normal hematopoiesis and red blood cell production [80], completely depleting them is not a perfect way to deplete microglia. An alternative genetic approach used CX3CR1-CreER::DTA and CX3CR1-CreER::DTR mouse lines to induce diphtheria toxin-dependent microglial cell death [81]. However, DTR models kill only short-lived microglia [81]. Pharmacological microglial depletion can be achieved by targeting the Csf1r gene. The CSF1R inhibitors PLX5622, PLX3397, GW5622, and PLX647 are able to cross the brain-blood barrier and induce microglial depletion. Administration of PLX5622 in rodent chow depletes microglia by up to 99% after 2 weeks in adult mice [22]. However, there are still some potential toxic or adverse effects of microglial depletion and replacement. In the report by Rubino et al., acute and synchronous microglial depletion in adult mice triggered gray matter gliosis and progressive ataxia-like neurological behavior [82]. Bruttger et al. reported that microglial ablation in DTRMG mice leads to a cytokine storm and astrogliosis [81]. Nevertheless, Peng et al. reported normal motor performance after diphtheria toxin (DT) treatment in DTR mice [83]. Several papers have reported long-term observations in the PLX depletion model and did not observe motor deficits [21]. The reasons for these different behaviors may involve the dose, concentration, and duration of drug treatment. Therefore, when choosing microglial replacement therapies, we should take into account the potential risks of the method and try to minimize them.
Current Microglial Replacement Strategies
Microglial replacement therapy has undergone several attempts and failures. Traditional bone marrow transplantation enables the partial replacement of endogenous microglia with donor cells [84]. However, the replacement efficiency is usually <5%–20%, due to the lack of a cell niche [85]. Recent studies established an experimental strategy for transplanting microglia into RAG2-/- IL2Rg-/- recipient mice with clear success. However, the treatment is limited in terms of time and clinical application. In light of these challenges, Xu et al. recently developed three effective strategies for microglial replacement either throughout the CNS or focusing only on brain regions of interest [86]: microglial replacement by bone marrow transplantation (Mr BMT) [87], microglial replacement by peripheral blood (Mr PB) [88], and microglial replacement by microglial transplantation (Mr MT) [89]. By choosing an appropriate strategy, the rate of microglial replacement can be effectively boosted. In this protocol, PLX5622 is used to clear microglia, and 9 Gy of whole body irradiation is administered to open the blood-brain barrier. Mr PB is able to induce peripheral blood cells to differentiate into microglia-like cells (MLCs) and can replace 80.74% of resident microglia in the brain. Mr BMT is capable of inducing allografted bone marrow cells (BMCs) to differentiate into microglia-like cells in the entire CNS, replacing 92.66% of resident microglia in the brain. The engrafted microglia after Mr MT exhibit the natural characteristics of naive microglia.
Similar but not the Same
Microglial replacement combining genetic engineering with cell transplantation represents a cutting-edge approach for the precise treatment of microglia-associated neurodegenerative diseases. Whether transplanted microglia can act as normal microglia is the key issue. Microglial cells in the normal brain are called “true” microglia or bona fide microglia, while transplanted microglial cells are often called “microglia-like cells” [90]. This is because of not only their different origins but also their slightly different characteristics. Bone marrow-derived MLCs, when engrafted into the CNS, share ~90% of the transcriptome with host microglia, including the expression of some key microglial genes, such as Tgfb receptor 1 (Tgfbr1). In addition, MLCs exhibit their characteristics, as they show reduced gene expression of Tmem119 and P2ry12 [91]. Similarly, the MLC population is also maintained by local proliferation [18]. Reports suggest that microglia and MLCs are very similar, and their phenotypes are constantly shaped by the microenvironment in a time- and context-dependent manner. For instance, under pathological conditions, during the acute phase of EAE, microglia shift to a pro-inflammatory macrophage phenotype, while during the chronic phase, macrophages turn down their pro-inflammatory signature to acquire a resting microglia phenotype [92]. However, to our knowledge, the functional differences between microglia and MLCs are not well established; for example, whether they have similar functions in parenchymal surveillance and neuronal circuit refinement remains to be elucidated. We summarize the main benefits and limitations as well as the caveats of each proposed strategy in Table 1. Therefore, these methods offer important rationales for further clinical applications, and each tactic has its own merits and limitations, which provide more choices. Importantly, these methods allow for the genetic modification of replacement microglia and the compensation of functional defects.
Microglial Replacement Therapy in CNS Disease
Microglial replacement therapy has been effectively used to treat other diseases. For instance, transplantation of wild-type bone marrow into mecp2-deficient hosts leads to the implantation of bone marrow-derived cells with a microglial phenotype in the brain parenchyma, which arrests disease progression [84]. Another in-depth study indicated that whole bone marrow is heterogeneous and ill-defined, and hematopoietic stem cells play a very important role after Mr BMT transplantation [93]. BM-derived immature monocytic cells can commit to a microglia-like phenotype, and they harbor several features and functions of native microglia [94]. Since resident microglia appear to degenerate in AD, Mr BMT compensates for the deficient functions of senescent resident microglia in AD [95]. ALS is a progressive, fatal neurodegenerative disease. BMT of mSOD1-transgenic mice with BMCs altered the functional properties of microglia and improved the neural cell microenvironment [96].
Moreover, transplantation of genetically modified BM cells can reduce symptoms of CNS diseases. Mr BMT methods are also being continuously improved. Recently, a study developed an approach for rapid and near-complete replacement of microglia in mice with circulation-derived myeloid cells, eliminating the substantial variability that occurs after conventional BMT, which slows neurodegeneration and ameliorates motor dysfunction in prosaposin-mutant mice [93]. However, Mr BMT has its limitations: for example, Mr BMT is a method for whole-brain microglial replacement, not for specific brain regions. Mr MT enables microglial replacement via microglial transplantation into the brain area of interest. The method of targeted removal of microglia in specific regions has been studied [97]. Although the application of Mr MT in diseases is relatively rare, microglial recolonization in a brain region of interest will certainly have better application prospects. We speculate that therapeutic genes can be transduced into the stem or progenitor cells with lentiviral vectors, leading to stable integration of genes such as MeCP2 in Rett syndrome or TREM2 in AD mice. It is conceivable that by genetically modifying the extracted normal microglia, we can treat more intractable genetic defects and avoid immune rejection at the same time.
Targeting Microglial Therapeutics: Challenges and Opportunities
In this review, we summarized the critical pathological role of microglia in several neurodegenerative diseases, mostly focused on AD. We envision microglial replacement as a potential medical intervention for many neurological diseases such as PD and MS, but further research is needed.
What factors affect the replacement of microglia? Xu et al. reported that Mr BMT cells are mainly derived from CCR2-positive BMCs [86]. CCR2-positive cells are migratory cells that respond to neural insults [98]. Therefore, the function of CCR2 may be related to the mechanism of microglial replacement in the brain. In addition, a study has indicated that engrafted BM-derived myeloid cells display significantly increased levels of CD68, a lysosomal marker associated with a heightened activation or phagocytic state [99]; microglial engraftment affects astrocyte activation and neuronal communication [99]. Thus, molecules that affect microglial activation or interactions with other cells, such as CX3CR1/CX3CL1 and CD200/CD200R [100], might also affect microglial replacement. The removal rate of resident microglia directly affects the replacement efficiency of external them. Our recent research found that microglial debris is primarily removed by astrocytes in the brain via the opsonization of C4b [105]. Microglia engraftment likely affects astrocyte reactivation. Additional research into the mechanisms of microglial replacement would be beneficial for developing clinical therapies.
Change history
13 February 2023
A Correction to this paper has been published: https://doi.org/10.1007/s12264-023-01026-9
References
Prinz M, Jung S, Priller J. Microglia biology: One century of evolving concepts. Cell 2019, 179: 292–311.
Zhang L, Cao Y, Zhang X, Gu X, Mao Y, Peng B. The origin and repopulation of microglia. Dev Neurobiol 2022, 82: 112–124.
Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, Wollscheid-Lengeling E. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun 2019, 10: 3215.
Zhou N, Liu K, Sun Y, Cao Y, Yang J. Transcriptional mechanism of IRF8 and PU.1 governs microglial activation in neurodegenerative condition. Protein Cell 2019, 10: 87–103.
Cowan M, Petri WA Jr. Microglia: Immune regulators of neurodevelopment. Front Immunol 2018, 9: 2576.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308: 1314–1318.
Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol 2014, 32: 367–402.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011, 91: 461–553.
Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 2019, 101: 207-223.e10.
Rao Y, Du S, Yang B, Wang Y, Li Y, Li R, et al. NeuroD1 induces microglial apoptosis and cannot induce microglia-to-neuron cross-lineage reprogramming. Neuron 2021, 109: 4094-4108.e5.
Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159: 1327–1340.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169: 1276-1290.e17.
Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47: 566-581.e9.
Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The major risk factors for Alzheimer’s disease: Age, sex, and genes modulate the microglia response to aβ plaques. Cell Rep 2019, 27: 1293-1306.e6.
Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep 2017, 21: 366–380.
Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep 2018, 22: 832–847.
Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience 1992, 48: 405–415.
Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep 2017, 18: 391–405.
Tay TL, Mai D, Dautzenberg J, Fernández-Klett F, Lin G, Sagar, et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci 2017, 20: 793–803.
Füger P, Hefendehl JK, Veeraraghavalu K, Wendeln AC, Schlosser C, Obermüller U, et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat Neurosci 2017, 20: 1371–1376.
Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82: 380–397.
Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci 2018, 21: 530–540.
Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, et al. A combination of ontogeny and CNS environment establishes microglial identity. Neuron 2018, 98: 1170-1183.e8.
Zhang L, Zhang J, You Z. Switching of the microglial activation phenotype is a possible treatment for depression disorder. Front Cell Neurosci 2018, 12: 306.
Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, et al. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron 2011, 71: 833–844.
Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett 1994, 180: 147–150.
Giannetti P, Politis M, Su P, Turkheimer F, Malik O, Keihaninejad S, et al. Microglia activation in multiple sclerosis black holes predicts outcome in progressive patients: An in vivo [(11)C](R)-PK11195-PET pilot study. Neurobiol Dis 2014, 65: 203–210.
Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci 2013, 16: 543–551.
Zhang J, Rong P, Zhang L, He H, Zhou T, Fan Y, et al. IL4-driven microglia modulate stress resilience through BDNF-dependent neurogenesis. Sci Adv 2021, 7: eabb9888.
Marín-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron 2004, 41: 535–547.
Takahashi K, Rochford CDP, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 2005, 201: 647–657.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333: 1456–1458.
Parkhurst CN III, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155: 1596–1609.
Hughes AN, Appel B. Microglia phagocytose myelin sheaths to modify developmental myelination. Nat Neurosci 2020, 23: 1055–1066.
Nemes-Baran AD, White DR, DeSilva TM. Fractalkine-dependent microglial pruning of viable oligodendrocyte progenitor cells regulates myelination. Cell Rep 2020, 32: 108047.
Araki T, Ikegaya Y, Koyama R. The effects of microglia- and astrocyte-derived factors on neurogenesis in health and disease. Eur J Neurosci 2021, 54: 5880–5901.
Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl Neurodegener 2020, 9: 42.
Scheltens P, Blennow K, Breteler MMB, de Strooper B, Frisoni GB, Salloway S, et al. Alzheimer’s disease. Lancet 2016, 388: 505–517.
Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol 2018, 25: 59–70.
Long JM, Holtzman DM. Alzheimer disease: An update on pathobiology and treatment strategies. Cell 2019, 179: 312–339.
Martin E, Boucher C, Fontaine B, Delarasse C. Distinct inflammatory phenotypes of microglia and monocyte-derived macrophages in Alzheimer’s disease models: Effects of aging and amyloid pathology. Aging Cell 2017, 16: 27–38.
Femminella GD, Ninan S, Atkinson R, Fan Z, Brooks DJ, Edison P. Does microglial activation influence hippocampal volume and neuronal function in Alzheimer’s disease and Parkinson’s disease dementia? J Alzheimers Dis 2016, 51: 1275–1289.
Qiu WQ, Ye Z, Kholodenko D, Seubert P, Selkoe DJ. Degradation of amyloid beta-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J Biol Chem 1997, 272: 6641–6646.
Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci 2003, 23: 2665–2674.
Pal A, Rani I, Pawar A, Picozza M, Rongioletti M, Squitti R. Microglia and astrocytes in Alzheimer’s disease in the context of the aberrant copper homeostasis hypothesis. Biomolecules 2021, 11: 1598.
Fakhoury M. Microglia and astrocytes in Alzheimer’s disease: Implications for therapy. Curr Neuropharmacol 2018, 16: 508–518.
Babcock AA, Ilkjær L, Clausen BH, Villadsen B, Dissing-Olesen L, Bendixen ATM, et al. Cytokine-producing microglia have an altered beta-amyloid load in aged APP/PS1 Tg mice. Brain Behav Immun 2015, 48: 86–101.
Steeland S, Gorlé N, Vandendriessche C, Balusu S, Brkic M, Van Cauwenberghe C, et al. Counteracting the effects of TNF receptor-1 has therapeutic potential in Alzheimer’s disease. EMBO Mol Med 2018, 10: e8300.
Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, et al. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J Immunol 2011, 187: 6539–6549.
Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med 2019, 216: 2546–2561.
Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 2015, 77: 43–51.
Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570: 332–337.
Del-Aguila JL, Li Z, Dube U, Mihindukulasuriya KA, Budde JP, Fernandez MV, et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res Ther 2019, 11: 71.
Esaulova E, Cantoni C, Shchukina I, Zaitsev K, Bucelli RC, Wu GF, et al. Single-cell RNA-seq analysis of human CSF microglia and myeloid cells in neuroinflammation. Neurol Neuroimmunol Neuroinflamm 2020, 7: e732.
Stefanis L. α-synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 2012, 2: a009399.
Burré J, Sharma M, Südhof TC. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A 2014, 111: E4274–E4283.
Cookson MR. α-Synuclein and neuronal cell death. Mol Neurodegener 2009, 4: 1–14.
Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 2010, 42: 781–785.
McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38: 1285–1291.
Bartels T, De Schepper S, Hong S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 2020, 370: 66–69.
Rabaneda-Lombarte N, Serratosa J, Bové J, Vila M, Saura J, Solà C. The CD200R1 microglial inhibitory receptor as a therapeutic target in the MPTP model of Parkinson’s disease. J Neuroinflammation. 2021, 18: 88.
Jyothi HJ, Vidyadhara DJ, Mahadevan A, Philip M, Parmar SK, Manohari SG, et al. Aging causes morphological alterations in astrocytes and microglia in human substantia nigra pars compacta. Neurobiol Aging 2015, 36: 3321–3333.
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with[11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 2006, 21: 404–412.
Ahmed S, Kwatra M, Ranjan Panda S, Murty USN, Naidu VGM. Andrographolide suppresses NLRP3 inflammasome activation in microglia through induction of parkin-mediated mitophagy in in-vitro and in-vivo models of Parkinson disease. Brain Behav Immun 2021, 91: 142–158.
George S, Rey NL, Tyson T, Esquibel C, Meyerdirk L, Schulz E, et al. Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol Neurodegener 2019, 14: 34.
Croisier E, Moran LB, Dexter DT, Pearce RKB, Graeber MB. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J Neuroinflammation 2005, 2: 14.
Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 2015, 14: 183–193.
Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: Mechanisms and immunotherapy. Neuron 2018, 97: 742–768.
Sosa RA, Murphey C, Ji N, Cardona AE, Forsthuber TG. The kinetics of myelin antigen uptake by myeloid cells in the central nervous system during experimental autoimmune encephalomyelitis. J Immunol 2013, 191: 5848–5857.
Merson TD, Binder MD, Kilpatrick TJ. Role of cytokines as mediators and regulators of microglial activity in inflammatory demyelination of the CNS. Neuromol Med 2010, 12: 99–132.
Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140: 1900–1913.
Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132: 1175–1189.
Hagan N, Kane JL, Grover D, Woodworth L, Madore C, Saleh J, et al. CSF1R signaling is a regulator of pathogenesis in progressive MS. Cell Death Dis 2020, 11: 904.
Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 2005, 11: 146–152.
Cignarella F, Filipello F, Bollman B, Cantoni C, Locca A, Mikesell R, et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol 2020, 140: 513–534.
Rice RA, Pham J, Lee RJ, Najafi AR, West BL, Green KN. Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia 2017, 65: 931–944.
Hoeffel G, Ginhoux F. Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol 2018, 330: 5–15.
McGrath KE, Frame JM, Palis J. Early hematopoiesis and macrophage development. Semin Immunol 2015, 27: 379–387.
Guilliams M, Scott CL. Does niche competition determine the origin of tissue-resident macrophages? Nat Rev Immunol 2017, 17: 451–460.
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49: 489–502.
Bruttger J, Karram K, Wortge S, Regen T, Marini F, Hoppmann N, et al. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity 2015, 43: 92–106.
Rubino SJ, Mayo L, Wimmer I, Siedler V, Brunner F, Hametner S, et al. Acute microglia ablation induces neurodegeneration in the somatosensory system. Nat Commun 2018, 9: 4578.
Peng J, Zou Q, Chen MJ, Ma CL, Li BM. Motor deficits seen in microglial ablation mice could be due to non-specific damage from high dose diphtheria toxin treatment. Nat Commun 2022, 13: 3874.
Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SBG, Guyenet PG, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 2012, 484: 105–109.
Priller J, Flügel A, Wehner T, Boentert M, Haas CA, Prinz M, et al. Targeting gene-modified hematopoietic cells to the central nervous system: Use of green fluorescent protein uncovers microglial engraftment. Nat Med 2001, 7: 1356–1361.
Xu Z, Rao Y, Huang Y, Zhou T, Feng R, Xiong S, et al. Efficient strategies for microglia replacement in the central nervous system. Cell Rep 2020, 32: 108041.
Xu Z, Zhou X, Peng B, Rao Y. Microglia replacement by bone marrow transplantation (Mr BMT) in the central nervous system of adult mice. STAR Protoc 2021, 2: 100666.
Xu Z, Rao Y, Peng B. Protocol for microglia replacement by peripheral blood (Mr PB). STAR Protoc 2021, 2: 100613.
Xu Z, Peng B, Rao Y. Microglia replacement by microglia transplantation (Mr MT) in the adult mouse brain. STAR Protoc 2021, 2: 100665.
Sevenich L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front Immunol 2018, 9: 697.
Shemer A, Grozovski J, Tay TL, Tao J, Volaski A, Süß P, et al. Engrafted parenchymal brain macrophages differ from microglia in transcriptome, chromatin landscape and response to challenge. Nat Commun 2018, 9: 5206.
Grassivaro F, Menon R, Acquaviva M, Ottoboni L, Ruffini F, Bergamaschi A, et al. Convergence between microglia and peripheral macrophages phenotype during development and neuroinflammation. J Neurosci 2020, 40: 784–795.
Shibuya Y, Kumar KK, Mader MMD, Yoo Y, Ayala LA, Zhou M, et al. Treatment of a genetic brain disease by CNS-wide microglia replacement. Sci Transl Med 2022, 14: eabl9945.
Kobashi S, Terashima T, Katagi M, Urushitani M, Kojima H. Bone marrow-derived inducible microglia-like cells ameliorate motor function and survival in a mouse model of amyotrophic lateral sclerosis. Cytotherapy 2022, 24: 789–801.
Qin C, Wang K, Zhang L, Bai L. Stem cell therapy for Alzheimer’s disease: An overview of experimental models and reality. Animal Model Exp Med 2022, 5: 15–26.
Lee JC, Seong J, Kim SH, Lee SJ, Cho YJ, An J, et al. Replacement of microglial cells using Clodronate liposome and bone marrow transplantation in the central nervous system of SOD1G93A transgenic mice as an in vivo model of amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2012, 418: 359–365.
Willis EF, Vukovic J. Protocol for brain-wide or region-specific microglia depletion and repopulation in adult mice. STAR Protoc 2020, 1: 100211.
Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci 2007, 10: 1544–1553.
Hohsfield LA, Najafi AR, Ghorbanian Y, Soni N, Hingco EE, Kim SJ, et al. Effects of long-term and brain-wide colonization of peripheral bone marrow-derived myeloid cells in the CNS. J Neuroinflammation 2020, 17: 279.
Wang L, Liu Y, Yan S, Du T, Fu X, Gong X, et al. Disease progression-dependent expression of CD200R1 and CX3CR1 in mouse models of Parkinson’s disease. Aging Dis 2020, 11: 254–268.
Varvel NH, Grathwohl SA, Baumann F, Liebig C, Bosch A, Brawek B, et al. Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc Natl Acad Sci U S A 2012, 109: 18150–18155.
Cronk JC, Filiano AJ, Louveau A, Marin I, Marsh R, Ji E, et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J Exp Med 2018, 215: 1627–1647.
Li B, Gonzalez-Toledo ME, Piao CS, Gu A, Kelley RE, Zhao LR. Stem cell factor and granulocyte colony-stimulating factor reduce β-amyloid deposits in the brains of APP/PS1 transgenic mice. Alzheimer’s Res Ther 2011, 3: 1–8.
Krivit W, Sung JH, Shapiro EG, Lockman LA. Microglia: The effector cell for reconstitution of the central nervous system following bone marrow transplantation for lysosomal and peroxisomal storage diseases. Cell Transplant 1995, 4: 385–392.
Zhou T, Li Y, Li X, Zeng F, Rao Y, He Y, et al. Microglial debris is cleared by astrocytes via C4b-facilitated phagocytosis and degraded via RUBICON-dependent noncanonical autophagy in mice. Nat Commun 2022, 13: 6233. https://doi.org/10.1038/s41467-022-33932-3
Acknowledgments
We thank Jiachen Zhu and Fang Lei (Fudan University) for their excellent laboratory management. This review was supported by STI2030-Major Projects (2022ZD0204700), the National Natural Science Foundation of China (31922027 and 32170958), the Shanghai Academic/Technology Research Leader Program (21XD1420400), the Shanghai Pilot Program for Basic Research (21TQ014), the Shanghai Municipal Science and Technology Major Project, The Innovative Research Team of High-Level Local Universities in Shanghai, the Shanghai Municipal Science and Technology Major Project (2018SHZDZX01), ZJ Lab, and the China Postdoctoral Science Foundation (2021M690687).
Author information
Authors and Affiliations
Contributions
Lijuan Zhang, Yafei Wang, Taohui Liu contributed equally to this review.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that there are no conflicts of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, L., Wang, Y., Liu, T. et al. Novel Microglia-based Therapeutic Approaches to Neurodegenerative Disorders. Neurosci. Bull. 39, 491–502 (2023). https://doi.org/10.1007/s12264-022-01013-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-022-01013-6