
Abstract
The World Health Organization (WHO) guidelines on evaluation of similar biotherapeutic products (SBPs; also called biosimilars) were adopted by the WHO Expert Committee on Biological Standardization (ECBS) in 2009. In 2019, the ECBS considered that a more tailored and potentially reduced clinical data package may be acceptable in cases where this was clearly supported by the available scientific evidence. The goal of this publication is to review the current clinical experience and scientific evidence and to provide an expert perspective for updating the WHO guidelines to provide more flexibility and clarity. As the first step, the relevant guidelines by other regulatory bodies were reviewed in order to identify issues that might help with updating the WHO guidelines. Next, a literature search was conducted for information on the long-term efficacy, safety, and immunogenicity of biosimilars to identify possible long-term problems. Finally, a search for articles concerning the role of clinical studies in the benefit–risk evaluation of biosimilars was conducted. The analysis of other guidelines suggested that the WHO guidelines may need more emphasis on the importance of the state-of-the-art physicochemical and structural comparability exercise and in vitro functional testing. The use of “foreign” reference product will also need clarifications. The value of in vivo toxicological tests in the development of biosimilars is questionable, and the non-clinical part needs revisions accordingly. The concepts of “totality of evidence,” “stepwise development,” and “residual uncertainty” were applied in the evaluation of the clinical sections of the guideline. The review of long-term safety and efficacy demonstrated the robustness of the current biosimilar development concept. The analysis of the roles of different development phases suggested that the large efficacy, safety, and immunogenicity studies are, in most cases, redundant. The residual uncertainty of safety, immunogenicity, and efficacy of biosimilars that has shaped the current regulatory guidelines is now substantially reduced. This will allow the re-evaluation of the non-clinical and clinical requirements of the current WHO main guideline. The shift of the relative impact of the development phases towards physico-chemical and in vitro functional testing will provide a relief to the manufacturers and new challenges to the regulators.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
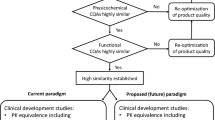
The World Health Organization (WHO) is revising its 2009 guidelines for the development of biosimilars according to current scientific knowledge. This publication provides the background for this revision. |
Long-term safety, efficacy, and immunogenicity data of licensed biosimilars since 2006 do not raise concerns. |
Current data suggest that state-of-the-art analytical and functional testing and robust pharmacokinetic and pharmacodynamic studies are sufficient to demonstrate biosimilarity, whereas in vivo animal studies and large confirmatory efficacy and safety studies are generally not needed. |
1 Introduction
The World Health Organization (WHO) has a mandate to support regulatory authorities in its 194 Member States, and one of the WHO core functions is “setting norms and standards and promoting and monitoring their implementation.” WHO guidelines are considered as WHO written standards to provide globally agreed principles that serve as a basis for establishing national regulatory requirements, and they also serve as a basis for WHO prequalification.
The WHO guidelines on evaluation of similar biotherapeutic products (SBPs; also called biosimilars) (hereafter referred to as “the Guidelines”) were adopted by the WHO Expert Committee on Biological Standardization (ECBS) in 2009 [1]. It was noted that the Guidelines have contributed significantly to setting the regulatory framework for SBPs within WHO countries, increasing international regulatory convergence and improving consistency in the terminology used in the evaluation of SBPs [2].
In the meantime, the main principles in the Guidelines have been re-assessed on a regular basis. As a result, the WHO main guidelines have been complemented by guidance on the evaluation of biosimilar monoclonal antibodies (mAbs) [3] and a Q&A document [4]. Finally, at its meeting in 2018, the ECBS confirmed that the Guidelines remain valid and provide guidance on the evaluation of biosimilars together with the Q&A document and mAb guidelines.
In 2019, the ECBS considered that a more tailored and potentially reduced clinical data package may be acceptable in cases where this was clearly supported by the available scientific evidence. The committee supported the review of current scientific evidence to consider updating the Guidelines to provide more flexibility and clarity. Thus, the WHO initiated a review of scientific evidence and experience to identify issues/cases for further reducing non-clinical and clinical data, and the progress was reported to the committee in 2020 (72nd report [5]).
This article describes the outcomes of the review for exploring possibilities to streamline the clinical development of biosimilars on the basis of revised guidelines, documented long-term use of licensed biosimilars, and reports of the role of non-clinical and clinical studies in biosimilar comparability exercises.
2 Methodology
As the first step, the relevant guidelines of the US Food and Drug Administration (FDA), Health Canada, and European Medicines Agency (EMA) were reviewed in order to seek possibilities for reducing the non-clinical and clinical data package in already established and continuously updated guidelines [6,7,8]. The current pre-licensing requirements demand a complete quality package including physicochemical, structural and functional similarity testing, in vitro and in vivo non-clinical testing, comparable pharmacokinetics (PK)/pharmacodynamics (PD), and usually clinical efficacy and safety studies using equivalence design. Thus, biosimilars have undergone a robust assessment of comparability at the time of licensing. In addition, post-marketing efficacy and safety are continuously monitored through adverse effect reporting, pharmaco-epidemiological studies and other risk-management measures.
Thus, the second step was to review the literature for long-term experience with biosimilars. A literature search was conducted for long-term efficacy, long-term safety, and immunogenicity of biosimilars for the years 2017–2020 in PubMed, Elsevier ScienceDirect, Wiley online library, and SpringerLink. Older reports were covered by systematic reviews published in 2017–2020.
The third step evaluated the roles and relevance of clinical studies for the benefit–risk assessment of biosimilars on the basis of recent scientific reports identified by using PubMed.
3 Results
3.1 Step 1: Review of the EMA, Health Canada and FDA guidelines
The pre-licensing requirements for biosimilars of the FDA 2015–2019, Health Canada 2016, and the EMA 2012–2017 have been revised on the basis of scientific progress and experience with licensed biosimilars [6,7,8]. Thus, they may be used as a source of information for the revision of the WHO 2009 guidelines. In general, the WHO guidelines are still in line with the newer, revised guidelines of other jurisdictions. However, possibilities have been identified to improve the WHO guidance with regard to analytical, structural, and in vitro functional tests as well as to simplify clinical development. As it is already indicated in the Guidelines, WHO guidelines ‘should be viewed as “living” documents that will be developed further in line with advances in scientific knowledge and experience.’
3.1.1 Biosimilarity: Totality of the Evidence, Risk-Based Approach, and Residual Uncertainty
The FDA guideline “Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product” [9] presents some helpful principles in the re-evaluation of the regulatory approach to biosimilarity. The numerous comparisons between the candidate biosimilars and their reference products (Reference Biotherapeutic Products, RBPs) will inevitably result in some, usually small differences, the relevance of which may not always be known. Thus, the assessment of biosimilarity is based on the totality of the data from the physico-chemical, structural, and functional tests as well as clinical data including PK and PD studies, immunogenicity testing, and a dedicated efficacy and safety study, where necessary. Ideally, the development of a biosimilar proceeds in a stepwise, risk-based manner. Thus, the developer can estimate the degree of residual uncertainty after critical steps in order to plan for appropriate studies to address uncertainties [10]. In the revision of the WHO guidelines, the most critical step is the transition from clinical PK/PD studies to the most resource-intensive step, an additional efficacy and safety study.
3.1.2 Use of a Reference Product
The RBP provides the basis of the abbreviated development of SBPs. It has been licensed on the basis of a full (stand-alone) registration package. In some jurisdictions, it is expected that the domestically licensed product is used as the RBP. However, this is not always possible or feasible, for example, in the countries that lack a particular nationally or regionally licensed originator product.
National regulatory authorities (NRAs) have used a foreign RBP in the following scenarios [11]:
-
1.
The RBP is not authorized locally. In this case, the NRA may allow the use of a product that is licensed by an experienced NRA that follows the WHO or corresponding regulatory standards. Several NRAs have selected suitable “reference” countries for this scenario.
-
2.
The RBP is licensed locally but sourced from another jurisdiction. In this case, some NRAs require “bridging” studies that ensure comparability of the local and foreign-sourced RBPs. This approach is mandatory because of legal requirements in, e.g., the EU and the US.
In order to facilitate the global development of biosimilars, the revision of the WHO guidelines may need to further clarify these scenarios.
Scenario 1 The current WHO 2009 guidelines [1] state, “Traditionally, NRAs have required the use of a nationally licensed reference product for licensing of generic medicines. This practice may not be feasible for countries lacking nationally licensed RBPs. NRAs may need to consider establishing additional criteria to guide the acceptability of using a RBP licensed in or sourced from other countries. The use of reference products with proven efficacy and safety in a given population will be one of the factors to consider.”
Health Canada permits the use of a foreign(-sourced) RBP licensed in an International Conference on Harmonization of Technical Requirements for Pharmaceuticals of Pharmaceuticals for Human Use (ICH) country [6] when it does not have locally sourced product.
Scenario 2 A foreign-sourced RBP can be used in clinical studies. This is the case for the EU and US, where a biosimilar must always make reference to a local RBP for legal reasons, but clinical studies can be performed with a non-European Economic Area (EEA)/non-US version of the reference medicinal product, provided this has been authorized by a regulatory authority with similar scientific and regulatory standards. In this case, the FDA guidelines require analytical and PK/PD “bridging” studies by default, whereas the EMA and Health Canada guidelines require analytical bridging, but PK/PD bridging only if analytical bridging alone is not sufficient [7, 12, 13].
The bridging studies, especially clinical PK/PD studies, have been criticized since they complicate global development of biosimilars. Webster and Woollett (2017) propose that “bridging studies will not be conducted if the reference comparator… has been approved in any ICH jurisdiction and there is evidence in the public domain that the reference product has been approved in both jurisdictions upon some of the same phase III clinical data” [14]. Tu et al. showed that it is unlikely that the EU- and US-sourced RBPs have meaningful differences [15]. In addition, the efficacy and safety of both versions of the RBPs are backed with more than 10 years of clinical use.
3.1.3 Quality Evaluation
The relative importance of the analytical and in vitro functional data will increase if clinical data requirements are reduced. EMA and FDA guidelines describe the current state of the art for analytical, structural, and functional similarity assessment between the RBP and the biosimilar candidate. The method of defining the acceptance ranges of critical quality attributes is well described in EMA and FDA quality guidelines [16,17,18]. In particular, the FDA biosimilar guidelines describe thoroughly the risk assessment of quality attributes, while EMA guidelines refer to other guiding documents. In general, both FDA, Health Canada, and EMA highlight the importance of state-of-the art orthogonal analytical methods and in vitro functional/potency tests in the characterization of biosimilars. The quality section of the WHO guidelines needs to be updated to be in line with the current expectations for analytical characterization and for demonstration of biosimilarity.
3.1.4 Non-clinical Evaluation
The FDA generally expects both comparative in vitro and in vivo studies. The FDA has the mandate to waive in vivo studies if justified by the applicant [10]. The EMA guideline provides the concept of stepwise progression in non-clinical testing and reduction of animal work in accordance with the 3 R principles according to Directive 2010/63/EU [19]. Health Canada states that in vivo toxicological studies are generally not needed [7].
The EMA guideline emphasizes in vitro testing. In vivo toxicological testing is important for products with a new active substance, but usually has no relevance for comparing two highly similar biological products with a well-known active substance.
The WHO guideline still requests a comparative subacute toxicity study. Such a study is highly unlikely to be helpful due to limitations in sensitivity and species specificity. Thus, in vivo testing may be needed only in exceptional circumstances. The Canadian guideline also allows the omission of in vivo toxicological testing of highly similar products. This approach is supported by several publications [20,21,22,23,24].
The non-clinical part of the WHO Guideline should put more emphasis on the in vitro functional tests. The standard in vivo toxicological studies should be discouraged.
3.1.5 Clinical Evaluation
The US, Canadian, and EU guidelines require comparative PK and, if relevant, PD studies between the candidate biosimilar and its RBP by default [7, 9, 19]. This approach is also recommended by the current WHO guidelines because PK(PD) studies are considered sensitive to detect potential product-related differences in vivo. In fact, scientific reports underline the importance of well-performed and robust PK studies with proper power calculation avoiding too optimistic calculation of the inter-individual variability. Such PK studies may already provide sufficient data on safety, including immunogenicity. In any case, EMA, Health Canada, and FDA guidelines request safety and immunogenicity data for all clinical studies [9, 19, 25,26,27].
FDA, Health Canada, and EMA guidelines [7, 9, 19] provide some flexibility in regard to the phase III-type “confirmatory” clinical efficacy and safety studies if certain requirements are met, especially availability of PD markers that are relevant markers or even surrogates for efficacy. The Health Canada guideline [7] (Health Canada) states, “The non-clinical and clinical programs should be designed to complement the structural and functional studies and address potential areas of residual uncertainty.” The FDA guideline presents points that need to be addressed if the confirmatory efficacy and safety study is considered dispensable [10].
However, for most products, especially for biosimilar mAbs, resource-intensive, phase III-type confirmatory studies with an equivalence design are still expected. The guidance by the WHO is still in line with these updated guidelines.
The US guidelines advise that confirmatory efficacy and safety studies be performed if residual uncertainties remain after the previous steps of development [9]. However, experience has shown a temporal overlap of the development steps. For example, confirmatory studies are frequently performed in parallel with the pivotal analytical tests using the biosimilar product from the final manufacturing process. This approach may shorten the development but does not reduce the costs. The issue of residual uncertainty after analytical, structural, functional, and human PK/PD studies should be re-visited in the revision of WHO guidelines, since the degree of residual uncertainty may not be as high in 2021 as it was at the time of the publication of the WHO guidelines in 2009.
3.2 Step 2: Scientific Considerations: Long-Term Safety and Residual Uncertainties
The EU has approved the largest number of biosimilars. In April 2022, there were 86 licensed biosimilars with European Public Assessment reports [28] (Fig. 1). The short-term safety has been established by extensive pre-licensing studies that have seldomly lasted longer than 12 months at the time of licensing. Therefore, literature searches were conducted for long-term efficacy, safety, and immunogenicity data in order to explore the post-marketing experience with biosimilars. Biosimilars are sometimes divided to “first-generation” and “second-generation” products. mAbs and fusion proteins (FCs) belong to the latter category since they are claimed to have more risks due to the presumed difficulties in their characterization [29].

Marketing authorization applications of biosimilars in the EU (as at 8 April 2022). EC European Commission, EMA European Medicines Agency, MA marketing authorization, MAA marketing authorization application
3.2.1 First-Generation Biosimilars
Biosimilar somatropin The first biosimilar product, biosimilar human growth hormone, was approved in the EU in 2006. There were several safety concerns among prescribers, especially relating to disturbances in glucose metabolism, cancer, and immunogenicity. Therefore, the safety of biosimilar growth hormone products was carefully followed not only by routine pharmacovigilance but also by individual centers, networks of investigators, and registers, especially PATRO. The safety and efficacy profile turned out to be similar to the originator product [30,31,32,33].
Biosimilar filgrastims The first biosimilar filgrastim was approved in 2008 and the first pegfilgrastim in 2018. Mobilization of hematopoietic stem cells by biosimilar filgrastims was regarded risky for healthy donors by the World Marrow Donor Association (WMDA). The efficacy and safety of biosimilar filgrastims were investigated in models of chemotherapy-induced neutropenia, PD studies, and registries, including also donors of hematopoietic progenitor cells. The safety profiles of biosimilar filgrastims have been shown to be similar, including mobilization of hematopoietic stem cells [34,35,36,37,38]. The WMDA now recommends the use of biosimilar filgrastims [38].
Biosimilar epoetin α The first biosimilar epoetin α product was licensed in 2007. The main concern was the risk of neutralizing antibodies cross-reacting with endogenous erythropoietin, which have caused pure red cell aplasia in patients treated with the RBP [39]. Indeed, neutralizing antibodies to erythropoietin were detected in the confirmatory clinical trial of a biosimilar epoetin α product in patients with renal anemia. This led to the discontinuation of the development of the product for subcutaneous administration until the underlying problem (which was not related to the quality of the active substance itself, but to tungsten leaching from the needle of the syringe) was eliminated [40, 41]. The licensed biosimilar epoetin α products have not shown excess immunogenicity as compared to the RBP [42, 43].
The other initial concern was the safety of biosimilars in anemia associated with cancer chemotherapy because the efficacy and safety were extrapolated from studies of renal anemia and the dose used in chemotherapy-induced anemia is higher. Later on, clinical studies and real-world studies have demonstrated comparable safety of the biosimilar epoetinα and the RBP in the oncology setting [44].
Biosimilar insulin glargine The first biosimilar insulin glargine was licensed in 2014. The main concern was an increased risk of hypoglycemia. A limited number of post-marketing clinical studies and limited real-world data are available. The current data suggest that the biosimilars and their RBP have comparable safety and efficacy profiles [45, 46].
3.2.2 Second-Generation Biosimilars
Biosimilar monoclonal antibodies and fusion proteins Monoclonal antibodies and related FCs represent the latest major group of biosimilars. In the EU, 37 biosimilar mAb or FP products are licensed and 30 are marketed by April 8 2022 [47] (Fig. 1).
Tumor necrosis factor-α inhibitors In the EU, the first biosimilar was licensed in 2013 for infliximab, 2017 for adalimumab, and 2017 for etanercept. These products are used to treat (auto)inflammatory diseases. Besides routine pharmacovigilance and patient registries, their post-marketing safety data consist mainly of extensions of pivotal confirmatory trials and post-marketing clinical trials in rheumatologic diseases, psoriasis, and inflammatory bowel diseases. In spite of the initial safety and immunogenicity concerns, amplified by manufacturers of original biologicals [48], biosimilar mAbs and FPs have been proven to have comparable safety and efficacy profiles to their references in all licensed therapeutic indications [49,50,51,52,53,54,55,56,57,58,59,60,61,62,63]. The estimated global exposure to biosimilar anti–tumor necrosis factor (TNF)α inhibitors until 2020 was 1,286,578 patient treatment years [27]. Extrapolation of safety and efficacy from one therapeutic indication to another/others and immunogenicity were presented as potential problems of biosimilar mAbs [29]. However, theoretical considerations suggest, and clinical data confirm, the therapeutic equivalence of biosimilar mAbs and their RBPs [27, 64, 65].
Biosimilar anti-cancer monoclonal antibodies In the EU, the first biosimilar was licensed in 2017 for rituximab, 2018 for bevacizumab, and 2017 for trastuzumab. Thus, the post-marketing follow-up is generally rather short, and difficult because of the underlying malignant diseases and concomitant chemotherapy. Nevertheless, data from clinical studies and post-marketing surveillance do not raise concerns [27]. Long-term safety data of biosimilar bevacizumabs and rituximabs are scarce. Nevertheless, the available post-marketing follow-up studies confirm comparable safety and immunogenicity established in pre-marketing clinical studies [66,67,68,69]. The exposure to biosimilar trastuzumabs is already considerable, and long-term safety, immunogenicity, and efficacy studies have raised no concerns [70,71,72].
3.2.3 Conclusions on Long-Term Safety Data
Long-term safety and immunogenicity data were available for most biosimilar products. For certain products, long-term data were scarce or absent because of recent approval or small sales volumes. It is estimated that the cumulative exposure to EU-approved biosimilars was more than 2 billion patient treatment days in 2020 [73]. In the EU, no biosimilar products have been withdrawn from the market for safety reasons and no biosimilar-specific adverse effects have been added to the product information [27, 74].
The analysis of scientific reports on long-term safety substantiates the data at the time of licensing. Thus, the uncertainty that prevailed among prescribers and learned societies at the time of licensing has vanished or at least greatly diminished for most biosimilar products. The current data on long-term safety validate the current concept of biosimilar development and create a foundation for analyzing redundancies, especially the need for large “confirmatory” efficacy and safety studies.
3.3 Step 3: Possibilities for Reducing Clinical Data Requirements
There are reports arguing that the development concept of biosimilars in the current regulatory guidelines, including the WHO main guidelines, may not correspond to the current scientific evidence and clinical experience. The need for confirmatory efficacy and safety studies is questioned based on the assumption that analytical assessments alone or in combination with human PK/PD studies can demonstrate comparable efficacy and safety. Even large molecules can be thoroughly analyzed by state-of-the art analytical and in vitro functional testing [75,76,77,78].
3.3.1 The Roles of Different Development Phases
The goal of biosimilar development is to establish a manufacturing process that produces a highly similar product when compared to the RBP. State-of-the art analytical orthogonal methods must be applied to detect all relevant differences between the active substances of biosimilars and their RBPs.
Comparative in vitro functional testing will confirm the similarity of the higher order structures and function of active substances. PK studies prove the similar exposure from the final (formulated) product and provide information on safety and immunogenicity. These are the essential elements of a biosimilar development, whereas the phase III-type efficacy and safety studies are regarded as “confirmatory” [25, 79, 80].
3.3.2 Reduction of Confirmatory Efficacy and Safety Data
The 2009 WHO guidelines already offer the possibility to waive the phase III-type safety and efficacy study in situations where suitable PD markers are available. In the EU, several types of biosimilars have been approved on the basis of pivotal PD studies, and further reduction of the burden of clinical data is anticipated [25]. Three publications have performed a retrospective analysis of the role of confirmatory efficacy and safety studies in regulatory decision-making.
Webster et al. [24] analyzed data on biosimilars submitted in the EU, US, Canada, and Australia from 2006 to March 2019 and concluded that no submissions that were rejected or withdrawn were concluded to be comparable at the analytical level and bioequivalent in PK studies and that no products shown to be comparable in analytical testing and PK studies had failed in the confirmatory efficacy and safety study.
Schiestl et al. [81] conducted a retrospective review of publicly available assessment reports of EMA and the US FDA from 2006 to November 2019 to clarify the role of clinical trials in biosimilar development. They pointed out that several biosimilars were approved without large safety and efficacy studies. Instead, PD-markers were used as surrogates of efficacy. They emphasize the role of PK studies. Interestingly, several PK trials initially failed to show comparable kinetics because of inadequate stratification and small sample size. However, subsequent PK studies demonstrated the comparable PK, an observation also made by other authors and leading to the conclusion that the successful and robust PK trial is a necessary condition for approval of a biosimilar [25].
Out of 42 development programs, 38 included a confirmatory phase III-type study with clinical endpoints. Three out of these studies failed to meet the predefined equivalence criteria. However, the three products were approved after results of post-hoc analyses and when the totality of the evidence from the complete comparability exercise had been considered. In two programs, the applicant had to perform additional studies to reach regulatory approval. In all cases of (initially) failed phase III studies, the underlying root cause was established.
The authors point out that the development programs have given valuable lessons of the structure–function relationship that will help to avoid similar problems in the future. Three products were rejected. In two cases, the comparability could not be established at the analytical and clinical level, and in the third case, comparative PK could not be established.
The conclusion was that in all but two cases, comparative phase III-type efficacy and safety studies did not change the outcome of the regulatory decision. In these two cases, the problem was increased immunogenicity caused by impurities, excess host cell contaminants, and leachables. Such problems are currently under careful scrutiny by developers and regulators and are unlikely to occur in the future. The authors conclude that phase III-type efficacy and safety studies contributed to the decision-making only by revealing increased immunogenicity.
Bielsky et al. [82] reviewed European Public Assessment Reports of 20 different complex biosimilar products related to six active substances [five mAbs (infliximab, rituximab, adalimumab, bevacizumab, and trastuzumab) and an FC (etanercept)] for possible differences in analytical tests, PK studies, and efficacy and safety studies.
Analysis of chemical and post-translational differences revealed minor differences that were considered to have no impact on safety, immunogenicity, or efficacy. These data will be useful in the interpretation of the analytical tests of future applications.
Two biosimilar candidates had major manufacturing and analytical comparability problems and were not approved even though, in both cases, the efficacy trials met their primary and secondary endpoints. This case demonstrates that a clinical phase III-type efficacy and safety study is less sensitive and discriminatory than state-of-the-art analytical testing.
In three cases, the first PK study failed to demonstrate comparable PK. Analysis of PK studies revealed problems in randomization and underestimation of inter-subject variability and sample size analysis. Avoiding those problems in additional PK studies led to demonstration of comparability.
The retrospective analyses found some difference in either immunogenicity, secondary efficacy endpoints, or in a subgroup analysis of efficacy. The problems were solved by clarifications and additional data or analyses.
The authors also reviewed data of previously withdrawn or rejected applications and found that efficacy and safety studies were never the only reason for failure to reach approval.
They conclude that the confirmatory efficacy and safety studies have not contributed to the benefit–risk assessment. Therefore, these costly and time-consuming studies should only be required in special circumstances, such as unknown main mode of action of the RBP or where there are difficulties in predicting the impact of observed analytical differences. The risk of known but unpredictable serious adverse effects is an additional reason for asking for additional safety data. Finally, the authors stress the importance of a risk-management plan for pharmacovigilance, enabling traceability and post-marketing studies. The Medicines and Healthcare products Regulatory Agency (MHRA) guideline for development of biosimilars is based on this reasoning [83].
The conclusions of the abovementioned studies are supported by Webster et al. [84]. They also point out that comparability exercises in biosimilar development and after a manufacturing change of a given product are based on the same principles. Thus, the data on manufacturing changes over 3 decades support the safety of biosimilars.
4 Conclusions
-
Biosimilars developed according to WHO and similar guidelines issued by other bodies are therapeutic equivalents to their RBPs. However, in view of the advancement in the analytical sciences and the experience gained, the development scheme of biosimilars is increasingly criticized for demanding unnecessary clinical studies. Therefore, the ECBS of the WHO asked for an analysis of possibilities to add flexibility according to the type of product and, thus, reduce the requirements for clinical data.
-
It should be noted that the aim of a biosimilar development program is to establish the similarity of a candidate biosimilar to the RBP. It is not conducted to independently establish the safety and efficacy of a candidate biosimilar.
-
The analysis of guidelines from several jurisdictions revealed possibilities for clarifications and for a reduction of the regulatory burden. In fact, the present guidelines include flexibility that is not often utilized because of the rigid regulatory approach. The possibility of using PD markers as surrogates of clinical endpoints is already mentioned in the current WHO guideline. The use of biomarkers reflecting the mechanism of action of the active substance should always be considered.
-
Additional flexibility may be introduced by re-estimating the “residual uncertainty” after completing analytical, in vitro functional testing, and clinical PK/PD studies.
-
Reduction of the clinical data requirements can be built on current overall regulatory principles but requires a new perspective on the role of “confirmatory” clinical efficacy and safety studies.
-
Three research groups analyzed the contribution of efficacy and safety studies to the benefit–risk assessment of biosimilars by comparing the outcomes of analytical, in vitro functional, and PK/PD studies to the outcome of confirmatory efficacy and safety studies. The results question the added value of phase III-type efficacy and safety studies on top of analytical, in vitro functional, and clinical PK studies.
-
However, the results do not definitely prove that the confirmatory safety and efficacy studies are useless. There were two cases, where increased immunogenicity was observed during the confirmatory efficacy and safety study. However, in the first case, unacceptable high levels of host cell impurities were already observable at the analytical level. The second case concerned epoetin α, a product with a high immunogenic risk, for which immunogenicity data will likely be necessary also in the future. In addition, the regulatory evaluation of biosimilars is an interactive process where regulators and manufacturers, on one hand, and assessors of quality, safety, and efficacy, on the other hand, discuss the “totality of the evidence.” Therefore, it is possible that the results of confirmatory efficacy and safety studies have contributed to the benefit–risk discussion by alleviating concerns of observed analytical or functional differences. We recommend that manufacturers consult relevant regulatory agencies when considering development plans without a confirmatory efficacy and safety study.
-
Nevertheless, regulators’ risk aversion should not suppress the development of important medicines [85]. Adequately powered PK and/or PD trials will provide sufficient clinical safety and immunogenicity data in most cases. Thus, the need for confirmatory efficacy and safety studies should be evaluated on a case-by-case basis according to criteria in the revised guideline.
-
Reduction of clinical efficacy and safety data requirements will increase the relative emphasis on physico-chemical, structural, and in vitro functional data. The manufacturers need to use state-of-the-art analytical methods and in vitro functional tests and robustly apply these for assessing similarity. Likewise, regulators need to have the competence to critically evaluate such data. PK/PD studies need to be robust in order to support the reduction of confirmatory efficacy and safety trials. A functional pharmacovigilance system is also a cornerstone of the safe reduction of pre-marketing requirements.
5 The Way Forward
It is proposed that the revision of the WHO guidelines will include the following main issues:
-
Additional clarity in the quality section is needed, including analytical characterization of a biosimilar and its RBP, risk assessment of quality attributes, the use of sensitive orthogonal analytical methods in characterization studies, and the establishment of comparability ranges for quality attributes and the use of the ranges in the similarity exercise.
-
Introduction of a stepwise progression of non-clinical studies that reduces or obviates the need for in vivo non-clinical tests, but will allow animal tests in exceptional cases. Nevertheless, the risk of unexpected adverse effects, different from those already known, from the RBP is negligible after demonstration of analytical and functional comparability. In addition, the current standard in vivo toxicological tests are not suitable for detecting potential small differences between a biosimilar and its RBP.
-
The use of a foreign RBP instead of a domestic product should be clarified for the two scenarios:
-
A suitable RBP is not licensed locally. In this case, the NRA may accept an RBP that has been licensed in another jurisdiction provided that:
-
The foreign RBP is licensed and marketed in a jurisdiction that has a well-established regulatory framework and considerable experience in the evaluation of biotherapeutic products developed according to WHO and similar guidelines, as well as an adequate post-marketing surveillance system.
-
The RBP has been marketed for a suitable duration and has a volume of marketed use such that the demonstration of similarity to it makes it possible to refer to substantial efficacy and safety data. However, this requirement should be used in a flexible way, e.g., in the case of orphan medicines.
-
The manufacturer will demonstrate that the chosen foreign RBP is suitable to support the application for marketing authorization of an SBP.
-
-
RBP is licensed locally but sourced from another jurisdiction.
-
If required by the legislation in place, the comparability of the local and foreign-sourced versions of the product should be demonstrated by analytical “bridging” studies and, where needed, complemented by additional PK/PD data.
-
-
-
Reduction of confirmatory efficacy and safety studies.
The need to conduct “confirmatory” efficacy and safety studies should not be a common requirement. The revision should include criteria for situations where data generated in a confirmatory efficacy and safety study are needed, e.g., a lack of in vitro test for relevant function(s). It may also be prudent not to waive the efficacy and safety study when the RBP has common or unpredictable serious adverse effects that cannot be merely explained by exaggerated pharmacological action.
References
WHO Expert Committee on Biological Standardization.2013. Annex 2. Guidelines on evaluation of similar biotherapeutic products (SBPs). WHO Technical Report Series no. 977. http://who.int/biologicals/publications/trs/areas/biological_therapeutics/TRS_977_Annex_2.pdf Accessed 12 Nov 2021.
Kang HN, Thorpe R, Knezevic I, Survey participants from 19 countries. The regulatory landscape of biosimilars: WHO efforts and progress made from 2009 to 2019. Biologicals. 2020;65:1–9. https://doi.org/10.1016/j.biologicals.2020.02.005.
World Health Organization Expert Committee on Biological Standardization. Guidelines on evaluation of monoclonal antibodies as similar biotherapeutic products (SBPs). In: sixty-seventh report, WHO technical report series 1004, 93–127. http://apps.who.int/iris/bitstream/10665/255657/1/9789241210133-eng.pdf. Accessed 12 Nov 2021.
World Health Organization. Questions and Answers: similar biotherapeutic products. 2018. https://www.who.int/biologicals/expert_committee/QA_for_SBPs_ECBS_2018.pdf?ua=. Accessed 12 Nov 2021.
WHO Expert Committee on Biological Standardization: Report of the seventy-second and seventy-third meetings. Geneva: World Health Organization; 2021 (WHO Technical Report Series, No. 1030; https://www.who.int/publications/i/item/9789240024373) Accessed 13 May 2022.
FDA. Biosimilars guidelines. https://www.fda.gov/vaccines-blood-biologics/general-biologics-guidances/biosimilars-guidances. Accessed 7 Dec 2021.
Health Canada. Information and Submission Requirements for Biosimilar Biologic Drugs. https://www.canada.ca/content/dam/hc-sc/migration/hc-sc/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/seb-pbu/seb-pbu-2016-eng.pdf Accessed 8 Dec 2021.
EMA: Multidisciplinary scientific guidelines: biosimilars. https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/multidisciplinary/multidisciplinary-biosimilar. Accessed 7 Dec 2021
FDA. Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product. https://www.fda.gov/media/88622/download. Accessed 23 Feb 2021.
FDA. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. https://www.fda.gov/media/82647/download. Accessed 15 July 2021
Kang HN, Thorpe R, Knezevic I, Casas Levano M, Chilufya MB, Chirachanakul P, et al. Regulatory challenges with biosimilars: an update from 20 countries. Ann N Y Acad Sci. 2021;1491(1):42–59.
FDA. Questions and Answers on Biosimilar Development and the BPCI Act. Guidance for Industry. Rev 2, 2021. https://www.fda.gov/media/119258/download. Accessed 7 Dec 2021.
EMA. Guideline on similar biological medicinal products. Revision 1 https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf. Accessed 8 Dec 2021.
Webster CJ, Woollett GR. A ‘global reference’ comparator for biosimilar development. BioDrugs. 2017;31(4):279–86. https://doi.org/10.1007/s40259-017-0227-4.
Tu CL, Wang YL, Hu TM, Hsu LF. Analysis of pharmacokinetic and pharmacodynamic parameters in EU-versus US-licensed reference biological products: are in vivo bridging studies justified for biosimilar development? BioDrugs. 2019;33(4):437–46. https://doi.org/10.1007/s40259-019-00357-2.
EMA. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). 2021. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf. Accessed 8 Dec 2021
EMA. Committee for Medicinal Products for Human Use (CHMP) Reflection paper on statistical methodology for the comparative assessment of quality attributes in drug development. 2021. https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-statistical-methodology-comparative-assessment-quality-attributes-drug-development_en.pdf. Accessed 7 Dec 2021.
FDA. Development of Therapeutic Protein Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations. Draft guidance https://www.federalregister.gov/documents/2019/05/22/2019-10667/development-of-therapeutic-protein-biosimilars. Accessed 10 Nov 2021.
EMA. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Revision 1 https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf. Accessed 8 Dec 2021.
Pipalava P, Patel R, Mehta M, Dahiya M, Singh I, Jose V. An update on the animal studies conducted for biosimilar approvals—regulatory requirement vs actual scenario. Regul Toxicol Pharmacol. 2019;107: 104415. https://doi.org/10.1016/j.yrtph.2019.104415 (Epub 2019 Jun 26 PMID: 31254556).
Chapman K, Adjei A, Baldrick P, et al. Waiving in vivo studies for monoclonal antibody biosimilar development: National and global challenges. MAbs. 2016;8(3):427–35. https://doi.org/10.1080/19420862.2016.1145331 (PMID: 26854177; PMCID: PMC4966840).
van Aerts L, De Smet K, Gabriele R, van der Laan JW, Schneider CK. Biosimilars entering the clinic without animal studies. MAbs. 2014;6(5):1155–62. https://doi.org/10.4161/mabs.29848.
van Meer PJK, Ebbers H, Kooijman M, et al. Contribution of animal studies to evaluate the similarity of biosimilars to reference product. Drug Discov Today. 2015;20:483–90.
Webster CJ, Wong AC, Woollett GR. An Efficient development paradigm for biosimilars. BioDrugs. 2019;33:603–11. https://doi.org/10.1007/s40259-019-00371-4.
Wolff-Holz E, Tiitso K, Vleminckx C, Weise M. Evolution of the EU biosimilar framework: past and future. BioDrugs. 2019;33(6):621–34. https://doi.org/10.1007/s40259-019-00377-y.
EMA. Guideline on Immunogenicity assessment of therapeutic proteins. Rev 1 https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revis12.11.ion-1_en.pdf. Accessed 12 Nov 2021
Kurki P, Barry S, Bourges I, Tsintili P, Wolff-Holz E. Safety, Immunogenicity and interchangeability of biosimilar monoclonal antibodies and fusion proteins: a regulatory perspective. Drugs. 2021;81:1881–96. https://doi.org/10.1007/s40265-021-01601-21.
EMA. Biosimilars https://www.ema.europa.eu/en/medicines/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar/ema_group_types/ema_withdrawn_applications/ema_group_types/ema_medicine. Accessed 9 Dec 2021.
Blandizzi C, Galeazzi M, Valesini G. Transitioning from first- to second-generation biosimilars: an appraisal of regulatory and post-marketing challenges. Pharmacol Res. 2018;128:306–14.
Pfäffle R, Bidlingmaier M, Kreitschmann-Andermahr I, et al. Safety and effectiveness of Omnitrope®, a biosimilar recombinant human growth hormone: more than 10 years’ experience from the PATRO Children Study. Horm Res Paediatr. 2020;93(3):154–63. https://doi.org/10.1159/000508190 (Epub 2020 Aug 19 PMID: 32814319).
Beck-Peccoz P, Höybye C, Murray RD, et al. No increased risk of glucose metabolism disorders in adults with growth hormone deficiency undergoing long-term treatment with biosimilar somatropin (Omnitrope®): data from an observational, longitudinal study. BMC Endocr Disord. 2019;19:138. https://doi.org/10.1186/s12902-019-0464-2.
López-Siguero JP, Pfäffle R, Chanson P, Szalecki M, Höbel N, Zabransky M. Ten years’ clinical experience with biosimilar human growth hormone: a review of efficacy data. Drug Des Dev Ther. 2017;11:1489–95. https://doi.org/10.2147/DDDT.S130320.PMID:28553079;PMCID:PMC5439972.
Beck-Peccoz P, Höybye C, Murray RD, et al. Malignancy risk in adults with growth hormone deficiency undergoing long-term treatment with biosimilar somatropin (Omnitrope®): data from the PATRO Adults study. Ther Adv Endocrinol Metab. 2020;11:1–12. https://doi.org/10.1177/2042018820943377 (PMID: 32973992; PMCID: PMC7491215).
Abboud CN, Lang N, Fung H, et al. Real-world safety experience of tevagrastim/ratiograstim/biograstim and tbo-filgrastim, short-acting recombinant human granulocyte colony-stimulating factors. Support Care Cancer. 2019;27:2569–77. https://doi.org/10.1007/s00520-018-4522-5.
Aapro M, Bokemeyer C, Ludwig H, et al. Chemotherapy-induced (febrile) neutropenia prophylaxis with biosimilar filgrastim in elderly versus non-elderly cancer patients: patterns, outcomes, and determinants (MONITOR-GCSF study). J Geriatr Oncol. 2017;8:86–95.
Becker P, Schwebig A, Brauninger S, et al. Healthy donor hematopoietic stem cell mobilization with biosimilar granulocyte-colony-stimulating factor: Safety, efficacy, and graft performance. Transfusion. 2016;56:3055–64.
Kang K-W, Lee B-H, Jeon MJ, et al. Efficacy and safety of two pegfilgrastim biosimilars: Tripegfilgrastim and pegteograstim. Cancer Med. 2020;9:6102–10.
Pahnke S, Egeland T, Halter J, et al. Working Group Medical of the World Marrow Donor Association. Current use of biosimilar G-CSF for haematopoietic stem cell mobilisation. Bone Marrow Transplant. 2019;54(6):858–66. https://doi.org/10.1038/s41409-018-0350-y.
Casadevall N, Nataf J, Viron B, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346:469–75.
Haag-Weber M, Eckardt KU, Hörl WH, Roger SD, Vetter A, Roth K. Safety, immunogenicity and efficacy of subcutaneous biosimilar epoetin-α (HX575) in non-dialysis patients with renal anemia: a multi-center, randomized, double-blind study. Clin Nephrol. 2012;77(1):8–17. https://doi.org/10.5414/cn107304 (PMID: 22185963).
Seidl A, Hainzl O, Richter M, et al. Tungsten-induced denaturation and aggregation of epoetin alfa during primary packaging as a cause of immunogenicity. Pharm Res. 2012;29(6):1454–67. https://doi.org/10.1007/s11095-011-0621-4.
Wish JB, Rocha MG, Martin NE, et al. Long-term safety of epoetin alfa-epbx for the treatment of anemia in ESKD: pooled analyses of randomized and open-label studies. Kidney Med. 2019;1(5):271–80. https://doi.org/10.1016/j.xkme.2019.06.009 (PMID: 32734207; PMCID: PMC7380401).
Goldsmith D, Dellanna F, Schiestl M, Krendyukov A, Combe C. Epoetin biosimilars in the treatment of renal anemia: what have we learned from a decade of european experience? Clin Drug Investig. 2018;38:481–90. https://doi.org/10.1007/s40261-018-0637-1.
Yang J, Yu S, Yang Z et al. Efficacy and safety of supportive care biosimilars among cancer patients: a systematic review and meta-analysis. BioDrug. 2019; 33(4):373–389. https://doi.org/10.1007/s40259-019-00356-3. https://www.researchgate.net/publication/333581066_Efficacy_and_Safety_of_SupportiveCare_Biosimilars_Among_Cancer_Patients_A_Systematic_Review_and_Meta-Analysis
Shingaki T, Taki K, Koyanagi M, et al. Long-term safety and effectiveness of biosimilar insulin glargine in Japanese patients with diabetes mellitus in routine clinical practice: results of a post-marketing safety study. Curr Med Res Opin. 2020;36(6):947–58. https://doi.org/10.1080/03007995.2020.1754182 (Epub 2020 Apr 23 PMID: 32271092).
Tieu C, Lucas EJ, DePaola M, Rosman L, Caleb AG. Efficacy and safety of biosimilar insulins compared to their reference products: a systematic review. PLoS One. 2018;13(4):e0195012. https://doi.org/10.1371/journal.pone.0195012 (Published 2018 Apr 18).
EMA. Medicines, https://www.ema.europa.eu/en/medicines. Accessed 11 Nov 2021
Cohen HP, McCabe D. The importance of countering biosimilar disparagement and misinformation. BioDrugs. 2020;34(4):407–14. https://doi.org/10.1007/s40259-020-00433-y.
Kim TH, Lee SS, Park W, et al. A 5-year retrospective analysis of drug survival, safety, and effectiveness of the infliximab biosimilar CT-P13 in patients with rheumatoid arthritis and ankylosing spondylitis. Clin Drug Investig. 2020;40(6):541–53. https://doi.org/10.1007/s40261-020-00907-5 (PMID: 32328979).
Lee SJ, Baek K, Lee S, Lee YJ, Park JE, Lee SG. Post-marketing pooled safety analysis for CT-P13 treatment of patients with immune-mediated inflammatory diseases in observational cohort studies. BioDrugs. 2020;34(4):513–28. https://doi.org/10.1007/s40259-020-00421-2.PMID:32356239;PMCID:PMC7223987.
Cohen SB, Radominski SC, Kameda H, et al. Long-term efficacy, safety, and immunogenicity of the infliximab (IFX) biosimilar, PF-06438179/GP1111, in patients with rheumatoid arthritis after switching from reference IFX or continuing biosimilar therapy: week 54–78 data from a randomized, double-blind, phase III trial. BioDrugs. 2020;34:197–207. https://doi.org/10.1007/s40259-019-00403-z.
Fischer S, Cohnen S, Klenske E, et al. Long-term effectiveness, safety and immunogenicity of the biosimilar SB2 in inflammatory bowel disease patients after switching from originator infliximab. Therap Adv Gastroenterol. 2021. https://doi.org/10.1177/1756284820982802 (Published online January 14).
Bellinvia S, Cummings JRF, Ardern-Jones MR, Edwards CJ. Adalimumab biosimilars in Europe: an overview of the clinical evidence. BioDrugs. 2019;33:241–53. https://doi.org/10.1007/s40259-019-00355-4.
Park HM, Kim J, Park S-H, et al. Long-term efficacy, safety and immunogenicity in patients with rheumatoid arthritis continuing on an etanercept biosimilar (LBEC0101) or switching from reference etanercept to LBEC0101: an open-label extension of a phase III multicentre, randomised, double-blind, parallel-group study. Arthritis Res Ther. 2019;21:122. https://doi.org/10.1186/s13075-019-1910-2.
Ebbers HC, Pieper B, Issa A, Addison J, Freudensprung U, Rezk MF. Real-world evidence on etanercept biosimilar SB4 in etanercept-naïve or switching patients: a systematic review. Rheumatol Ther. 2019;6:317–38. https://doi.org/10.1007/s40744-019-00169-4.
Martelli L, Peyrin-Biroulet L. Efficacy, safety and immunogenicity of biosimilars in inflammatory bowel diseases: a systematic review. Curr Med Chem. 2019;26(2):270–9. https://doi.org/10.2174/0929867323666161014153346 (PMID: 27758715).
Numan S, Faccin F. Non-medical switching from originator tumor necrosis factor inhibitors to their biosimilars: systematic review of randomized controlled trials and real-world studies. Adv Ther. 2018;35:1295–332. https://doi.org/10.1007/s12325-018-0742-9.
Egeberg A, Ottosen MB, Gniadecki R, et al. Safety, efficacy and drug survival of biologics and biosimilars for moderate-to-severe plaque psoriasis. Br J Dermatol. 2018;178(2):509–19. https://doi.org/10.1111/bjd.16102 (Epub 2018 Jan 9 PMID: 29094341).
Avila-Ribeiro P, Fiorino G, Danese S. The experience with biosimilars of infliximab in inflammatory bowel disease. Curr Pharm Des. 2017;23(44):6759–69. https://doi.org/10.2174/1381612824666171204095342 (PMID: 29205114).
Radin M, Sciascia S, Roccatello D, Cuadrado MJ. Infliximab biosimilars in the treatment of inflammatory bowel diseases: a systematic review. BioDrugs. 2017;31(1):37–49. https://doi.org/10.1007/s40259-016-0206-1 (PMID: 28035633).
Moots R, Azevedo V, Coindreau JL, et al. Switching between reference biologics and biosimilars for the treatment of rheumatology, gastroenterology, and dermatology inflammatory conditions: considerations for the clinician. Curr Rheumatol Rep. 2017;19:37. https://doi.org/10.1007/s11926-017-0658-4.
Calleja-Hernández MÁ, Martínez-Sesmero JM, Santiago-Josefat B. Biosimilars of monoclonal antibodies in inflammatory diseases and cancer: current situation, challenges, and opportunities. Farm Hosp. 2020;44(3):100–8. https://doi.org/10.7399/fh.11280 (PMID: 32452309).
Huizinga TWJ, Torii Y, Muniz R. Adalimumab biosimilars in the treatment of rheumatoid arthritis: a systematic review of the evidence for biosimilarity. Rheumatol Ther. 2021;8:41–61. https://doi.org/10.1007/s40744-020-00259-8.
Weise M, Kurki P, Wolff-Holz E, Bielsky M-C, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124:3191–6.
Doevendans E, Schellekens H. Immunogenicity of innovative and biosimilar monoclonal antibodies. Antibodies. 2019;8(21):1–10.
Yang J, Yu S, Yang Z, et al. Efficacy and safety of anti-cancer biosimilars compared to reference biologics in oncology: a systematic review and meta-analysis of randomized controlled trials. BioDrugs. 2019;33:357–71. https://doi.org/10.1007/s40259-019-00358-1.
Tony HP, Krüger K, Cohen SB, et al. Brief report: safety and immunogenicity of rituximab biosimilar GP 2013 after switch from reference rituximab in patients with active rheumatoid arthritis. Arthritis Care Res (Hoboken). 2019;71(1):88–94. https://doi.org/10.1002/acr.23771 (PMID: 30295429).
Suh CH, Yoo DH, Berrocal KA, et al. Long-term efficacy and safety of biosimilar CT-P10 versus innovator rituximab in rheumatoid arthritis: 48-week results from a randomized phase III trial. BioDrugs. 2019;33:79–91. https://doi.org/10.1007/s40259-018-00331-4.
Shim SC, Božić-Majstorović L, Berrocal Kasay A, et al. Efficacy and safety of switching from rituximab to biosimilar CT-P10 in rheumatoid arthritis: 72-week data from a randomized Phase 3 trial. Rheumatology. 2019;58:2193–202. https://doi.org/10.1093/rheumatology/kez152.
Esteva FJ, Lee S, Yu S, Kim M, Kim N, Stebbing J. 24 months results from a double-blind, randomized phase III trial comparing the efficacy and safety of neoadjuvant then adjuvant trastuzumab and its biosimilar candidate CT-P6 in HER2 positive early breast cancer (EBC). https://doi.org/10.1158/1538-7445.SABCS18-P6-17-03. Accessed 8 Dec 2021.
Stebbing J, Baranay Y, Baryash V, et al. 3-year follow-up of a phase III trial comparing the efficacy and safety of neoadjuvant and adjuvant trastuzumab and its biosimilar CT-P6 in HER2 positive early breast cancer (EBC). European Society for Medical Oncology (ESMO) congress 2019. Annals of Oncology Vol 30, Supplement 5 v64, Breast Cancer, Early Stage, Abstract 190P.Medicines for Europe. Lancet Oncol. 2017;18(7):917–28. https://doi.org/10.1016/S1470-2045(17)30434-5.
Pivot X, Pegram MD, Cortes J, et al. Final survival analysis of a phase 3 study comparing SB3 (trastuzumab biosimilar) and reference trastuzumab in HER2-positive early or locally advanced breast cancer. Presented at: 2021 San Antonio Breast Cancer Symposium; December 7-10, 2021. Poster P2-13-04.
Medicines for Europe. Biosimilar Medicines. The total clinical experience with biosimilar medicines https://www.medicinesforeurope.com/wp-content/uploads/2020/12/BIOS5.pdf. Accessed 12 Nov 2021.
Biosimilars in the EU—Information guide for healthcare professionals. https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf. Accessed 12 Nov 2021.
González CPV, Muñoz CG. The controversy around technical standards for similar biotherapeutics: barriers to access and competition? Pharmacoepidemiol Drug Saf. 2020;29(11):1518–22. https://doi.org/10.1002/pds.5100 (Epub 2020 Sep 22 PMID: 32964695).
Frapaise FX. The end of phase 3 clinical trials in biosimilars development? BioDrugs. 2018;32(4):319–24. https://doi.org/10.1007/s40259-018-0287-0 (PMID: 29943088).
McCamish M, Woollett G. The continuum of comparability extends to biosimilarity: how much is enough and what clinical data are necessary? Clin Pharmacol Ther. 2013;93(4):315–7. https://doi.org/10.1038/clpt.2013.17 (Epub 2013 Jan 25. PMID: 23443756; PMCID: PMC3604642).
Liu J, Eris T, Li C, et al. Assessing analytical similarity of proposed amgen biosimilar ABP 501 to adalimumab. BioDrugs. 2016;2016(30):321–38. https://doi.org/10.1007/s40259-016-0184-3.
McCamish M, Woollett G. The state of the art in the development of biosimilars. Clin Pharmacol Ther. 2012;91(3):405–17. https://doi.org/10.1038/clpt.2011.343 (Epub 2012 Feb 8 PMID: 22318617).
Christl L. FDA’s Overview of the regulatory guidance for the development and approval of biosimilar products in the US. https://www.fda.gov/files/drugs/published/FDA%E2%80%99s-Overview-of-the-Regulatory-Guidance-for-the-Development-and-Approval-of-Biosimilar-Products-in-the-US.pdf. Accessed 12 Nov 2021.
Schiestl M, Ranganna G, Watson K, Jung B, Roth K, Capsius B, Trieb M, Bias P, Maréchal-Jamil J. The path towards a tailored clinical biosimilar development. BioDrugs. 2020;34(3):297–306. https://doi.org/10.1007/s40259-020-00422-1.PMID:32266678;PMCID:PMC7211192.
Bielsky MC, Cook A, Wallington A, Exley A, Kauser S, Hay JL, et al. Streamlined approval of biosimilars: moving on from the confirmatory efficacy trial. Drug Discov Today. 2020. https://doi.org/10.1016/j.drudis.2020.09.006 (Epub ahead of print. PMID: 32916269).
MHRA. Guidance on the licensing of biosimilar products. 2019. https://www.gov.uk/government/publications/guidance-on-the-licensing-of-biosimilar-products/guidance-on-the-licensing-of-biosimilar-products. Accessed 8 Apr 2022.
Webster CJ, George KL, Woollett GR. Comparability of biologics: global principles, evidentiary consistency and unrealized reliance. BioDrugs 2021;35(4):379–387. https://doi.org/10.1007/s40259-021-00488-5. Accessed 12 Nov 2021.
Eichler HG, Bloechl-Daum B, Brasseur D, Breckenridge A, Leufkens H, Raine J, et al. The risks of risk aversion in drug regulation. Nat Rev Drug Discov. 2013;12:907–16. https://doi.org/10.1038/nrd4129.
Acknowledgements
The authors thank Dr. Hans-Karl Heim (Federal Institute for Drugs and Medical Devices (BfArM), Bonn, Germany), Dr. Jeewon Joung (Ministry of Food and Drug Safety, Osong, Republic of Korea), Dr. Maria Savkina (Ministry of Health, Russia), Dr. Robin Thorpe (Individual consultant, Welwyn, United Kingdom), and Dr. Meenu Wadhwa (National Institute for Biological Standards and Control, Potters Bar, United Kingdom), who provided valuable advice and comments during the literature review process. Sanna Saarinen (Finnish Medicines Agency Fimea) performed literature searches for the efficacy, safety, and immunogenicity of biosimilars.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The Ministry of Health and Welfare of the Republic of Korea has provided funds to the WHO for this project through a voluntary contribution for the period of 1 December 2020–30 September 2022. The open access of this article is also supported by the Norwegian Agency for Development Co-operation (Award number 73182 - 11).
Author contributions
PK and H-NK planned the review. PK reviewed the materials and prepared a draft manuscript. H-NK designed and coordinated the work. NE, IK, MW, and EW-H provided technical comments to finalize the manuscript. The co-authors are listed in alphabetical order in the author section after the two primary authors.
Disclaimer
The authors alone are responsible for the views expressed in this article and they do not necessarily represent the views, decisions, or policies of the institutions with which they are affiliated.
Conflict of interest
The authors declare no conflicts of interest.
Ethics approval
Not applicable.
Consent to participate/publish
Not applicable.
Availability of data and material (data transparency)
All data generated or analyzed during this study are included in this published article.
Code availability (software application or custom code)
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kurki, P., Kang, HN., Ekman, N. et al. Regulatory Evaluation of Biosimilars: Refinement of Principles Based on the Scientific Evidence and Clinical Experience. BioDrugs 36, 359–371 (2022). https://doi.org/10.1007/s40259-022-00533-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-022-00533-x