
Abstract
Three new compounds, including two diterpenoids, nemoralisins H and I (1 and 2), and a limonoid, 2-methoxy khayseneganin E (3), along with four known constituents (4–7), were isolated from the leaves and twigs of Swietenia mahagoni. Their chemical structures were elucidated by means of spectroscopic analysis. The cytotoxities of these isolated constituents were assayed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Swietenia mahagoni, which is an economically important timber tree, has been used as a folk medicine for the treatment of hypertension, diabetes, and malaria [1, 2]. Chemical investigations conducted previously on S. mahagoni had led to the isolation of various B,D-seco limonoids such as mexicanolides, phragmalins, andirobins, gedunins, and rearranged phragmalins [3–7]. Modern pharmacological studies demonstrated that limonoids from Swietenia displayed insecticide, antitumor, antibacterial, antidiabetic, and antidyslipidemic activities [8–12].
With the aim of searching for structurally unique and bioactive chemical constituents, we isolated the leaves and twigs of S. mahagoni and gained two new diterpenoids (1 and 2), one new limonoid (3) and four known compounds (4–7) (Fig. 1). In this paper, we reported the isolation and structural elucidation of new compounds, as well as cytotoxicities of all the compounds against five human cancer cell lines.

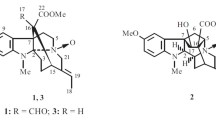
The structure of compounds 1–7
2 Results and Discussion
The air-dried powder of leaves and twigs of S. mahagoni was extracted with 70 % aqueous acetone at room temperature three times to give the residue, which was then partitioned between EtOAc and water to get the EtOAc-soluble fraction. Then, three new constituents together with four known compounds were acquired by a series of chromatographic methods. Herein, we described the isolation and structural elucidation of these new compounds.
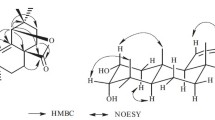
Compound 1, colorless oil, was assigned a molecular formula C20H28O5 according to its positive HREIMS peak at m/z 348.1932 [M]+ (calcd for 348.1937), suggesting seven degrees of unsaturation. The IR spectrum revealed characteristic bands corresponding to hydroxyl (3,434 cm−1), α,β-unsaturated-δ-lactone (1,761 cm−1) and double-bond (1,639 cm−1) groups. The 1H NMR spectrum of 1 (Table 1) showed the presence of three typical olefinic protons δH (5.80, s-like; 5.42, s; 5.26, dd, J = 8.7, 1.2 Hz), three methyl singlets at δH 1.35 (6H), 2.05 (3H) and two methyl doublets at δH (1.23, J = 7.0 Hz; 1.75, J = 1.2 Hz). The 13C DEPT displayed five methyls, three methylenes, six methines, and six quaternary carbons (Table 1). 1D-NMR data of 1 exhibited high resemblance with nemoralisin A isolated from Aphanamixis grandifolia (Meliaceae) [13], and they showed the same molecular formula and the similar skeletal structure (α,β-unsaturated ketone was connected with one α,β-unsaturated δ-lactone by an aliphatic chain). The only difference between 1 and nemoralisin A was that the hydroxyl group located at C-4 in 1 but at C-8 in nemoralisin A. And this was confirmed by HMBC correlations of H-4 (δH 4.04, d, J = 7.2 Hz) with C-2 (δC 117.1), C-3 (δC 162.6), C-5 (δC 80.5), C-6 (δC 121.9), and of Me-20 (δH 2.05) with C-4 (δC 69.7), and together with by the 1H-1H COSY cross-peaks between H-4 and H-5 (δH 4.99, d, J = 8.7, 7.2 Hz) (Fig. 2)

1H-1H COSY ( ) and key HMBC (
) and key HMBC ( ) correlations of 1–3 and key ROESY (
) correlations of 1–3 and key ROESY ( ) correlations of 1
) correlations of 1
.
In the ROSEY spectrum, the strong correlation from H-6 (δH 5.26, dd, J = 8.7, 1.2 Hz) to H-8 (δH 2.10, m) inferred E-geometry of the Δ6 double bond. This was confirmed by the upfield resonance of vinylic Me-19 group at δC 17.1 [14]. The ROESY correlations of H-4/H-6 and H-5/Me-19 (δH 1.75, d, J = 1.2 Hz) suggested that H-4 and H-5 was trans oriented. However, the absolute configuration was not determined on the basis of 1D and 2D NMR data. Accordingly, compound 1 was established and named as nemoralisin H.
Compound 2 was isolated as colorless oil. The molecular formula of C20H28O5 was determined by the positive ion peak at m/z 348.1927 [M]+ (calcd for 348.1937) in the HREIMS. Extensive analysis of the 1D-NMR spectroscopic data (Table 1) of 2 exhibited a close resemblance with nemoralisin [14]. However, downfield shifts of the lactone carbon (δC 176.3), the β-sp2 carbon (δC 170.7), and the oxygenated methine (δC 89.3) were observed in the 13C DEPT spectrum of 2, which implied the α,β-unsaturated δ-lactone (ring A) in nemoralisin was replaced by an α,β-unsaturated γ-lactone in 2. This deduction was confirmed by the observed HMBC correlations of H-4 (δH 4.89) with C-1 (δC 176.3), C-2 (δC 118.9), C-3 (δC 170.7), C-6 (δC 124.3), and of Me-20 (δH 2.13) with C-4 (δC 89.3) (Fig. 2). Meanwhile, on the basis of 1H-1H COSY correlations of H-4/H-5/H-6, a hydroxyl group at C-5 and Δ6 double bond were established. The ROESY correlations of H-6 (δH 5.20, d, J = 8.8 Hz) with H-8 (δH 2.03, m) inferred E-geometry of the Δ6 double bond. Therefore, the structure of 2 was determined and gave the name nemoralisin I.
Compound 3, white amorphous powder, was found to possess the molecular formula of C28H36O11 as inferred by HREIMS peak at m/z 548.2250 [M]+ (calcd for 548.2258). The 1D NMR data (Table 2) of 3 indicated that it was a phragmalin-type limonoid and similar with khayseneganin E [15–17]. However, a methoxy in 3 replaced the hydroxyl group at C-2 in khayseneganin E, which was supported by HMBC correlation between OMe (δH 3.68) and the ketal carbon (δC 106.9) (Fig. 2). The relative configuration of 3 was determined by analysis of the ROESY spectrum, in which correlations of H-5/H-6, H-5/H-12β, H-12β/H-17, and H-6/Me-28 suggested that these groups are all β-oriented. In addition, Me-19, Me-18, H-9, and H-3 were assigned as α-oriented on the basis of the ROESY correlations between Me-19/H-9 and between H2-29/H-3, Me-18/H-3, suggesting that OH-3 is β-oriented. Consequently, the structure of 3 was determined as shown, named 2-methoxy khayseneganin E.
Four known constituents: nemoralisin C (4) [13], khayanolide B (5) [18], khayseneganin G (6) [17], and deacetylkhayanolide E (7) [19], were identified by the comparison of their spectroscopic data with those reported in the literature.
Compounds 1–7 were tested for in vitro inhibitory activities against HL-60, SMMC-7721, A549, MCF-7 and SW480 human tumor cell lines. All the tested samples showed no activities against the mentioned cell lines with IC50 > 40 μM. Much to our delight, Liu J. et al. [20] reported that aphadilactones A−D, which can be considered as the dimers of diterpenoid compounds (1, 2 and 4) showed potent and selective inhibition against the diacylglycerol o-acyltransferase-1 (DGAT-1) enzyme, and are the strongest natural DGAT-1 inhibitors discovered to date. So emphasis can be laid on this class of compounds in our future search of potential DGAT-1 inhibitors.
3 Experiment Section
3.1 General Experimental Procedures
Optical rotations were obtained with a Jasco P-1020 polarimeter. UV (in MeOH) and IR (in CHCl3) spectra were measured on Shimadzu UV-2401 PC spectrophotometer and Bruker Tensor-27 infrared spectrophotometer, respectively. ESIMS spectra were recorded on an API QSTAR Pulsar spectrometer. EIMS and HREIMS were performed on a Waters Autospec Premier P776. 1D and 2D NMR spectra were recorded on Bruker DRX-500 and Bruker Avance III-600 MHz spectrometers. Chemical shifts (δ) were expressed in ppm with reference to the TMS resonance. Semi-preparative HPLC studies were carried out on an Agilent 1100 liquid chromatograph with a Zorbax SB-C18 (9.4 mm × 25 cm) column. Column chromatography was performed using Silica gel [(200–300) mesh, Qingdao Marine Chemical, Inc, Qingdao, China]. Fractions were monitored by TLC and spots were visualized by heating the silica gel plates sprayed with 10 % H2SO4 in EtOH. Lichroprep RP-18 [(40–63) μm, Merck] and Sephadex LH-20 [(20–150) μm, Pharmacia] were also used for column chromatography.
3.2 Plant Material
The leaves and twigs of S. mahagoni were collected from Xishuangbanna, Yunnan Province, China. A voucher sample has been deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and Isolation
The air-dried powdered leaves and twigs of S. mahagoni (10 kg) were extracted with 70 % aqueous acetone (each 30 L, 3 days) at room temperature three times to give a dark green residue (650 g), which was then partitioned between EtOAc and water to give the EtOAc-soluble fraction (120 g). The EtOAc extract was chromatographed by silica gel column eluted with CHCl3–MeOH as a gradient (100:1, 50:1, 20:1, 5:1) to afford four fractions. The CHCl3–MeOH (100:1) portion was evaporated to obtain a residue (20 g), which was subjected to silica gel chromatograph column with petroleum ether–EtOAc (10:1, 6:1, 3:1, 1:1) as elution, to give fractions (A, B, C, D). Fraction B was further subjected to RP-18 chromatograph column, eluting with MeOH–H2O (50:50, 65:35, 80:20) to afford subfraction (E), which was then purified by semi-preparation HPLC with MeCN–H2O (50:50) to give compound 3 (6 mg), 5 (12 mg), 6 (10 mg), 7 (15 mg). Fraction C was subjected to silica gel chromatograph column with petroleum ether–EtOAc (8:1, 5:1, 3:1, 1:1) as elution, to give fractions F, which was successively subjected to RP-18, Sephadex LH-20 and semi-preparation HPLC, compound 1 (1.5 mg), 2 (1.3 mg), and 4 (6 mg) were obtained.
3.4 Nemoralisin H (1)
Colorless oil; [α] 25D − 0.48 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 202 (3.75), 261(3.37) nm; IR (KBr) νmax 3434, 2976, 2930, 1761, 1639, 1585, 1457, 1383, 1270, 1177, 1088, and 1036 cm−1, 1H and 13C-DEPT data see Table 1; EIMS m/z 371 [M + Na]+; HREIMS m/z 348.1932 (calcd for C20H28O5 [M]+, 348.1937).
3.5 Nemoralisin I (2)
Colorless oil; [α] 25D − 14.3 (c 0.18, MeOH); UV (MeOH) λmax (log ε) 204 (3.87), 261(3.70) nm; IR (KBr) νmax 3432, 2977, 2933, 1761, 1698, 1584, 1458, 1383, 1299, 1267, 1177, 1034 and 938 cm−1, 1H and 13C-DEPT data see Table 1; ESIMS m/z 371 [M + Na]+; HREIMS m/z 348.1927 (calcd for C20H28O5 [M]+, 348.1937).
3.6 2-Methoxy khayseneganin E (3)
White amorphous powder; [α] 18D + 26.3 (c 0.03, MeOH); UV (MeOH) λmax(log ε) 209 (3.83) nm; IR (KBr) νmax 3440, 2952, 1727, 1632, 1462, 1441, 1387, 1280, 1245, 1159, 1053, 1024, and 600 cm−1, ESIMS m/z 571 [M + Na]+; HREIMS m/z 548.2250 (calcd for C28H36O11 [M]+, 548.2258).
3.7 Cytotoxicity Assay
The cytotoxic activities of compounds 1–7 against HL-60, SMMC-7721, A549, MCF-7 and SW480 cell lines were determined by the MTT method [21].
References
Y.Y. Chen, X.N. Wang, C.Q. Fan, S. Yin, J.M. Yue, Tetrahedron Lett. 48, 7480–7484 (2007)
A.M.A. Samir, D. Matsumi, N. Munehiro, Phytochemistry 96, 312–317 (2013)
Q.G. Tan, X.D. Luo, Chem. Rev. 111, 7437–7522 (2011)
S. Abdelgaleil, M. Doe, Y. Morimoto, M. Nakatani, Phytochemistry 67, 452–458 (2006)
B.D. Lin, T. Yuan, C.R. Zhang, L. Dong, B. Zhang, Y. Wu, J.M. Yue, J. Nat. Prod. 72, 2084–2090 (2009)
A.K.M.S. Rahman, A.K.A. Chowdhury, H.A. Ali, S.Z. Raihan, M.S. Ali, L. Nahar, S.D. Sarker, J. Nat. Med. 63, 41–45 (2009)
J.Q. Liu, X.R. Peng, W.M. Zhang, L. Shi, X.Y. Li, J.C. Chen, M.H. Qiu, RSC. Adv. 3, 4890–4893 (2013)
J. Li, M.Y. Li, G. Feng, Q. Xiao, J. Sinkkonen, T. Satyanandamurty, J. Wu, Phytochemistry 71, 1917–1924 (2010)
W. Yang, L. Kong, Y. Zhang, G.H. Tang, F. Zhu, S.F. Li, L.L. Guo, Y.Y. Cheng, X.J. Hao, H.P. He, Planta Med. 78, 1676–1682 (2012)
A. Sakamoto, Y. Tanaka, T. Inoue, T. Kikuchi, T. Kajimoto, O. Muraoka, T. Yamada, R. Tanaka, Fitoterapia 90, 20–29 (2013)
S.E. Okhale, J.O. Amupitan, I.G. Ndukwe, R.G. Ayo, P.O. Oladosu, J.I. Okogun, Afr. J. Pure Appl. Chem. 7, 157–163 (2013)
V. Lakshmi, A.K. Srivastava, Nat. Prod. 8, 284–288 (2012)
Y. Zhang, J.S. Wang, D.D. Wei, Y.C. Gu, X.B. Wang, L.Y. Kong, J. Nat. Prod. 76, 1191–1195 (2013)
X.F. He, X.N. Wang, C.Q. Fan, L.S. Gan, S. Yin, J.M. Yue, Helv. Chim. Acta 90, 783–791 (2007)
C.M. Yuan, Y. Zhang, G.H. Tang, S.L. Li, Y.T. Di, L. Hou, J.Y. Cai, H.M. Hua, H.P. He, X.J. Hao, Chem. Asian J. 7, 2024–2027 (2012)
X. Fang, Y.T. Di, X.J. Hao, Curr. Org. Chem. 15, 1363–1391 (2011)
C.M. Yuan, Y. Zhang, G.H. Tang, Y.T. Di, M.M. Cao, X.Y. Wang, G.Y. Zuo, S.L. Li, H.M. Hua, H.P. He, J. Nat. Prod. 76, 327–333 (2013)
S.A.M. Abdelgaleil, H. Okamura, T. Iwagawa, A. Sato, I. Miyahara, M. Doe, M. Nakatani, Tetrahedron 57, 119–126 (2001)
H. Zhang, O.A. Odeku, X.N. Wang, J.M. Yue, Phytochemistry 69, 271–275 (2008)
J. Liu, X.F. He, G.H. Wang, E.F. Merino, S.P. Yang, R.X. Zhu, L.S. Gan, H. Zhang, M.B. Cassera, H.Y. Wang, D.G.I. Kingston, J.M. Yue, J. Org. Chem. 79, 599–607 (2014)
J.Q. Liu, Y.F. Yang, X.Y. Li, E.Q. Liu, Z.R. Li, L. Zhou, Y. Li, M.H. Qiu, Phytochemistry 96, 265–272 (2013)
Acknowledgments
This work was supported financially by the National Special Program of Basic Research (SB2007FY400), the Knowledge Innovation Program of CAS (Grant No. Qian-2011, KSCX2-YW-G-038), as well as Foundation of Yunnan Tobacco Industry Group (2010–2012) and State Key Laboratory of Phytochemistry and Plant Resources in West China. The authors also were sincerely grateful to Professor Li Yan for cytotoxicity bioassay.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Zhang, WM., Liu, JQ., Deng, YY. et al. Diterpenoids and Limonoids from the Leaves and Twigs of Swietenia mahagoni. Nat. Prod. Bioprospect. 4, 53–57 (2014). https://doi.org/10.1007/s13659-014-0006-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-014-0006-6