
Abstract
Although KIT mutations are present in 20–25% of cases of t(8;21)(q22;q22) acute myeloid leukemia (AML), concurrent development of systemic mastocytosis (SM) is exceedingly rare. We examined the clinicopathologic features of SM associated with t(8;21)(q22;q22) AML in ten patients (six from our institutions and four from published literature) with t(8;21) AML and SM. In the majority of these cases, a definitive diagnosis of SM was made after chemotherapy, when the mast cell infiltrates were prominent. Deletion 9q was an additional cytogenetic abnormality in four cases. Four of the ten patients failed to achieve remission after standard chemotherapy and seven of the ten patients have died of AML. In the two patients who achieved durable remission after allogeneic hematopoietic stem cell transplant, recipient-derived neoplastic bone marrow mast cells persisted despite leukemic remission. SM associated with t(8;21) AML carries a dismal prognosis; therefore, detection of concurrent SM at diagnosis of t(8;21) AML has important prognostic implications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The t(8;21)(q22;q22) occurs in about 5% of all acute myeloid leukemias (AML) and in 10% of cases of AML with FAB-M2 morphology [1]. The Runt domain transcription factor RUNX1 (AML1) is essential for differentiation of hematopoietic stem cells. The t(8;21) generates the RUNX1-RUNX1T1 (AML1-ETO) fusion protein that acts as transcriptional repressor and inhibits the expression of AML1 responsive genes. This in turn causes a block in hematopoietic stem cell differentiation which in conjunction with additional genetic events leads to leukemic transformation [2, 3]. Studies have shown that t(8;21) by itself is incapable of leukemogenesis and additional genetic events are necessary for development of AML [4, 5]. Among these additional genetic changes that cooperate with RUNX1-RUNX1T1 fusion to cause AML, activating KIT mutations appear to be the most common. Data suggest that activating mutations in the receptor tyrosine kinases such as KIT play a role in the development of overt t(8;21)AML [5–8].
Systemic mastocytosis (SM) occurs due to an activating KIT mutation in a hematopoietic progenitor that leads to abnormal proliferation and accumulation of neoplastic mast cells in the bone marrow and other organs. In a subset of SM termed systemic mastocytosis with associated hematological non-mast cell disease (SM-AHNMD) in the current WHO classification, SM coexists with hematologic malignancies that are usually of myeloid origin [1, 9]. SM-AHNMD comprises 20% of all cases of SM and the most common associated hematologic malignancies are acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), chronic myeloproliferative disorders, and myelodysplastic/myeloproliferative syndromes such as chronic myelomonocytic leukemia [10–13].
In previous studies, the frequency of KIT mutations involving exon 17 as represented by a single amino acid substitution at D816 in patients with t(8;21)(q22;q22) AML have ranged from 12% to 24.2% [14–17]. Most common mutations described were at D816 and involved the Asp to Val substitution (D816V) [14–17]. Other substitutions included D816H, D816Y, and those at N822 including N822K and N822T [14–17]. Despite the fact that activating exon 17 KIT mutations are not uncommon in t(8;21) AML, a review of published literature suggests that SM associated with t(8;21) AML is exceedingly rare. Data on such cases are limited and largely confined to a handful of single case reports. We report our experience with SM associated with t(8;21)(q22;q22) AML and review the current literature on this rare entity.
Materials and methods
Patients with t(8;21) AML who also met WHO criteria for SM either at initial diagnosis or at relapse were identified from the databases of our institutions. Additional cases were identified by PubMed search of the English language literature as well as from the reference list of the reported cases [17–20]. Immunohistochemistry for CD117, tryptase, and CD25 staining was performed using standard techniques. Polymerase chain reaction (PCR) amplification and direct sequencing of the exon 17 of the KIT gene were performed by methods previously described [9]. Briefly, genomic DNA was isolated from bone marrow specimens using a QiAmp kit (Qiagen, Valencia, CA, USA). The codon 816-containing region of the KIT gene was amplified using the primers: 5′-TGT GAA CAT CAT TCA AGG CGT AC-3′ (forward) and 5′-ACT CAG CCT GTT TCT GGG AAA CTC-3′ (reverse). PCR conditions were each cycle of 30 s at 93°C, 1 min at 50°C, and 5 min at 72°C for a total of 40 cycles. The resulting 322-bp product was purified from a 2.5% agarose gel using a gel extraction kit (Qiagen) and directly sequenced. On case 3, fluorescence in situ hybridization (FISH) analysis for t(8;21)(q22;q22) on morphologically identified bone marrow mast cells (target FISH) was performed. Briefly, Wright-Giemsa stained bone marrow aspirate smears were scanned with a Bioview Duet Image Analyzer (Bioview, Rehovot, Israel) to identify mast cells for FISH. Following destaining with Carnoy’s fixative and digestion with pepsin, slides were co-denatured with t(8;21) probe (Vysis, Downer’s Grove, IL, USA). Slides were counterstained with DAPI and analyzed on a BioView Duet Image analyzer. Details of the target FISH technique were described previously [21].
Results
The clinical and pathologic features of the patients are summarized in Table 1. Of the ten patients who are included in this report, cases 1 through 6 were treated at our institutions and four additional cases (cases 7–10) were identified from the literature [17–20]. Cases 1, 2, and 3 of this series were included in previous reports [9, 21].
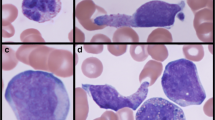
The patients ranged in age from 26 to 67 years and included three males and seven females. None of the patients had urtricaria pigmentosa or other evidence of extramedullary mast cell involvement except for one published case that showed hepatic infiltration with mast cells after AML induction therapy [17]. Symptoms related to mast cell mediator release were not reported in any of the patients. All cases had morphologic and immunophenotypic features of AML with maturation (FAB-M2). Leucocytosis was present at diagnosis in six of seven patients in whom initial blood counts were available. Patient 4 had metastatic breast cancer and had undergone anthracycline-containing combination chemotherapy. She developed SM with t(8;21)(q22;q22) AML 1 year after initiation of chemotherapy. In nine of the ten cases, increased bone marrow mast cells were noted at the time of AML diagnosis In case 4, SM was diagnosed following three cycles of consolidation chemotherapy for AML. In that case, the bone marrow biopsies at diagnosis were reevaluated, but failed to demonstrate coexisting SM. An activating exon 17 KIT mutation was detected in seven of nine cases in whom this data is available. Three patients had D816V, two had D816Y, and one carried the D816H mutation. In one case (case 6) the exact mutation was not analyzed while another (case 4) carried an A814S mutation in addition to D816V. It is noteworthy that four of the ten cases had chromosome 9q deletion as an additional cytogenetic abnormality. Of the eight cases where immunophenotype data on the myeloblasts was available by flow cytometry, four of eight cases reported dim expression of CD19 and five of eight cases showed CD56 expression (Table 1). Bone marrow pathology of a representative case is shown in Fig. 1.

a Pretreatment bone marrow aspirate (case 3) showing predominantly myeloblasts admixed with rare mast cells. Note the hypergranular cytoplasm within the mast cells (Wright-Giemsa, ×500). b Day 14 post-treatment bone marrow aspirate (case 3) with prominent mast cell infiltrate (Wright-Giemsa, ×200). c Immunohistochemistry for tryptase (case 3) highlights the mast cell infiltrate (×200). d Mast cells show expression of CD25 (case 3), a characteristic feature of neoplastic mast cells (×200)
All patients were initially treated with AML induction chemotherapy that contained an anthracycline and cytarabine. Four of the ten patients failed to achieve a morphologic remission after induction chemotherapy. Six of the ten patients have died of progressive or relapsed leukemia and one patient remains with active disease awaiting allogeneic HSCT. The two long-term survivors (cases 3 and 10) in this report had both undergone allogeneic HSCT from matched siblings after radiation-based high-dose conditioning. In these two patients, recipient-derived bone marrow mast cells were detected up to a year after HSCT, but the leukemia remained in durable remission.
Discussion
In this report, we have summarized our experience and reviewed the published literature on SM that coexists with t(8;21)(q22;q22) AML. KIT mutations are the most common additional genetic abnormality in t(8;21) AML and range in their incidence from 26% to 47% in various reports [22–24]. However, among the t(8;21) AML with KIT mutations, the number of cases that have concurrent SM appears to be extremely rare. Hence, it appears that specific genetic events in addition to activating KIT mutations and t(8;21) are required for development of mastocytosis. Although it can be postulated that these additional genetic events may promote mast cell differentiation from leukemic progenitors, the nature of these additional genetic aberrations remains unknown.
Studies examining the relationship between mast cells and leukemic blasts have shown that the neoplastic mast cells of SM associated with t(8;21) carry the RUNX1-RUNX1T1 translocation, thereby proving their derivation from the leukemic clone. This was demonstrated in case 3 of this series by target FISH, the details of which have been published elsewhere [21]. Similar findings were noted in another published case that is included in the current series (case 10, Nagai et al.) where KIT mutation as well as RUNX1-RUNX1T1 translocation was detected in recipient-derived clonal mast cells that persisted after allogeneic HSCT [20]. In both these cases, the recipient-derived mast cells gradually declined and the AML remained in remission. It is unclear if this phenomenon represents gradual apoptosis of these mast cells or progressive elimination of leukemic progenitors due to graft-versus-leukemia effect.
We sought to examine if there were other distinct cytogenetic or pathologic features that were associated with SM and t(8;21) AML. Deletion of chromosome 9q was an additional cytogenetic finding in four of the ten cases. Del (9q) has been previously reported as part of a complex phenotype in another case of t(8;21) positive myelomastocytic leukemia (AML with increased bone marrow mast cells not meeting criteria for SM) [26]. In addition, del (9q) is the most common additional cytogenetic abnormality in t(8;21) AML and has been reported in 7–14% of pediatric cases and 9.7% of adult cases [27]. In comparison, del (9q) appears to be more frequent in t(8;21) with associated SM, suggesting that this deletion may play a role in the pathogenesis of SM. Recently, TLE1 and TLE4 have been identified as critical genes in the commonly deleted 9q region in t(8;21) AML [28]. In vitro experiments using Kasumi-1 cell line showed that these genes behave as tumor suppressors and knockdown of TLE1 or TLE4 increased the rate of cell division of the AML1-ETO expressing Kasumi-1 cell line while forced expression of either caused apoptosis and cell death [28].
The precise incidence of SM coexisting with t(8;21) AML is unknown. In previous reports, authors have cautioned that in some cases of AML with coexisting SM, the diagnosis of SM maybe missed on the initial bone marrow evaluation due to the excess numbers of blasts which mask the underlying mast cells and the tendency of mast cells to localize within stroma of bone marrow particles in aspirate smears [9]. The bone marrow mast cell infiltrate appears to become more evident when the leukemic blasts decrease after therapy as was seen in this report (Fig. 1b). This prominence of the mast cell infiltrate after chemotherapy may also be due to the poor sensitivity of the mast cells to leukemia chemotherapy as evidenced by their persistence even after high-dose chemotherapy conditioning for allogeneic HSCT [20]. Infiltration of extramedullary tissues with mast cells, as well as symptoms due to mast cell mediator release, appear to be distinctly uncommon in cases of SM associated with t(8;21) AML. This may be due to the lack of functionality of these neoplastic MC as a result of the leukemic aberrations they carry.
AML with t(8;21)(q22;q22) as the sole cytogenetic abnormality in general have a favorable outcome when treated with consolidation regimens containing high-dose cytarabine [29, 30]; however, studies have shown that only 50% of t(8;21) AML patients are alive at 5 years [27]. D816 KIT mutations were detected in 10.5% of patients with t(8;21) AML in one study [17]. The presence of KIT mutations (D816 and others) may explain the poor prognosis of a subset of t(8;21) patients as activating exon 17 KIT mutations have now been shown to be a major adverse prognostic factor for event-free and overall survival in t(8;21) AML [14–17] and a predictor of higher relapse risk [15]. The number of patients with mutant KIT who had SM in addition has not been reported in any of these studies, which may be an indicator of its extreme rarity. Interestingly, in previous studies it has been reported that 91–100% of t(8;21) AML patients with KIT mutations achieved complete remission after chemotherapy [15, 25]. However, in our current series induction failure occurred in four of ten patients. This observation again re-emphasizes the fact that SM associated with AML has a grave prognosis.
It is worth noting that of the ten patients (Table 1) only two patients achieved a durable remission of AML. Both of these patients had undergone allogeneic HSCT. One of our patients (case 3) is in continuous remission 4 years after allogeneic HSCT. Sperr et al. have reported a patient with myelomastocytic leukemia and t(8;21) AML who relapsed after a reduced intensity HSCT, but achieved a lasting remission following high-dose conditioning and a second HSCT from the same donor, thereby suggesting that conditioning intensity may be important [26]. Although data are very limited, the dismal results with conventional chemotherapy suggests that allogeneic HSCT should be an early consideration in SM associated with t(8;21) AML in patients who are suitable candidates. The D816V KIT mutation is resistant to the tyrosine kinase inhibitor imatinib [31]. Dasatinib, to which KIT D816V is sensitive, may have a role in the therapy of these cases as evidenced by in vitro data as well as its in vivo activity of this drug in recent reports of SM, including a patient with SM-AML [32–34]. Other tyrosine kinase inhibitors like midostaurin (PKC 412) and nilotinib (AMN 107)) that are capable of inhibiting activating KIT D816V may also have potential in the treatment of t(8;21) AML that carry this mutation [35, 36].
In conclusion, SM associated with t(8;21) AML is extremely rare and carries a dismal prognosis. During evaluation of t(8;21)(q22;q22) AML, care should be taken to look for coexisting SM in the initial and subsequent bone marrow specimens since the mast cell infiltrate may be subtle and easily overlooked. This will help identify a subset of t(8;21) AML patients with a particularly poor prognosis.
References
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW eds (2008) WHO classification of tumors of haematopoietic and lymphoid tissues. IARC, Lyon
Asou N (2003) The role of a Runt domain transcription factor AML1/RUNX1 in leukemogenesis and its clinical implications. Crit Rev Oncol Hematol 45:129–150
Tenen DG (2003) Disruption of differentiation in human cancer. AML shows the way. Nat Rev Cancer 3:89–101
Yuan Y, Zhou L, Miyamoto T et al (2001) AML-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc Natl Acad Sci USA 98:10398–10403
Wang YY, Zhou GB, Yin T et al (2005) AML1-ETO and C-KIT mutation/overexpression in t(8;21) leukemia: implication in stepwise leukemogenesis and response to Gleevec. Proc Natl Acad Sci USA 102:1104–1109
Kuchenbauer F, Feuring-Buske M, Buske C (2005) AML1-ETO needs a partner. Cell Cycle 4:1716–1718
Grisolano JL, O’Neal J, Cain J, Tomasson MH (2001) An activated receptor tyrosine kinase, TEL/PDGF cooperates with AML1/ETO to induce acute myeloid leukemia in mice. Proc Natl Acad Sci USA 100:9506–9511
Rhoades KL, Hetherington CJ, Harakawa N et al (2000) Analysis of the role of AML-ETO in leukemogenesis using an inducible transgeneic mouse model. Blood 96:2108–2115
Pullarkat VA, Bueso-Ramos C, Lai R et al (2003) Systemic mastocytosis with associated clonal hematological non-mast cell lineage disease: analysis of clinicopathologic features and activating c-kit mutations. Am J Hematol 73:12–17
Sperr WR, Horny H-P, Valent P (2002) Spectrum of associated clonal hematologic non-mast cell lineage disorders occurring in patients with systemic mastocytosis. Int Arch Allergy Immunol 127:140–142
Sperr WR, Horny HP, Lechner K, Valent P (2000) Clinical and biologic diversity of leukemias occurring in patients with mastocytosis. Leuk Lymphoma 37:473–486
Travis WD, Li CY, Yam LT, Bergstralh EJ, Swee RG (1988) Significance of systemic mast cell disease with associated hematologic disorders. Cancer 62:965–972
Horny HP, Parwaresch MR, Lennert K (1985) Bone marrow findings in systemic mastocytosis. Human Pathol 16:808–814
Cairoli R, Beghini A, Grillo G et al (2006) Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood 107:3463–3468
Paschka P, Marcucci G, Ruppert AS et al (2006) Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv (16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol 24:3904–3911
Schnittger S, Kohl TM, haferlach T, Kern W, Hiddemann W, Spiekermann K, Schoch C (2006) KIT-D816 mutation in AML1-ETO positive AML are associated with impaired event-free and overall survival. Blood 107:1791–1799
Wong KF, Chan JKC, Chan JCW, Kwong YL, Ma SK, Chow TC (1991) Concurrent acute myeloid leukemia and systemic mastocytosis. Am J Hematol 28:243–244
Escribano L, Garcia-Montero A, Nunez-Lopez R et al (2004) Systemic mastocytosis associated with acute myeloid leukemia: case report and implications for disease pathogenesis. J Allergy Clin Immunol 114:28–33
Bernd HW, Sotlar K, Lorenzen J et al (2004) Acute myeloid leuke mia with t(8;21) associated with “occult” mastocytosis. Report of an unusual case and review of the literature. J Clin Pathol 57:324–328
Nagai S, Ichikawa M, Takahashi T, Sato H, Yokota H, Oshima K, Izutsu K et al (2007) The origin of neoplastic mast cells in systemic mastocytosis with AML1/ETO positive acute myeloid leukemia. Exp Hematol 35:1747–1752
Pullarkat V, Bedell V, Kim Y et al (2007) Neoplastic mast cells in systemic mastocytosis associated with t(8;21) acute myeloid leukemia are derived from the leukemic clone. Leuk Res 31:261–265
De J, Zanjani R, Hibbard M et al (2007) Immunophenotype profile predictive of KIT activating mutations in AML-ETO leukemia. Am J Clin Path 128:550–557
Beghini A, Cairoli R, Morra E, Larriza L (1998) In vivo differentiation of mast cells from acute myeloid leukemia blasts carrying novel activating ligand-independent c-kit mutation. Blood Cells Mol Dis 24:262–270
Beghini A, Peterlongo P, Ripamonti CB et al (2000) C-Kit mutations in core binding factor leukemias [letter]. Blood 95:726–727
Nanri T, Matsuno N, Kawakita T, Suzushima H, Kawano F, Mitsuya H, Asou N (2005) Mutations in the receptor tyrosine kinase pathway are associated with clinical outcome in patients with acute myeloblastic leukemia harboring t(8;21) (q22;q22). Leukemia 19:1361–1366
Sperr WR, Drach J, Hauswirth AW et al (2005) Myelomastocytic Leukemia: evidence for the origin of mast cells from the leukemia clone and eradication by allogeneic stem cell transplantation. Clin Cancer Res 11:6787–6792
Marcucci G, Mrozek K, Ruppert AS et al (2003) Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t(8;21) differ from those of patients with inv(16): a Cancer and Leukemia Group B study. J Clin Oncol 23:5705–5717
Dayyani F, Wang J, Yeh J-R J (2008) Loss of TLE1 and TLE4 from the del(9q) commonly deleted region in AML cooperate with AML1-ETO to affect myeloid cell proliferation and survival. Blood 111(8):4338–4347
Bloomfield CD, Lawrence D, Byrd JC et al (1998) Frequency of prolonged remission duration after high dose cytorabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res 58:4173–4179
Byrd JC, Dodge RK, Carroll A et al (1999) Patients with t(8;21)(q22;q22) and acute myeloid leukemia have superior failure free and overall survival when repetitive cycles of high-dose cytarabine are administered. J Clin Oncol 17:3767–3775
Frost MJ, Ferrao PT, Hughes TP, Ashman LK (2002) Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI 571) compared with wild-type c-KIT whereas the kinase domain mutant D816VKit is resistant. Mol Cancer Ther 1:1115–1124
Shah NP, Lee FY, Luo R et al (2006) Dasatinib (BMS-354825) inhibits KITD816V, an imatinib-resistant activating mutation that triggers neoplastic growth in most patients with systemic mastocytosis. Blood 108:286–291
Purtill D, Cooney J, Sinnah R et al (2008) Dasatinib therapy for systemic mastocytosis: four cases. Eur J Haematol 80:456–458
Ustun C, Corless CL, Savage N et al (2008) Chemotherapy and dasatinib induce long-term hematologic and molecular remission in systemic mastocytosis with acute myeloid leukemia with KIT D816V. Leuk Res (Epub ahead of print)
Gotlib J, Berube C, Growney JD et al (2005) Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood 106:2865–2870
Von Bubnoff N, Gorantla SHP, Kancha RK et al (2005) The systemic mastocytosis-specific activating cKit mutation D816V can be inhibited by the tyrosine kinase inhibitor AMN107. Leukemia 19:1670–1671
Acknowledgements
The authors declare that there are no sources of support to acknowledge.
Conflicts of interest statement
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pullarkat, S.T., Pullarkat, V., Kroft, S.H. et al. Systemic mastocytosis associated with t(8;21)(q22;q22) acute myeloid leukemia. J Hematopathol 2, 27–33 (2009). https://doi.org/10.1007/s12308-009-0023-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12308-009-0023-2