
Abstract
Chronic kidney disease (CKD) is increasingly recognized as a global health problem, and new and effective strategies are needed for the management of this condition. Recently, there has been renewed interest in the relationship between serum uric acid (SUA) levels and CKD, and several recent trials have demonstrated a possible link between SUA and the development and/or progression of CKD in patients with and without diabetes. The identification of key urate transporters such as urate transporter 1 and glucose transporter 9 has provided not only insights into the pathophysiology of hyperuricemia, but also possible links to other processes, such as glucose homeostasis. The renewed interest in the role of SUA in CKD has coincided with the development of sodium glucose co-transporter 2 inhibitors for the treatment of diabetes. In addition to improving glycemic control, these agents, acting via the kidneys in an insulin-independent manner, have also been shown to reduce SUA levels and potentially improve some measures of renal function. This review will discuss the role of uric acid in CKD treatment, and how SUA-lowering therapies may prevent or delay the progression of CKD.
Funding: Janssen Scientific Affairs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic kidney disease (CKD) is a key determinant of poor health outcomes in major non-communicable diseases such as cardiovascular (CV) disease and diabetes, and it is increasingly recognized as a global public health problem [1, 2]. The burden of CKD includes not only progression to end-stage renal disease (ESRD), but also complications related to renal impairment and increased risk of CV disease [2]. The increased morbidity and mortality related to CKD may be reduced with early detection and effective management to prevent or delay further progression of renal dysfunction and its associated complications [1, 2].
Interest in the relationship between serum uric acid (SUA) and CKD has been renewed recently, due in part to new experimental and clinical data [3, 4]. The debate continues regarding whether SUA plays a causal role in CKD or is simply a marker of renal dysfunction [3]. Emerging evidence continues to support a pathogenic role for SUA in CKD [5–9]. Notably, this renewed interest has coincided with the development of sodium glucose co-transporter 2 (SGLT2) inhibitors for type 2 diabetes mellitus (T2DM); these agents, acting via the kidneys in an insulin-independent manner, reduce SUA and potentially improve renal outcomes [10–13]. In this review, we will discuss the currently available evidence on the relationship between SUA and CKD, and how the uric acid-lowering effects of SGLT2 inhibitors may contribute to renoprotection. This article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by the author.
Uric Acid and Hyperuricemia
Uric acid is a weak organic acid that under physiologic conditions exists mainly as monosodium urate [14]; therefore, uric acid and urate, although chemically different, are generally interchangeable terms. In higher primates, uric acid is the end product of the metabolism of unwanted purine nucleotides [4, 10, 15]. Unlike other mammals, higher primates lack the enzyme uricase that converts uric acid to allantoin [15–17]. Beneficial effects of uric acid, such as its ability to scavenge harmful radicals, may have been evolutionarily advantageous [15, 16]. However, increased SUA levels can lead to urate crystallization and associated complications, such as kidney stones and gout. There is also increasing evidence that increased SUA levels may negatively affect CV and renal health in a crystal-independent manner [18].
About two-thirds of uric acid is produced endogenously, primarily in the liver, muscle, and intestine, and one-third is derived from dietary sources, including fructose, alcohol, and purine-rich foods like certain meats and seafood [4]. Uric acid is produced from fructose via an endogenous pathway as well as from dietary sources (e.g., corn syrup), and a high fructose intake can cause intracellular adenosine triphosphate depletion and nucleotide turnover, with uric acid formation [19]. In diabetes, fructose is generated endogenously via the polyol pathway; blocking the metabolism of fructose in a diabetic mouse model has been found to mitigate the development of tubulointerstitial injury through blocking the production of tubular uric acid [20].
Overall, the SUA level is determined primarily by a balance of uric acid generation, reabsorption, and excretion in the kidney, but it can also be influenced by other factors, like dietary purine intake, hydration status, and use of diuretics [4, 15]. Volume depletion will reduce uric acid excretion independently of urine flow [21], while a comparison of mannitol- and glucose-associated osmotic diuresis showed that, for each osmolar unit, glucosuric diuresis had a threefold greater uricosuric effect [22].
Uric acid is excreted primarily via the kidneys; therefore, the kidneys play an important role in regulating SUA levels [4, 23]. In the kidneys, urate reabsorption and secretion are regulated by multiple urate transporters. For example, urate transporter 1 (URAT1) is expressed in the apical membrane of proximal tubule cells and is responsible for urate reabsorption [15, 24]. In addition, two isoforms of the urate transporter SLC2A9 (glucose transporter 9 [GLUT9]) are known, one expressed at the baso-lateral side of the proximal tubule and the other on the apical membranes in the collecting duct. Normally, no glucose will reach the collecting ducts, but in diabetes (and especially with SGLT2 inhibition) large amounts of glucose can be excreted: GLUT9 is now exchanging glucose and fructose for uric acid, and therefore plays an important role in regulating urate homeostasis [23, 25–29]. Only a small fraction of uric acid (8–10%) is excreted (net urinary urate excretion is 250–750 mg/day), as about 90% of filtered urate is reabsorbed [4]. Obesity, insulin resistance, hypertension, and diuretic use have been associated with increased urate reabsorption in renal tubules and hyperuricemia [4]. It is currently not known how tubuloglomerular feedback or fructose-induced uric acid formation can affect uricosuria in the setting of SGLT2 inhibition.
The definition of hyperuricemia in adults is not universally agreed upon, but commonly used thresholds are in the range of 6–7 mg/dL (350–400 μM/L), and are usually higher for men than for women [23]. The presence of hyperuricemia has been associated with a number of disorders including CV and renal disease, metabolic syndrome, diabetes, and hypertension [30]. In patients with T2DM, hyperuricemia is associated with an increased risk of developing gout, kidney stones, vascular complications [31, 32], and death [32].
Establishing the exact relationship between SUA level and diabetes has been complicated by the fact that both hypouricemia and hyperuricemia have been linked to diabetes [33]. SUA levels may be low in patients with recently diagnosed diabetes due to increased urate excretion [34]. Hyperuricemia, on the other hand, has been associated with the development of diabetes, especially in the context of insulin resistance and impaired glucose tolerance [35]. In a large population-based study, increased SUA levels were shown to be an independent risk factor for developing T2DM, and one-quarter of diabetes cases could be attributed to high SUA levels [36]. These findings may have a strong public health impact, as lowering SUA may decrease the incidence of diabetes, if the relationship is causal [37].
Chronic Kidney Disease
CKD is defined as abnormalities of kidney structure or function that persist for more than 3 months with health implications [37, 38]. Abnormalities include decreased glomerular filtration rate (GFR; <60 mL/min/1.73 m2) or evidence of one or more markers of kidney damage (albuminuria, urine sediment abnormalities, electrolyte abnormalities, abnormalities detected by histology or imaging, or history of kidney transplantation [37]). CKD is becoming increasingly recognized as a worldwide public health problem that may be more common in developing countries and in disadvantaged and minority populations [1]. In the USA, CKD is present in over 13% of the population, making it more prevalent than diabetes [38, 39]. Using an updated Kidney Disease: Improving Global Outcomes (KDIGO) classification system for CKD to include the level of both renal function and urinary albumin excretion, recent estimates suggest that the prevalence of CKD among patients with diabetes is 43.5%, and the prevalence of mildly decreased renal function or worse [estimated GFR (eGFR) <60 mL/min/1.73 m2] is 22.0% [40]. Diabetic kidney disease is the single strongest predictor of mortality in patients with diabetes [41, 42]. Furthermore, CKD is associated with an eight- to tenfold increase in CV mortality, and its presence is a negative risk factor in patients with diabetes and hypertension [1]. In developed countries like the USA, diabetic kidney disease is the leading cause of ESRD [41].
Prognosis is determined primarily by GFR and albuminuria category (albumin excretion rate <30 [A1], 30–300 [A2], or >300 [A3] mg/24 h for normal, microalbuminuria, or macroalbuminuria [clinical proteinuria], respectively) [43], but other factors, such as the cause of CKD and presence of comorbidities, may also be taken into account. Progression of CKD, as defined by a decline in category (e.g., from CKD stage 3 to 4) or a loss of 5 mL eGFR or more per year, can potentially lead to ESRD. It has been suggested that early detection and management of CKD, when patients are typically seen by general internists, can slow or prevent progression of the disease using simple and relatively inexpensive interventions, including control of blood pressure and, in diabetes, blood glucose level [1, 44]. However, while glycemic control has been shown to reduce the initial development of micro- and macroalbuminuria, there are limited data on its effects on GFR [45, 46]. Control of CV risk factors, such as hyperlipidemia, plays also an important role in the management of patients with diabetic kidney disease [38]. Despite evidence of significant reductions in CV events with statin therapy in patients with and without albuminuria, no significant effect on slowing CKD progression was found with statin use [45, 47, 48]; these findings highlight the need for more effective strategies for managing CKD.
Relationship Between Hyperuricemia and CKD
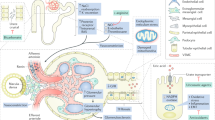
SUA levels are usually increased in patients with CKD, but whether SUA plays a causal role in CKD remains a topic of debate [3, 4]. A recent renal biopsy study of 167 Japanese patients with CKD (eGFR <60 mL/min/1.73 m2) found that, after adjusting for age, sex, hypertension, diabetes, and eGFR, an SUA level above 7.2 mg/dL was associated with renal arterial wall thickening and hyalinosis consistent with renal arteriolopathy [49]. The suggestion that hyperuricemia is a risk factor for CKD is supported by many studies. Preclinical data suggest that hyperuricemia induces renal injury through multiple crystal-independent mechanisms involving renal vasoconstriction mediated by endothelial dysfunction, inflammation, and activation of the renin–angiotensin system (Fig. 1) [8, 17, 18, 50–53]. Hyperuricemia has also been associated with afferent renal arteriolopathy, tubulointerstitial fibrosis, and renal inflammation [54].

Potential mechanisms of uric acid-induced kidney disease [4]. COX-2 Cyclooxygenase-2, ECM extracellular matrix, EMT epithelial-to-mesenchymal transition, NO nitric oxide, RAS renin–angiotensin system, VEC vascular endothelial cells, VSMC vascular smooth muscle cells. Reprinted from Seminars in Nephrology, Vol. 32, Issue 5. Kang DH, Chen W, Uric acid and chronic kidney disease: new understanding of an old problem. pp. 447–52. Copyright 2011, with permission from Elsevier
Data from most epidemiological studies suggest that uric acid is a major independent risk factor for the development and progression of renal disease in patients both with [9, 55–57] and without diabetes [56, 58–68]. In a recent meta-analysis of 13 observational trials involving over 190,000 patients [56], the presence of hyperuricemia was an independent predictor of new-onset CKD development. The effect was seen in subsets of patients with and without diabetes, and the magnitude of the effect was comparable in both subsets. Possibly reflecting dietary differences, this effect was more pronounced in Western populations than in Asian populations [56]. Notably, in patients with increased SUA levels, the risk of developing new-onset CKD increased as the duration of follow-up increased, suggesting that hyperuricemia may play a role in the long-term progression of chronic renal dysfunction [56]. In a recent cross-sectional study enrolling 3212 Chinese patients with T2DM, the prevalence of diabetic kidney disease was significantly elevated in hyperuricemia subjects and increased with increasing uric acid levels [55]. The presence of a mutation in the urate transporter GLUT9 strongly predicted progression of CKD in a cohort of 755 patients with CKD [69]. Lastly, in a study of 4233 women, increased SUA during pregnancy was associated with a higher risk of subsequent hospitalization for significant atherosclerotic-related events, including renal failure, diabetic ketoacidosis, hypertensive crisis, and CV events [70].
Some epidemiological studies, however, have reported no significant relationship between hyperuricemia and progression of kidney disease and development of kidney failure [71, 72]. In a prospective analysis of the Mild to Moderate Kidney Disease study, 177 patients with non-diabetic primary CKD and various stages of renal impairment were followed for 7 years [71]. Increased SUA levels predicted CKD progression only when patients receiving uric acid-lowering drugs were excluded from the analysis. After adjusting for baseline GFR and proteinuria, hyperuricemia was no longer an independent predictor of CKD progression [71]. Similarly, in the Modification of Diet in Renal Disease study, 838 patients with stage 3–4 CKD were followed for median of 10 years [72]. In continuous analyses, each 1 mg/dL increase in SUA level was associated with an increased risk of all-cause mortality and CV disease-related mortality, but not with progression to kidney failure [72].
Diabetes
In patients with diabetes, a multifactorial treatment approach with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) is needed to control hypertension, i.e., reducing blood pressure below 140/90 mmHg, and to lower albuminuria. Tight glucose control and correction of hyperlipidemia have been shown to reduce CV risks, but currently no data exist to show that such interventions can slow down the progression of CKD [50, 73]. In patients with type 1 diabetes mellitus, elevated SUA levels at baseline were shown to predict the development of CKD in several trials [5–7, 57]. A recent multiple regression analysis performed on 1108 patients with type 1 diabetes identified SUA as the third most important risk factor after tumor necrosis factor receptor 1 and albuminuria, associated with early progressive renal decline [74]. Similar results have been reported in patients with T2DM. For example, in a study of 1449 patients with T2DM, normal kidney function, and no proteinuria, subjects were followed for 5 years to assess the occurrence of new-onset CKD [9]. During the 5-year period, the cumulative incidence of CKD was significantly higher in patients with baseline hyperuricemia (>7.0 mg/dL) compared with those without hyperuricemia (29.5% vs. 11.4%). A multivariate analysis in this population of patients with T2DM confirmed that hyperuricemia, together with older age, use of antihypertensive drugs, and lower eGFR and higher albuminuria at baseline, was an independent risk factor for the development of CKD [9].
IgA Nephropathy
IgA nephropathy (IgAN) is the most common type of primary glomerular disease characterized by variable tubulointerstitial lesions, and it can progress to ESRD over time [75]. In patients with IgAN, SUA levels have been shown to correlate with renal damage [76, 77] and progression of IgAN [58, 78, 79]. In a recent study of 93 patients with IgAN, baseline SUA levels significantly predicted the change in eGFR over a 6-month period after adjusting for multiple factors, including baseline GFR and albumin level [58]. In a long-term study (mean follow-up 5 years) of 348 consecutive cases of IgAN, patients with high SUA levels at baseline were significantly more likely to experience a decline in GFR (41% vs. 16%) and develop ESRD (65% vs. 35%) compared with patients with SUA levels within the normal range [78].
Acute Kidney Injury
Lastly, little is known about the relationship between SUA and acute kidney injury (AKI). Data from Atherosclerosis Risk in Communities, a population-based study of over 11,000 participants followed for an average of 12 years, indicated that increased plasma urate (>5.0 mg/dL) was independently associated with the risk of hospitalization for AKI [80]. For each 1 mg/dL increase in plasma urate beyond 5.0 mg/dL, the risk of AKI increased 16%. However, a causal role of urate in AKI could not be established.
Effects of Lowering SUA levels
SUA levels are an important risk factor for progression of renal dysfunction, hypertension, and lifestyle-related diseases such as metabolic syndrome or T2DM. Lowering SUA levels may represent a new therapeutic approach, but large clinical trials are warranted to evaluate the associated benefits and risks before implementation in clinical practice.
Allopurinol
Several small studies have explored the impact of treatment with the xanthine oxidase inhibitor allopurinol on renal outcomes [81–85]. Recent meta-analyses have produced variable results. In an analysis of randomized trials of patients with or without CKD at baseline, uric acid-lowering therapy with allopurinol had no effect on GFR compared with controls in five trials (n = 346) but abrogated increases in serum creatinine in three trials (n = 130) [85, 86]. In a separate analysis of 19 randomized trials involving a total of 992 patients with stage 3–5 CKD, treatment with allopurinol was associated with significant reductions in SUA levels and blood pressure and more favorable eGFR compared with controls [87]. Both meta-analyses reported significant heterogeneity among the trials of allopurinol in terms of design, duration of follow-up, and comparators used. It has been suggested that the potential renoprotective effect observed in one study of allopurinol may have been caused by lowering SUA levels from slightly above to slightly below the uric acid saturation point (6.8 mg/dL); mean SUA levels over the 7-year observation period were 7.2 mg/dL in the control group and 6.5 mg/dL in the allopurinol-treated group. Reducing the SUA level as low as possible may be less important than simply maintaining the level below the saturation point, in terms of preventing renal damage [84, 85]. Interpretation of results obtained with allopurinol can also be complicated by a secondary effect related to xanthine oxidase. Xanthine oxidase, which is inhibited by allopurinol, produces reactive oxygen species (ROS); inhibition of this ROS formation with allopurinol may have beneficial effects unrelated to SUA levels [4, 54, 87]. Two ongoing trials sponsored by the US government are evaluating the effects of allopurinol on the progression of IgAN [88] and diabetic nephropathy in patients with type 1 diabetes mellitus [89, 90].
Febuxostat and Topiroxostat
Febuxostat is a non-purine selective inhibitor of xanthine oxidase approved by the US FDA for the treatment of adult hyperuricemia with gout (not for hyperuricemia alone), and can be used in stage 3 CKD (eGFR 30–60 mL/min/1.73 m2). A 108-week prospective, double-blind, placebo-controlled study (FEATHER; UMIN identifier, UMIN000008343) is currently ongoing, investigating the effect of febuxostat on eGFR in 400 adult Japanese patients with stage 3 CKD and hyperuricemia (SUA 7.1–10.0 mg/dL) without gouty arthritis [91]. Topiroxostat is another selective xanthine oxidase inhibitor, approved for use in Japan. A recent 22-week double-blind study in 123 patients with stage 3 CKD and hyperuricemia with or without gout showed that treatment with topiroxostat significantly reduced SUA by about 45% and lowered the albumin to creatinine ratio by almost 30%, compared with placebo [92].
Losartan
Losartan is an ARB that has renoprotective effects in patients with diabetic nephropathy [93]. This effect is attributed primarily to its ability to reduce blood pressure and proteinuria [93], but evidence indicates that, unlike other ARBs, losartan also reduces SUA levels [94], possibly by inhibition of URAT1, leading to reduced urate reabsorption in the proximal tubule [95]. To determine the effect of SUA reduction on renal outcomes in patients with T2DM and nephropathy treated with losartan, a post hoc analysis was conducted using data from the Reduction of Endpoints in Non-Insulin-Dependent Diabetes Mellitus with the Angiotensin II Antagonist Losartan Trial (ClinicalTrials.gov identifier, NCT00308347) [96]. In 1342 patients, losartan treatment significantly reduced SUA levels over a 6-month period. Moreover, for every 0.5 mg/dL decrement in SUA, the risk of adverse renal outcomes (a doubling of serum creatinine or development of ESRD) decreased by 6% [96]. Excluding the effects of SUA on renal outcomes reduced the renoprotective effect of losartan from 22% to 17%, suggesting that the SUA-lowering effect of losartan accounts for approximately one-fifth of the agent’s renoprotective effects [96]. Results of this post hoc analysis provide evidence that strategies to reduce SUA levels may reduce renal risk.
SGLT2 Inhibitors
The kidney plays a key role in glucose homeostasis by contributing to endogenous glucose production via gluconeogenesis and by reabsorbing filtered glucose [97]. SGLT2 is a high-capacity, low-affinity glucose transporter that is found primarily in the proximal tubule of the kidney and is responsible for 90% of glucose reabsorption [98]. Inhibition of SGLT2 increases urinary glucose excretion, thereby improving glycemic control in an insulin-independent manner in patients with T2DM [99]. As a class, SGLT2 inhibitors have shown multiple beneficial effects in patients with T2DM, including reduction in HbA1C, decreased blood pressure, and weight loss [11–13, 98, 99]. They may also have some beneficial renal-related effects, including a reduction in intraglomerular pressure and amelioration of albuminuria [100, 101]. In addition, SGLT2 inhibitors also reduce SUA levels, possibly through mediating the GLUT9 urate transporter via induction of glycosuria: higher levels of glucose in the urine due to SGLT2 inhibition result in a higher level of exchange of uric acid across the apical membrane of tubular cells, so more uric acid is released from blood into the urine, ultimately reducing SUA levels [10, 102, 103]. In a pooled analysis of data from four phase 3 trials, canagliflozin was shown to reduce SUA levels compared with placebo [10]. In a subgroup with hyperuricemia at baseline, 20–30% of patients with hyperuricemia achieved normal SUA levels after 26 weeks of treatment with canagliflozin [10]. Similarly, in a meta-analysis of small, randomized, placebo-controlled trials, treatment with dapagliflozin at doses ≤20 mg significantly reduced SUA levels [13]. Results from individual phase 2 trials of the SGLT2 inhibitor empagliflozin have reported reductions in SUA [11, 12]. The uricosuric effect seen in phase 3 trials of all three FDA-approved SGLT2 inhibitors has not been associated with an increase in uric acid nephrolithiasis.
The beneficial effects of SGLT2 inhibition on renal parameters must be weighed against potential side effects [98]. As a class, SGLT2 inhibitors have been associated with an increased risk of genitourinary infections, both bacterial and fungal [11–13, 104]. For example, in clinical trials, canagliflozin was associated with increased risk of genital mycotic infections as well as events related to osmotic diuresis [104]. Renal-related adverse events were uncommon. Small, transient reductions in eGFR have been observed with SGLT2 inhibitors during the initial weeks of treatment [12, 104], but longer-term studies (104 weeks) have confirmed that eGFR remains stable after this initial reduction [101, 105]. Since the efficacy of SGLT2 inhibitors is dependent on renal function, it is not surprising that the HbA1C-lowering effects of SGLT2 inhibitors are reduced in patients with renal impairment [106–108]. In the USA, initiation of available SGLT2 inhibitors requires an eGFR >45 mL/min/1.73 m2 for empagliflozin and canagliflozin (100 mg), and >60 mL/min/1.73 m2 for dapagliflozin. Ongoing trials may provide further insight into the effects of SGLT2 inhibitors on SUA and renal outcomes. The CREDENCE trial (ClinicalTrials.gov identifier, NCT02065791), for example, is a phase 3 trial evaluating canagliflozin in patients with T2DM, stage 2 or 3 CKD (eGFR 60–89 and 45–59 mL/min/1.73 m2, respectively), and macroalbuminuria (albumin excretion >300 mg/24 h) [109]. The trial will assess whether canagliflozin has renal and vascular protective effects in this population. The ongoing phase 4 CANVAS-R trial (ClinicalTrials.gov identifier, NCT01989754) [110] will also study the effects of canagliflozin on renal endpoints.
Conclusions
The burden of CKD is increasingly recognized as a global health problem, underscoring the need for new and effective disease-management strategies. Renewed interest in the relationship between uric acid and CKD has led to new data that, on the whole, suggest that SUA may be a modifiable risk factor for CKD. Ongoing research continues to explore the exact relationship between hyperuricemia and CKD, and how SUA-lowering therapies may influence renal outcomes. Preliminary data suggest that SGLT2 inhibitors can reduce SUA levels, which may in turn contribute to a potential renoprotective effect. Ongoing clinical studies are prospectively evaluating the effect of SLGT2 inhibitors on SUA levels and renal parameters and outcomes in patients with CKD.
References
Couser WG, Remuzzi G, Mendis S, Tonelli M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. 2011;80:1258–70.
Levey AS, Atkins R, Coresh J, et al. Chronic kidney disease as a global public health problem: approaches and initiatives—a position statement from Kidney Disease Improving Global Outcomes. Kidney Int. 2007;72:247–59.
Johnson RJ, Nakagawa T, Jalal D, Sánchez-Lozada LG, Kang DH, Ritz E. Uric acid and chronic kidney disease: which is chasing which? Nephrol Dial Transpl. 2013;28:2221–8.
Kang DH, Chen W. Uric acid and chronic kidney disease: new understanding of an old problem. Semin Nephol. 2011;31:447–552.
Ficociello LH, Rosolowsky ET, Niewczas MA, et al. High-normal serum uric acid increases risk of early progressive renal function loss in type 1 diabetes: results of a 6-year follow-up. Diabetes Care. 2010;33:1337–43.
Hovind P, Rossing P, Tarnow L, Johnson RJ, Parving HH. Serum uric acid as a predictor for development of diabetic nephropathy in type 1 diabetes: an inception cohort study. Diabetes. 2009;58:1668–71.
Jalal DI, Rivard CJ, Johnson RJ, et al. Serum uric acid levels predict the development of albuminuria over 6 years in patients with type 1 diabetes: findings from the Coronary Artery Calcification in Type 1 Diabetes study. Nephrol Dial Transpl. 2010;25:1865–9.
Kang DH, Nakagawa T, Feng L, et al. A role for uric acid in the progression of renal disease. J Am Soc Nephrol. 2002;13:2888–97.
Zoppini G, Targher G, Chonchol M, et al. Serum uric acid levels and incident chronic kidney disease in patients with type 2 diabetes and preserved kidney function. Diabetes Care. 2012;35:99–104.
Davies MJ, Trujillo A, Vijapurkar U, Damaraju CV, Meininger G. Effect of canagliflozin on serum uric acid in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2015;17:426–9.
Ferrannini E, Berk A, Hantel S, et al. Long-term safety and efficacy of empagliflozin, sitagliptin, and metformin: an active-controlled, parallel-group, randomized, 78-week open-label extension study in patients with type 2 diabetes. Diabetes Care. 2013;36:4015–21.
Häring HU, Merker L, Seewaldt-Becker E, et al. Empagliflozin as add-on to metformin in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2014;37:1650–9.
Musso G, Gambino R, Cassader M, Pagano G. A novel approach to control hyperglycemia in type 2 diabetes: sodium glucose co-transport (SGLT) inhibitors: systematic review and meta-analysis of randomized trials. Ann Med. 2012;44:375–93.
Barr WG. Uric Acid. In: Walker HH, Hall WD, Hurst JW, editors. Clinical methods: the history, physical, and laboratory examinations. Boston: Butterworths; 1990. p. 770–2.
Hediger MA, Johnson RJ, Miyazaki H, Endou H. Molecular physiology of urate transport. Physiology (Bethesda). 2005;20:125–33.
Wu XW, Muzny DM, Lee CC, Caskey CT. Two independent mutational events in the loss of urate oxidase during hominoid evolution. J Mol Evol. 1992;34:78–84.
Preitner F, Pimentel A, Metref S, et al. No development of hypertension in the hyperuricemic liver-Glut9 knockout mouse. Kidney Int. 2015;87:940–7.
Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001;38:1101–6.
Johnson RJ, Nakagawa T, Sanchez-Lozada LG, et al. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes. 2013;62:3307–15.
Bjornstad P, Lanaspa MA, Ishimoto T, et al. Fructose and uric acid in diabetic nephropathy. Diabetologia. 2015;58:1993–2002.
Marangella M. Uric acid elimination in urine. Pathophysiological implications. Contrib Nephrol. 2005;147:132–48.
Skeith MD, Healey LA, Cutler RE. Urate excretion during mannitol and glucose diuresis. J Lab Clin Med. 1967;70:213–20.
Bobulescu IA, Moe OW. Renal transport of uric acid: evolving concepts and uncertainties. Adv Chronic Kidney Dis. 2012;19:358–71.
Enomoto A, Kimura H, Chairoungdua A, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–52.
Preitner F, Bonny O, Laverrière A, et al. Glut9 is a major regulator of urate homeostasis and its genetic inactivation induces hyperuricosuria and urate nephropathy. Proc Natl Acad Sci USA. 2009;106:15501–6.
Sakurai H. Urate transporters in the genomic era. Curr Opin Nephrol Hypertens. 2013;22:545–50.
Matsuo H, Chiba T, Nagamori S, et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet. 2008;83:744–51.
Kimura T, Takahashi M, Yan K, Sakurai H. Expression of SLC2A9 isoforms in the kidney and their localization in polarized epithelial cells. PLoS One. 2014;9:e84996. doi:10.1371/journal.pone.0084996.
Anzai N, Jutabha P, Amonpatumrat-Takahashi S, Sakurai H. Recent advances in renal urate transport: characterization of candidate transporters indicated by genome-wide association studies. Clin Exp Nephrol. 2012;16:89–95.
Soltani Z, Rasheed K, Kapusta DR, Reisin E. Potential role of uric acid in metabolic syndrome, hypertension, kidney injury, and cardiovascular diseases: is it time for reappraisal? Curr Hypertens Rep. 2013;15:175–81.
Ito H, Abe M, Mifune M, et al. Hyperuricemia is independently associated with coronary heart disease and renal dysfunction in patients with type 2 diabetes mellitus. PLoS One. 2001;6:e27817.
Xu Y, Zhu J, Gao L, et al. Hyperuricemia as an independent predictor of vascular complications and mortality in type 2 diabetes patients: a meta-analysis. PLoS One. 2013;8:e78206.
Kushiyama A, Tanaka K, Hara S, Kawazu S. Uric acid metabolism and diabetic complications. World J Diabetes. 2014;5:787–95.
Shichiri M, Iwamoto H, Shiigai T. Diabetic renal hypouricemia. Arch Intern Med. 1987;147:225–8.
Bo S, Cavallo-Perin P, Gentile L, Repetti E, Pagano G. Hypouricemia and hyperuricemia in type 2 diabetes: two different phenotypes. Eur J Clin Invest. 2001;31:318–21.
Dehghan A, van Hoek M, Sijbrands EJ, Hofman A, Witteman JC. High serum uric acid as a novel risk factor for type 2 diabetes. Diabetes Care. 2008;31:361–2.
Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3:1–150.
National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI). Chronic kidney disease: evaluation, classification, and stratification. 2002. http://www2.kidney.org/professionals/KDOQI/guidelines_ckd/toc.htm. Accessed Aug 2015.
Coresh J, Selvin E, Stevens LA. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038–47.
Bailey RA, Wang Y, Zhu V, Rupnow MF. Chronic kidney disease in US adults with type 2 diabetes: an updated national estimate of prevalence based on Kidney Disease: Improving Global Outcomes (KDIGO) staging. BMC Res Notes. 2014;7:415.
Saran R, Li Y, Robinson B, et al. US Renal Data System 2014 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis. 2015;66(suppl 1):S1–306.
Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124:2333–40.
Kowalski A, Krikorian A, Lerma EV. Diabetic nephropathy for the primary care provider: new understandings on early detection and treatment. Ochsner J. 2014;14:369–79.
Leveyand AS, Coresh J. Chronic kidney disease. Lancet. 2012;379:165–80.
Molitch ME, Adler AI, Flyvbjerg A, et al. Diabetic kidney disease: a clinical update from Kidney Disease: Improving Global Outcomes. Kidney Int. 2015;87:20–30.
Breyer JA, Bain RP, Evans JK, et al. Predictors of the progression of renal insufficiency in patients with insulin-dependent diabetes and overt diabetic nephropathy. The Collaborative Study Group. Kidney Int. 1996;50:1651–8.
Baigent C, Landray MJ, Reith C, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet. 2011;377:2181–92.
Colhoun HM, Betteridge DJ, Durrington PN, et al. Effects of atorvastatin on kidney outcomes and cardiovascular disease in patients with diabetes: an analysis from the Collaborative Atorvastatin Diabetes Study (CARDS). Am J Kid Dis. 2009;54:810–9.
Kohagura K, Kochi M, Miyagi T, et al. An association between uric acid levels and renal arteriolopathy in chronic kidney disease: a biopsy-based study. Hypertens Res. 2013;36:43–9.
Hovind P, Rossing P, Johnson RJ, Parving HH. Serum uric acid as a new player in the development of diabetic nephropathy. J Ren Nutr. 2011;21:124–7.
Sánchez-Lozada LG, Tapia E, Avila-Casado C, et al. Mild hyperuricemia induces glomerular hypertension in normal rats. Am J Physiol Ren Physiol. 2002;283:F1105–10.
Sánchez-Lozada LG, Tapia E, Santamaría J, et al. Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney Int. 2005;67:237–47.
Mazzali M, Kanellis J, Han L, et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Ren Physiol. 2002;282:F991–7.
Jalal DI, Maahs DM, Hovind P, Nakagawa T. Uric acid as a mediator of diabetic nephropathy. Semin Nephrol. 2011;31:459–65.
Yan D, Tu Y, Jiang F, et al. Uric acid is independently associated with diabetic kidney disease: a cross-sectional study in a Chinese population. PLoS One. 2015;10:e0129797.
Li L, Yang C, Zhao Y, Zeng X, Liu F, Fu P. Is hyperuricemia an independent risk factor for new-onset chronic kidney disease?: a systematic review and meta-analysis based on observational cohort studies. BMC Nephrol. 2014;15:122.
Rosolowsky ET, Ficociello LH, Maselli NJ, et al. High-normal serum uric acid is associated with impaired glomerular filtration rate in nonproteinuric patients with type 1 diabetes. Clin J Am Soc Nephrol. 2008;3:706–13.
Bakan A, Oral A, Elcioglu OC, et al. Hyperuricemia is associated with progression of IgA nephropathy. Int Urol Nephrol. 2015;47:673–8.
Bellomo G, Venanzi S, Verdura C, Saronio P, Esposito A, Timio M. Association of uric acid with change in kidney function in healthy normotensive individuals. Am J Kidney Dis. 2010;56:264–72.
Sonoda H, Takase H, Dohi Y, Kimura G. Uric acid levels predict future development of chronic kidney disease. Am J Nephrol. 2011;33:352–7.
Weiner DE, Tighiouart H, Elsayed EF, Griffith JL, Salem DN, Levey AS. Uric acid and incident kidney disease in the community. J Am Soc Nephrol. 2008;19:1204–11.
Chonchol M, Shlipak MG, Katz R, et al. Relationship of uric acid with progression of kidney disease. Am J Kidney Dis. 2007;50:239–47.
Obermayr RP, Temml C, Gutjahr G, Knechtelsdorfer M, Oberbauer R, Klauser-Braun R. Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol. 2008;19:2407–23.
Hsu CY, Iribarren C, McCulloch CE, Darbinian J, Go AS. Risk factors for end-stage renal disease: 25-year follow-up. Arch Intern Med. 2009;169:342–50.
Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol. 2000;10:403–9.
Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res. 2001;24:691–7.
Iseki K, Ikemiya Y, Kinjo K, Iseki C, Takishita S. Prevalence of high fasting plasma glucose and risk of developing end-stage renal disease in screened subjects in Okinawa, Japan. Clin Exp Nephrol. 2004;8:250–6.
Domrongkitchaiporn S, Sritara P, Kitiyakara C, et al. Risk factors for development of decreased kidney function in a southeast Asian population: a 12-year cohort study. J Am Soc Nephrol. 2005;16:791–9.
Testa A, Mallamaci F, Spoto B, et al. Association of a polymorphism in a gene encoding a urate transporter with CKD progression. Clin J Am Soc Nephrol. 2014;9:1059–65.
Wolak T, Shoham-Vardi I, Sergienko R, Sheiner E. High uric acid levels during pregnancy linked to increased risk for future atherosclerotic-related hospitalization. J Clin Hypertens (Greenwich). 2015;17:481–5.
Sturm G, Kollerits B, Neyer U, Ritz E, Kronenberg F, MMKD Study Group. Uric acid as a risk factor for progression of non-diabetic chronic kidney disease? The Mild to Moderate Kidney Disease (MMKD) Study. Exp Gerentol. 2008;43:347–52.
Madero M, Sarnak MJ, Wang X, et al. Uric acid and long-term outcomes in CKD. Am J Kidney Dis. 2009;53:796–803.
Voulgari C, Katsilambros N, Tentolouris N. Smoking cessation predicts amelioration of microalbuminuria in newly diagnosed type 2 diabetes mellitus: a 1-year prospective study. Metabolism. 2011;60:1456–64.
Krolewski AS. Progressive renal decline: the new paradigm of diabetic nephropathy in type 1 diabetes. Diabetes Care. 2015;38:954–62.
D’Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Q J Med. 1987;64:709–27.
Myllymäki J, Honkanen T, Syrjänen J, et al. Uric acid correlates with the severity of histopathological parameters in IgA nephropathy. Nephrol Dial Transpl. 2005;20:89–95.
Zhou J, Chen Y, Liu Y, et al. Plasma uric acid level indicates tubular interstitial lesions at early stage of IgA nephropathy. BMC Nephrol. 2014;15:11.
Cheng GY, Liu DW, Zhang N, Tang L, Zhao ZZ, Liu ZS. Clinical and prognostic implications of serum uric acid levels on IgA nephropathy: a cohort study of 348 cases with a mean 5-year follow-up. Clin Nephrol. 2013;80:40–6.
Shi Y, Chen W, Jalal D, et al. Clinical outcome of hyperuricemia in IgA nephropathy: a retrospective cohort study and randomized controlled trial. Kidney Blood Press Res. 2012;35:153–60.
Greenberg KI, McAdams-DeMarco MA, Köttgen A, Appel LJ, Coresh J, Grams ME. Plasma urate and risk of a hospital stay with AKI: the Atherosclerosis Risk in Communities Study. Clin J Am Soc Nephrol. 2015;10:776–83.
Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis. 2006;47:51–9.
Kanbay M, Ozkara A, Selcoki Y, et al. Effect of treatment of hyperuricemia with allopurinol on blood pressure, creatinine clearance, and proteinuria in patients with normal renal functions. Int Urol Nephrol. 2007;39:1227–33.
Goicoechea M, de Vinuesa SG, Verdalles U, et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol. 2010;5:1388–93.
Goicoechea M, Garcia de Vinuesa S, Verdalles U, et al. Allopurinol and progression of CKD and cardiovascular events: long-term follow-up of a randomized clinical trial. Am J Kidney Dis. 2015;65:543–9.
Bellomo G. The relationship between uric acid, allopurinol, cardiovascular events, and kidney disease progression: a step forward. Am J Kidney Dis. 2015;65:525–7.
Bose B, Badve SV, Hiremath SS, et al. Effects of uric acid-lowering therapy on renal outcomes: a systematic review and meta-analysis. Neph Dial Transpl. 2014;29:406–13.
Kanji T, Gandhi M, Clase CM, Yang R. Urate lowering therapy to improve renal outcomes in patients with chronic kidney disease: systematic review and meta-analysis. BMC Nephrol. 2015;16:58.
ClinicalTrials.gov identifier NCT00793585: A controlled study of uric acid on the progression of IgA nephropathy. https://clinicaltrials.gov/ct2/show/NCT00793585. Accessed Aug 2015.
Maahs DM, Caramori L, Cherney DZ, et al. Uric acid lowering to prevent kidney function loss in diabetes: the preventing early renal function loss (PERL) allopurinol study. Curr Diab Rep. 2013;13:550–9.
ClinicalTrials.gov identifier NCT02017171: A multicenter clinical trial of allopurinol to prevent kidney function loss in type 1 diabetes. https://clinicaltrials.gov/ct2/show/NCT02017171. Accessed Aug 2015.
Hosoya T, Kimura K, Itoh S, et al. The effect of febuxostat to prevent a further reduction in renal function of patients with hyperuricemia who have never had gout and are complicated by chronic kidney disease stage 3: study protocol for a multicenter randomized controlled study. Trials. 2014;15:26.
Hosoya T, Ohno I, Nomura S, et al. Effects of topiroxostat on the serum urate levels and urinary albumin excretion in hyperuricemic stage 3 chronic kidney disease patients with or without gout. Clin Exp Nephrol. 2014;18:876–84.
de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int. 2004;65:2309–20.
Daskalopoulou SS, Tzovaras V, Mikhailidis DP, Elisaf M. Effect on serum uric acid levels of drugs prescribed for indications other than treating hyperuricaemia. Curr Pharm Des. 2005;11:4161–75.
Hamada T, Ichida K, Hosoyamada M, et al. Uricosuric action of losartan via the inhibition of urate transporter 1 (URAT 1) in hypertensive patients. Am J Hypertens. 2008;21:1157–62.
Miao Y, Ottenbros SA, Laverman GD, et al. Effect of a reduction in uric acid on renal outcomes during losartan treatment: a post hoc analysis of the reduction of endpoints in non-insulin-dependent diabetes mellitus with the Angiotensin II Antagonist Losartan Trial. Hypertension. 2011;58:2–7.
DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab. 2012;14:5–14.
Ferrannini E, Solini A. SGLT2 inhibition in diabetes mellitus: rationale and clinical prospects. Nat Rev Endocrinol. 2012;8:495–502.
Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab. 2013;15:372–82.
Cherney DZ, Perkins BA, Soleymanlou N, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation. 2014;129:587–97.
Leiter LA, Yoon KH, Arias P, et al. Canagliflozin provides durable glycemic improvements and body weight reduction over 104 weeks versus glimepiride in patients with type 2 diabetes on metformin: a randomized, double-blind, phase 3 study. Diabetes Care. 2015;38:355–64.
Chino Y, Samukawa Y, Sakai S, et al. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm Drug Dispos. 2014;35:391–404.
Caulfield MJ, Munroe PB, O’Neill D, et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008;5:197.
Usiskin K, Kline I, Fung A, Mayer C, Meininger G. Safety and tolerability of canagliflozin in patients with type 2 diabetes mellitus: pooled analysis of phase 3 study results. Postgrad Med. 2014;126:16–34.
Bode B, Stenlöf K, Harris S, et al. Long-term efficacy and safety of canagliflozin over 104 weeks in patients aged 55–80 years with type 2 diabetes. Diabetes Obes Metab. 2015;17:294–303.
Yamout H, Perkovic V, Davies M, et al. Efficacy and safety of canagliflozin in patients with type 2 diabetes and stage 3 nephropathy. Am J Nephrol. 2014;40:64–74.
Kohan DE, Fioretto P, Tang W, List JF. Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int. 2014;85:962–71.
Barnett AH, Mithal A, Manassie J, et al. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2:369–84.
ClinicalTrials.gov identifier NCT02065791: Evaluation of the effects of canagliflozin on renal and cardiovascular outcomes in participants with diabetic nephropathy (CREDENCE). https://clinicaltrials.gov/ct2/show/NCT02065791. Accessed Aug 2015.
ClinicalTrials.gov identifier NCT01989754: A study of the effects of canagliflozin (JNJ-28431754) on renal endpoints in adult participants with type 2 diabetes mellitus (CANVAS-R). https://clinicaltrials.gov/ct2/show/NCT01989754. Accessed Aug 2015.
Acknowledgments
The article processing charges and open access fee for this publication were funded by Janssen Scientific Affairs, LLC. The author is the guarantor for this article and meets the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript. He takes responsibility for the integrity of the work as a whole, and has given final approval to the version to be published. Medical writing and editorial assistance in the preparation of this manuscript was provided by Adriana Stan, PhD, of Excerpta Medica. The manuscript was prepared following the “Good Publication Practice for Communicating Company-Sponsored Medical Research: the GPP3 Guidelines” of the International Society for Medical Publication Professionals. Support for this assistance was funded by Janssen Scientific Affairs, LLC. Canagliflozin has been developed by Janssen Research and Development, LLC, in collaboration with Mitsubishi Tanabe Pharma Corporation.
Disclosures
Christian Mende has nothing to disclose regarding the publication of this paper.
Compliance with ethics guidelines
This article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by the author.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mende, C. Management of Chronic Kidney Disease: The Relationship Between Serum Uric Acid and Development of Nephropathy. Adv Ther 32, 1177–1191 (2015). https://doi.org/10.1007/s12325-015-0272-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-015-0272-7