
Abstract
Intestinal metabolism can limit oral bioavailability of drugs and increase the risk of drug interactions. It is therefore important to be able to predict and quantify it in drug discovery and early development. In recent years, a plethora of models—in vivo, in situ and in vitro—have been discussed in the literature. The primary objective of this review is to summarize the current knowledge in the quantitative prediction of gut-wall metabolism. As well as discussing the successes of current models for intestinal metabolism, the challenges in the establishment of good preclinical models are highlighted, including species differences in the isoforms; regional abundances and activities of drug metabolizing enzymes; the interplay of enzyme-transporter proteins; and lack of knowledge on enzyme abundances and availability of empirical scaling factors. Due to its broad specificity and high abundance in the intestine, CYP3A is the enzyme that is frequently implicated in human gut metabolism and is therefore the major focus of this review. A strategy to assess the impact of gut wall metabolism on oral bioavailability during drug discovery and early development phases is presented. Current gaps in the mechanistic understanding and the prediction of gut metabolism are highlighted, with suggestions on how they can be overcome in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
A summary of current knowledge for the prediction of intestinal metabolism using in vivo, in situ, in vitro and mathematical models is provided. |
A strategy for the prediction of intestinal extraction that can be applied in drug discovery and early development is outlined. |
Gaps in current knowledge and technology that hamper the prediction of intestinal metabolism have been identified and a future direction proposed. |
1 Introduction
Oral dosing is the preferred route of administration as it is cheap, convenient and safe for patients [1, 2]. However, oral drug bioavailability is often limited by first-pass extraction in the gut and liver requiring higher doses compared with intravenous administration. Poor oral bioavailability has led to the failure of many drugs. As such, pharmaceutical companies aim to minimize hepatic and intestinal metabolism through drug design during lead optimization (LO). Oral bioavailability (F) is defined as:
where F a is the fraction of orally administered drug that is absorbed into the enterocytes, F G is the fraction of drug escaping first-pass metabolism in the enterocytes, and F H is the fraction of drug escaping first-pass hepatic metabolism and biliary secretion. Absolute oral bioavailability (F a × F G × F H) is determined by comparing the drug exposure (area under the plasma concentration–time profile) following oral administration with that after intravenous administration, assuming that first-pass metabolism from organs other than liver and gut can be neglected.
Intestinal metabolism can occur in the gut lumen as well as in enterocytes. In the gut lumen, microflora-mediated reduction [3] and hydrolysis [4–6] can be important but are often overlooked. Among the drug metabolizing enzymes (DMEs) expressed in human enterocytes [7], cytochrome P450 (CYP450), uridine diphospho-glucuronosyltransferases (UGTs) and sulphotransferases (SULTs) are the most important (Table 1). CYP3A is localized predominantly in functionally mature enterocytes at the villous tips, which result from migration of immature enterocytes in the crypts, a process that takes approximately 2–6 days [7]. Recently, meta-analysis has indicated that turnover of mature enterocytes in preclinical species and humans takes between 2.4 and 3.5 days [8]. The total mass of CYP3A in the entire small intestine based on homogenates from isolated enterocytes has been estimated to be less than 1 % of the liver [9, 10]. However, the intestine contributes significantly, or even equal, to hepatic CYP3A in the overall first-pass metabolism of several drugs (e.g. cyclosporine, midazolam and verapamil) [11–13]. Extensive presystemic intestinal metabolism is probably due to anterior positioning of the intestine with respect to the liver; the high luminal drug concentration and the long intestinal transit times that provide an increased opportunity for substrate–enzyme interaction, especially given the potential synergistic interplay of CYP3A and P-glycoprotein (P-gp) on dual substrates [14–16]. Additionally, unlike hepatic metabolism, presystemic metabolism is not limited by plasma protein binding or blood perfusion rates, but rather by permeability across enterocytes [17].
Poor oral bioavailability and extensive first-pass metabolism predisposes a patient population to potential toxicity arising from high doses as well as large interindividual variability [18] in exposures. In addition, extensive first-pass metabolism increases the risk of drug–drug interactions (DDIs) [19–23], the magnitude of which depends on the fraction escaping metabolism of both the substrate and the inhibitor [24] (if both the substrate and inhibitor are metabolized by the inhibited enzyme). The DDI risk arising from first-pass extraction can also be transporter-mediated [25–28]. These risks highlight the need for robust, quantitative models for predicting drug extraction through the gut wall and liver, backed by a sound understanding of the underlying mechanisms.
After decades of research, hepatic drug metabolism is well-understood. Only in recent years has there been an increased effort to understand intestinal first-pass extraction. Among the factors limiting intestinal availability (F a × F G), intestinal metabolism can be a major determinant [29] but is poorly understood. This is due to a number of confounding factors affecting it; namely, drug transit through the gastrointestinal tract, drug permeability, solubility and intestinal blood flow. Species differences in the isoforms, regional abundances and activities of DMEs [9, 30, 31] and transporters [32, 33], as well as the interplay of enzyme-transporter proteins and overlap of substrate and inhibitor specificity [34], make it difficult to establish good preclinical models for this process. Knowledge gaps on enzyme abundances and lack of empirical enzyme activity scaling factors are the main limitations of in vitro methods for the evaluation of intestinal metabolism. The large interindividual variability and inaccurate estimation of human intestinal extraction, even when intravenous pharmacokinetic (PK) data are available, pose additional challenges to the validation of both in vitro and in vivo models [35, 36]. Due to its broad specificity and high abundance in the intestine, CYP3A is the enzyme most frequently implicated in human gut metabolism, and therefore the most studied. Focusing on CYP-mediated processes, this review will summarize current knowledge on the advantages and limitations of preclinical models (in vivo, in situ, in vitro and mathematical) available for quantitative prediction of intestinal availability. This review will also outline a strategy applicable in drug discovery and early development, identify gaps in current understanding and propose future directions for the prediction of human gut-wall metabolism.
2 Human In Vivo F G
Seminal work from clinical studies with CYP3A drug substrates such as cyclosporine A [11, 37, 38] and midazolam [12, 39, 40] established the role of gut-wall metabolism in limiting human oral bioavailability. Substantial intestinal extraction was demonstrated in patients after sampling portal vein blood following intraduodenal drug administration during the anhepatic phase of liver transplantations. However, interpretation of this PK data is challenging given confounding factors attributable to the use of anaesthetics, surgery and the often poor condition of patients. These studies, as with in situ regional perfusions [41, 42], are rarely undertaken in humans for ethical, technical and cost reasons [21, 43].
Alternative clinical approaches rely on indirect estimation of human F G from intravenous/oral area under the curve (AUC) data or drug–food interaction data. Comparison of AUCs following intravenous and oral drug administration is relatively straightforward, although assumptions often hinder interpretation [44]. For example, the extent of metabolic extraction in the intestine can be over-emphasized if (1) notable extrahepatic systemic clearance is left unaccounted [45]; (2) the blood:plasma ratio deviates significantly from unity [46]; or (3) first-order elimination is not conserved at each site of drug administration [47, 48]. Additionally, calculated F G can be sensitive and biased according to the average value used for liver blood flow [49, 50]. When factors limiting oral absorption (efflux, low F a) are minimal and transporter-mediated uptake is negligible [46, 51], the drug–food interaction method offers an attractive, pragmatic model for estimating the extent of F G for CYP3A drug substrates. This is because certain fruit juices offer complete and exclusive presystemic inhibition of CYP3A in the small intestine, providing means for F G to be more readily identifiable and separated from hepatic first-pass elimination [52–58]. Advantageously, the interaction approach requires only measurement of oral AUC in the presence and absence of inhibitor, avoids many of the confounding factors associated with the intravenous/oral method, and allows researchers to benefit from the abundance of clinical data available [46, 51]. Clinical studies helped to delineate the metabolic component in liver and intestine for drugs such as alfentanil, cyclosporine, felodipine, midazolam, nifedipine and tacrolimus [46, 52, 54–56, 59, 60]. Grapefruit juice (GFJ) has been the most popular CYP3A inhibitor in food–drug interactions trialled thus far. The extent of clinical drug interactions with GFJ can be variable, depending on the GFJ strength and duration of administration [51, 61, 62]. Understanding underlying mechanisms and kinetics should aid study design and control exposure to inhibitory agents (furanocoumarins) mediating the interactions [57, 58, 61, 63–65]. In turn, this should address interstudy reproducibility and erroneous estimation of F G [21]. Detailed discussion of the advantages, limitations and underlying assumptions of these in vivo models are elaborated elsewhere [21, 46, 51]. Uncoupling F G and F a remains a significant challenge to any clinical approach, as does factoring out potential contributions from nonhepatic tissues [43].
3 Regional Differences in Intestinal Metabolism and Transport in the Human Gut
Compounds that have their maximum absorption in different regions of the gut are likely to have very different F G, even if they have comparable metabolic liabilities. This is probably due to regional differences in drug uptake into enterocytes, efflux and intestinal metabolism arising from regional variation in the luminal environment (e.g. pH, composition of intestinal fluids) [66–68] and epithelial membrane (e.g. surface area, expression of enzymes and transporters) [67]. Information on regional expression of enzymes and transporters is important for understanding the in vivo plasma PK profile and possible DDIs with co-medications.
It is well known that passive diffusion varies in humans and preclinical models according to the physicochemical properties of the drug in question and the region of intestine [69, 70]. Lipophilic compounds have the same or higher permeability coefficients in the lower bowel compared with the upper small intestine. With hydrophilic molecules the trend is reversed [69, 70], partly due to differences in the membrane lipid composition and the tightness of the tight junctional area [70]. Regional differences in drug uptake into the enterocytes can also occur due to differences in intracellular metabolism along the gut. Higher rates of intracellular metabolism can cause sink conditions inside the cells. A concentration gradient maintained across the membrane favors increased uptake, as has been suggested for indinavir [71].
Efflux proteins impact gut metabolism by reducing the intracellular drug concentrations exposed to DMEs in the enterocytes. At high doses, efflux proteins may become saturated, allowing a larger proportion of drug to pass the membrane efficiently. Expression of the main ABC transporters is heterogeneous along the gastrointestinal tract (Table 2) [72–74]. It is well known that multidrug resistance protein 1 (MDR1) [ABCB1, P-gp] is preferentially expressed toward the lower parts of the human small intestine (ileum) [74–76], while multidrug resistance-associated protein 3 (MRP3) [ABCC3] is expressed at higher levels than MDR1 along the small and large intestines [74]. In contrast, the efflux protein MRP2 (ABCC2) is expressed at relatively high levels in the small intestine but at extremely low levels in the colonic regions [74]. The other important efflux protein, breast cancer resistance protein (BCRP) (ABCG2), has higher levels than both MRP2 and MRP3 in the small intestine, but very low levels in the colonic regions [77, 78].
The solute carrier (SLC) proteins, including PepT1 (SLC15A), MCT1 (SLC16A), OATPs (OATP2B1; SLC0), OATs (SLC22A), OCT/OCTN (SLC22A) and the recently described PMAT (SLC29), may add significantly to uptake of the drug into enterocytes for many compounds with lower lipophilicities ([72], and refs. therein). These are also affected by food constituents as well as genetic polymorphisms and disease states [79, 80]. OATP2B1 has been found to have an unexpected influence on the absorption of the drug aliskiren [81]. The compound has affinity for MDR1 as well as CYP3A4, and inhibition of these mechanisms was expected to increase its bioavailability. However, in a study involving 11 healthy volunteers, a reduction of the absorption of aliskiren was found, when coadministered with GFJ, due to additional inhibition of the uptake transporter OATP2B1 by GFJ. Thus, prediction of drug absorption and DDI in the clinic gets further complicated for substrates of uptake transporters.
The regional distribution of these SLC proteins in the intestine, as well as species differences, are largely unknown. However, PepT1 is reported to be highly distributed in the proximal intestine of humans and many animal models [72, 82], and MCT1 is well known to be highly abundant along the whole gastrointestinal tract. The abundances of other SLC proteins tend not to be significant [78, 83, 155]. Further details can be found in the excellent review by Estudante et al., and references therein [72].
The abundance and catalytic activity of the main human CYP450 enzymes is generally highest in the proximal regions (i.e. duodenum and proximal jejunum), declining towards the lower ileum after a slight increase from the duodenum to the jejunum (Table 2) ([84–87], and refs. therein]). Data on regional gene expression and enzymatic activity are readily available for CYP3A4 and members of the CYP2C family, but are less well-characterized for other CYP450s. Paine et al., measured CYP450 protein levels along the gastrointestinal tract in 31 human donors and found that after CYP3A4, the most abundant enzyme was CYP2C9, then CYP2C19 with low levels of CYP2J2 and CYP2D6 [9]. Western blot data indicated that concentrations of CYP3A4 and CYP2C isoforms decreased dramatically towards the distal small intestine, with CYP2C levels falling faster compared with CYP3A4 [9, 86]. Information available on the expression of CYP450s in the human colonic enterocyte is limited, and is contradictory depending on the technique used [88–90]. Using messenger RNA (mRNA) and protein analysis, Bergheim et al., [91] found expression levels of CYP2C, CYP2E1 and CYP3A5 significantly differed between different regions of the large intestine, with CYP2C significantly higher in the ascending colon, and CYP2E1 and CYP3A5 significantly lower. In contrast, others failed to detect any significant levels of CYP4502C8–10 and CYP2E1 protein in colonic tissue [63, 64].
The most obvious difference between the small intestine and colonic tissue is the content of CYP3A4 and CYP3A5. In the small intestine, CYP3A5 is detected only at low levels [89] and may be absent in some individuals [9], while in colonic tissues, very low levels of CYP3A4 and higher relative expression of CYP3A5 were reported. Thus, CYP3A5 constituted the major CYP3A isoform in this tissue [88]. The overall lower rate of hydroxylation in the colonic region compared with the proximal jejunum was confirmed by van de Kerkhof et al., using a mixture of CYP450 substrates [CYP3A4/5 substrate midazolam (CYP3A4/5), followed by CYP2C9 (diclofenac) and CYP2D6 (bufuralol)] [90].
Regional differences in the abundance of phase II enzymes in the gut are not well-understood. van de Kerkhof et al. have reported similar activity for UGT and SULT enzymes [based on 7-hydroxy-coumarin (7-HC) conjugation] in both the proximal jejunum and the colon. They also report that glucuronidation efficiency in the gut was approximately sixfold higher than sulphation in both regions [90]. As with all quantitative approaches to measuring transporter and DME protein abundances, comparison of expression levels of different phase I and II enzymes between regions of the small intestine and colonic tissue, and between different studies, may be difficult given the different techniques used for quantification, i.e. protein quantitation using Western blot or liquid chromatography–tandem mass spectrometry (LC–MS/MS), as well as immunohistochemistry, mRNA and enzymatic activity using selected probes.
4 Combined Action of Drug Metabolizing Enzymes (DMEs) and Transporters in the Gut
Co-localization of CYP3A and MDR1 in the enterocyte along the crypt villus axis, overlapping substrate specificities and poor oral bioavailability of their joint substrates [15], have lead scientists to suspect an interplay between ABC transporters and members of the CYP450 enzyme families in the intestinal membrane affecting intestinal absorption and metabolism. It may also explain some DDIs which cannot be rationalized through either protein acting alone [15, 72].
P-gp is situated in the apical membrane of the enterocyte and efflux substrates from inside the cell towards the intestinal lumen. The CYP3A enzymes are located within the endoplasmic reticulum. It is therefore suggested that P-gp may regulate the intracellular concentration of dual substrates of P-gp and CYP3A. One might speculate that increasing P-gp levels in the proximal to distal direction serves to recycle its substrate and aid the efficient elimination of harmful molecules by reducing the intracellular drug concentration to levels below DME saturation in the proximal region, and increase the residence time of its substrate for metabolism in the distal gastrointestinal tract. Although demonstrated in vitro [90], some authors have pointed out that the extent of such synergistic effects in vivo is minimal [92]. The extent of activity of DMEs and P-gp are determined by the drug concentration at the site of the absorption, which in turn is related to solubility (biopharmaceutical classification) [73] and regional stability of the substrate in the gut lumen. Murakami and Takano suggested that the biopharmaceutics classification system (BCS) Class I compounds that are readily water-soluble and have high permeability will be rapidly absorbed in the upper part of the small intestine by passive diffusion [73]. Consequently, these compounds can be extensively metabolized due to higher CYP450 expression in the proximal small intestine and lower dependence on P-gp and/or BCRP [29]. However, BCS class II and III, as well as intravenous compounds with poor solubility and/or permeability, are likely to be absorbed in the lower parts of the intestine where metabolism can be substantial if they are also substrates of P-gp/BCRP [73]. In conclusion, compounds with high permeability that are typically absorbed in the upper region of the small intestine can escape the combined action and potentiation of metabolism and efflux, while moderate and low-soluble compounds have an increased potential to be involved in the recycling action of transporters.
Metabolites formed by CYP450s can be efficiently transported to either the mucosal or blood side of the enterocyte by the action of either MDR1, BCRP or the MRPs. The metabolite of ropivacain was secreted to a larger extent on the luminal side of the human jejunum compared with the ileum [93]. Although the transporters involved were not identified, examples such as this clearly highlight the need for a greater understanding of the interplay between enzymes and transporters in the intestinal tract.
5 Preclinical Models for the Prediction of F G
Various preclinical models of the intestine have been described (see Table 3). If appropriately integrated into DMPK strategies for optimization of drug absorption, distribution, metabolism and excretion (ADME) properties, it may provide more reliable prediction of human first-pass oral clearance, bioavailability and PK profile. Models should be considered in terms of their complexity, the mechanistic understanding they provide, and their clinical translation, e.g. quantitative prediction of the fraction of drug escaping metabolism in the human gut wall.
Often there is a trade-off between the different in vitro, in situ or in vivo models. On the one hand, in vivo models retain the native architecture of the small intestine and physiologically relevant expression profiles of DMEs, co-factors and transporter proteins. They offer integration of dynamic processes such as the mesenteric blood circulation and mucous layer coupled with function to study complexities arising from simultaneous metabolism–transporter interplay [15, 94, 95]. Notable attractions with in situ techniques are they closely mimic the in vivo situation yet provide a unique opportunity to study intestinal events in isolation, e.g. absence of biliary excretion and enterohepatic recirculation; however, this isolation could compromise the interpretability of the results. Furthermore, species differences in enzymes and transporters (Table 2) can make them unreliable for human F G prediction. In vitro models employing human-specific systems lack native architecture, but, in combination with mathematical models, hold the promise of robust prediction, provided there is a correlation between in vitro and human in vivo.
5.1 In Vivo and In Situ Animal Models
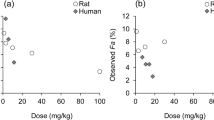
Gut-wall metabolism has been studied in several animal models [96–102]. By varying sites of drug administration (oral, intraportal and intravenous routes are common but intraperitoneal is also used) and PK sampling, the available fractions in liver and intestine can be calculated from comparison of AUCs under first-order conditions [103–107]; however, comparison of intravenous and oral AUCs after sampling at one site is more straightforward and routinely applied [108–110]. As such, similar issues described with the indirect approaches are to be expected. Despite differences in the CYP450 isoforms expressed in the rat compared with human (see the following paragraphs), a good correlation [root mean square error (RMSE) = 0.19] between rat and human F G has been reported using a set of 11 CYP3A-metabolized compounds that had both intravenous and oral PK data [111]. Ten of these 11 compounds studied had human F a of 0.8 or higher. As rat is a good model for human oral drug absorption, it follows that any differences in the intestinal availability should arise from intestinal efflux or metabolism. For BCS class I compounds, good solubility ensures sufficient concentrations for the saturation of efflux transporters. A high permeability rate ensures a low residence time within enterocytes, resulting in a rate of metabolic extraction that is limited by the rate of drug permeation through the enterocytes, rather than by the intrinsic ability of the intestinal DMEs. With permeability-limited intestinal extraction, any differences in enzyme isoform, abundance or activity have limited impact on the metabolic extraction, especially since CYP450s with broad substrate specificity abound in both human and rat, and their regional distributions are similar in both species. However, for hepatic extraction, lower hepatic concentrations compared with the gut lumen implies a greater role for transporters, while species differences in plasma protein binding and blood flow rate contribute to varying exposures to uptake and efflux transporters, as well as to DMEs, heightening the impact of differences in transporter and enzyme isoform, abundance and activities on hepatic extraction. This may explain why the rat is a good model for the prediction of human F G but is a poor predictor of human oral bioavailability [112]. A strong correlation was also reported between cynomolgus monkey and human F G for human CYP3A [113]. However, the cost and ethical concerns limit the availability of monkey PK studies. Animal models may be useful in the prediction of human F G for CYP3A-metabolized compounds, but not much is known about their utility for other intestinal DMEs.
In situ approaches include the perfused gut loop, single-pass and recirculating intestinal perfusions, portal vein cannulations, portacaval shunts and transpositions [114–121]. These techniques open the possibility of studying route-dependent intestinal metabolism following systemic and luminal drug presentation. This has been shown with acetaminophen, morphine and enalapril [96, 98, 106, 118, 122, 123]. Naturally-occurring tissue structure and physiology are retained, notwithstanding surgical manipulation and effect of anaesthetics [114, 124, 125]. Minimal interference of intestinal function and architecture means optimal tissue viability is maintained [106]. A full complement of endogenous DMEs and transporter proteins are present. Discrete segments of the small intestine can be evaluated to assess the impact of regional differences in DMEs or transporter expression and gut physiology. Intestinal metabolism can be more rigorously evaluated if drug concentrations are measured after sampling from mesenteric or portal veins [126]. Detailed methodologies are provided elsewhere [114, 127–130]. In situ models have several advantages over in vivo models. For instance, bypassing the stomach means acidic compounds are unlikely to precipitate, and therefore dissolution rates do not confound intestinal drug concentrations and resultant plasma levels [94]. These models can be exploited to investigate metabolism–transporter interplay, as exemplified with midazolam, indinavir and UK-343,664 [48, 131, 132]. Various aspects need to be controlled [133], including luminal flow rate and thickness of the unstirred water layer (UWL), which can be rate-limiting for rapidly absorbed compounds [106]. In spite of their utility, isolating perfused organs from, or within, the laboratory animals typically requires specialized surgical procedures. As such, these approaches are arguably more labour-intensive, time-consuming and costly compared with other in vivo and in vitro approaches.
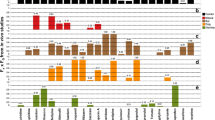
Whereas certain processes, such as passive diffusion, can be relatively well-predicted from animals [94, 106, 134–139], pronounced species differences in expression of DME isoforms, substrate selectivity and abundance along the gastrointestinal tract [30, 31, 113, 140–142] implies that human F G prediction from preclinical species is not always feasible. The differences in relative protein expression of individual CYP450 isoforms in rat, dog, monkey and human small intestines are presented in Fig. 1. These species differences have led to substantial differences in apparent F a × F G as well as F G [30, 143–145]. Metabolism studies, using drug substrates for human CYP450s [141, 143, 146] and UGT enzymes [142, 147], have generally reported poor correlation between human and animal F G. Certain DMEs appear to be selectively expressed in human intestines, including UGT1A8, UGT1A10 and SULT1A3 [140, 148, 149]. To the best of our knowledge, there is no known animal orthologue of SULT1A3 or UGT1A10 [147]. Additionally, there are known species differences for other enzyme classes expressed in the gut, such as the carboxylesterases [3, 4, 150, 151]. If a drug is shown to be a substrate for one of these enzymes then predicting human F G using animal data would be questionable.

Small intestine CYP450 pie charts for rat, dog, monkey and humans. The mean percentage contributions of individual CYP450 enzymes were calculated from total immunoquantified CYP450 protein (dog, monkey, human) or total spectrally determined CYP450 (rat). Analysis was based on published data: rat [154], dog [254, 255], monkey [268] and humans [9]. Additionally, CYP4F2 protein was detected (~7 pmol/mg protein) in human small intestine microsomal fractions [269]. This has been excluded from the human CYP450 pie chart because the total intestinal CYP450 content has not been reported, precluding comparison of relative abundances of individual enzyme isoforms between studies. CYP450 cytochrome P450
Species differences in regional enzymatic profiles exist within the intestine (Table 2). The regional decrease in CYP3A in humans, as a function of distance along the small intestine from the duodenum to the ileum [9, 85], is similar to the rat small intestine [152–154]. Interestingly, in the rat intestine Cyp2b1 is highly expressed, whereas the equivalent isoform in humans (CYP2B6) is not expressed [9]. Rat Cyp2b1 is present at much higher protein levels in the upper parts of the intestine, whereas in humans the equivalent isoform (CYP2D6) is not [154]. The trend in the regional expression of rat Cyp2c isoforms (such as Cyp2c6) is opposite to that of Cyp3a, with higher expression towards the lower bowel [154] (Table 2). In contrast to being absent in the human small intestine, the extrahepatic enzyme Cyp1a1 has been reported by some authors to be the predominant CYP450 isoform expressed in rat small intestine, together with Cyp3a [9, 154–156]. The glucuronidation/sulphation ratio can differ between regions according to individual species. In rat colon and proximal jejunum, the ratio was 16 and 23, respectively [157], showing a clear species difference in conjugation activity and regional difference compared with humans (see Sect. 3 and van de Kerkhof et al. [90]).
Regional expression of Mdr1b in rat intestine is similar to regional expression of MDR1 in the human small intestine and colon [74–76, 78, 158] (Table 2). Contrary to this, canine intestinal Mdr1 expression is highest in the jejunum and very low in the lower parts of the ileum and colon [159]. For the MRP2 transporter, humans are similar to rat, with high expression in the proximal small intestine and very low levels in the lower bowel [74, 78, 83]. Abcc3 (Mrp3) is highly expressed in the colon of rats [160], and mice, at both the mRNA and protein levels [161] (Table 2). Interestingly, MRP3 (the corresponding transporter gene in humans) has the highest level of all the efflux proteins expressed along the intestinal tract [74]. In mouse, the expression of bcrp is higher in the mid jejunum compared with the rest of the intestinal tract [162]. A similar cellular location (basolateral in enterocytes) found in mice, rats and humans further supports a high degree of conservation for ABC transporters amongst eukaryotes.
5.2 Knockout and Transgenic Mouse Models
In vivo rodent models with intestinal or hepatic enzyme gene knockdown or replacement have demonstrated the impact of intestinal versus hepatic elimination [163–168]. These knockout (KO) and/or genetically modified (GM), transgenic (TG) mouse models have been established to create more reliable in vivo systems to study and predict human response to novel chemical entities (NCEs) [163, 169–173]. For example, the importance of CYP3A metabolism in the intestine and liver has been illustrated with docetaxel in KO mice lacking all Cyp3a genes [Cyp3a (−/−)] [174]. When the CYP3A anticancer drug was administered intravenously to Cyp3a (−/−) mice, a sevenfold increase in systemic exposure was observed compared with wild-type. After oral dosing, an 18-fold higher systemic exposure was reported. Similar findings were reported for lopinavir and triazolam (CYP3A) [175, 176], debrisoquine (CYP2D6) [177] and tolbutamide (CYP2C9) [178]. This highlights the critical role intestinal CYP3A plays in human first-pass oral clearance and bioavailability.
Unfortunately, compensatory mechanisms arising from expression of host (murine) DMEs may confound data interpretation from KO models. For instance, clearance of midazolam was expected to be severely reduced in Cyp3a KO mice. In spite of this, metabolism was only marginally altered versus wild-type [179]. The revelation that several murine Cyp2c isoforms were significantly upregulated in Cyp3a KO mice, and could catalyze formation of midazolam 1′- and 4′-hydroxymidazolam, helped rationalize these results [179]. In contrast, no such effect was seen with triazolam, apparently a more selective Cyp3a substrate in mouse [176]. These compensatory mechanisms are likely to be drug- and species-dependent. In vitro studies characterizing background metabolism may prevent assessment of drugs susceptible to elevated host DMEs. Alternatively, exciting development of a viable mouse model in which all murine CYP450 genes have been deleted could avoid this issue altogether [180]. Interestingly, TG mice have been generated that are capable of expressing human CYP3A4 in the intestine and/or the liver on top of a mouse Cyp3a KO background [174]. As demonstrated with docetaxel and lopinavir, these humanized mice can provide mechanistic insight into the separate and combined roles of intestinal and hepatic human CYP3A, and the interplay with transporters such as P-gp in vivo [163, 175].
5.3 In Vitro and Mathematical Models
‘Bottom-up’ quantitative predictions that use in vitro data in conjunction with mathematical models describing processes within the gastrointestinal tract have been implemented [181–185]. Several of the in vitro assays highlighted in Table 3 are simplistic in nature, take less time to complete, and are a fraction of the cost of animal experiments. This makes them eminently suited for screening NCEs and designing out presystemic metabolic liabilities. Models vary in complexity, from simple membrane preparations of individually recombinantly expressed enzymes [186–190] to subcellular fractions [12, 43, 45, 85, 153, 191–195], intact cells [196–201] to tissue explants. The latter retain their in vivo tissue architecture, albeit lacking physiological surroundings. Although relatively labour-intensive and lower throughput (see Table 3), working with whole tissue preparations such as precision cut tissue slices (PCTS) [157, 202–206], Ussing chamber [43, 69, 90, 93, 207], everted sac [126, 208–212] and isolated tissue perfusions [94, 197, 213–216] brings with it several advantages. The cell–cell contacts remain intact, all cell types are present and the DME systems, co-factors and transporters are available at physiologically relevant concentrations that more closely mimic the in vivo situation. With the isolated perfusion technique, variables such as temperature, pH, osmolality, blood pressure and flow can be controlled [217]. Metabolism on both sides of the intestine (luminal and vascular) can be studied. However, maintaining tissue viability is the major issue with this approach [94]. As such, its application is generally limited to animal tissue, and short-term incubations (approximately 2 h post-excision) impeding assessment of slowly metabolized drugs or enzyme induction [43]. This can be circumvented in situ but is not without complication. Loss of tissue viability caused by insufficient oxygenation is also problematic for everted sacs (1–2 h) and Ussing chambers preparations (2–4 h) [218–220]. Nevertheless, everted sacs provide a fast and relatively inexpensive model for measuring regional differences in metabolism [106, 126]. The small volume inside the sacs offers analytical advantages over the isolated tissue perfusion and Ussing chamber, which due to sample dilution require sensitive bioanalysis to detect drug and/or metabolites [119, 126].
The Ussing chamber can be used with animal or human tissue providing a good model of drug absorption, transporter interactions, as well as metabolism, during passage across the gastrointestinal membrane [42, 69, 90, 219, 221–223]. Drug can be added to luminal or serosal sides, allowing bidirectional transport and metabolism kinetics to be studied in different sections of the intestine [106]. Detailed mechanistic interpretation from studies such as these may require additional insight from experimentation with enzyme inhibitors, radiolabelled drug, or separate consideration of metabolism and permeability, e.g. using intestinal microsomes and Caco-2 monolayers. Indeed the extent of drug extraction in human intestine (E g) has been successfully predicted for testosterone, midazolam and ropivacaine using human in vitro Ussing experiments [69, 93]. PCTS have received growing attention now that reproducible production of very thin slices (between 250 and 450 μm) is possible. These maintain better viability [203] and retain a high drug biotransformation capacity [43]. PCTS are also suitable for studying regional differences in intestinal metabolism, as well as regulation of enzymes and transporters involved in drug disposition [206, 224]. Applicable to all species, investigations have been reported in mouse [225], rat [157] and human [90]. Additionally, other tissues can be examined, allowing the extent of metabolism in different organs to be compared [204]. Potential drawbacks include poor penetration of highly metabolized drugs into the slices inner cell layers, lag time in phase II metabolism, and nonspecific binding to the slices [202, 226].
Intestinal subcellular fractions (S9 homogenates or microsomes) are one of the more established in vitro approaches used in drug discovery. More information has been published on the physiological scalars (Table 4) and several investigators have explored quantitative prediction of F G [49, 111, 227–231]. Commercial availability of animal and human samples, ease of storage and automation, make them an attractive option in terms of assay speed, capacity and cost. The fractions contain multiple enriched enzymes in their native configuration for assessment of phase I and selective phase II metabolism [45, 113, 195, 231, 232]; however, they do not contain a full complement of DMEs and lack potentially important interactions with uptake and efflux transporters. Incubations require the addition of expensive co-factors for optimal DME activity, often at higher nonphysiological concentrations. Others have suggested metabolic rates in S9 and microsomal fractions can be much lower compared with matrix such as PCTS [43]. This may be attributed to poor recovery of enzymes through suboptimal preparation procedures [195]. Presented in Table 5 are 6β-hydroxy testosterone rat data (normalized to units of pmol/min/mg rat intestinal protein) including intrinsic clearance (CLint) from in-house intestinal microsomes prepared under optimal conditions [233, 234]. Interestingly, in-house microsomes achieved broadly similar rates compared with PCTS and biopsies [43, 205, 225], and were much higher than previous microsomal preparations [212, 235]. This highlights progress made with DME extraction procedures, which, until recently, have limited the scalability of intestinal microsomes [192, 195]. Given the profound effect enzyme extraction procedure and incubation conditions can have on enzyme activity, consensus is needed on best practise before we can expect significant improvements to the accuracy and reproducibility of predictions.
In vitro models allow the function of metabolic enzymes, transporters and absorption processes to be studied in the gut wall. However, to apply this data to retrospectively explain, or prospectively predict, human oral PK ultimately requires insight into the mechanisms influencing drug behaviour in vivo [132, 236–238]. As such, several mechanistic approaches of varying complexity have been described for in vitro to in vivo extrapolation (IVIVE), some of which are available commercially, e.g. GastroPlus™ and Simcyp® [17, 92, 132, 238–241]. These mathematical translations (Fig. 2) can be relatively straightforward and ‘minimal’ models, such as Q Gut, require only in vitro metabolic CLint and cell permeability data to estimate F G. Several groups have reported successful prediction of F G in animals [242, 243] and humans [49, 227, 228, 244, 245] with this approach. Drugs with high in vivo extractions (F G values <0.5) were less accurately predicted and may reflect inability of the Q Gut model to account for changes in enterocyte drug concentration and therefore saturation of DME and efflux transporter processes [246]. However, to the authors’ knowledge, no critical assessment of possible systematic underprediction of IVIVE similar to that reported for hepatic metabolism [247–249] is available in the literature. Improved F G and oral clearance prediction was noted when the same set of drugs were evaluated using a physiological-based PK (PBPK) model. This was partly attributed to the model’s ability to account for saturation of intestinal metabolism by using maximum velocity (V max) and Michaelis-Menten constant K m, the substrate concentration at which the reaction rate is half of V max rather than CLint [246].

Bottom-up approach for prediction of F G using established in vitro systems and mathematical models of the gut. ACAT Advanced compartmental absorption and transit model [239, 275], ADAM advanced dissolution absorption metabolism model [92], GITA gastrointestinal transit absorption model [276, 277], Q gut minimal static model of intestinal metabolism [227], RAF relative activity factor, REF relative expression factor, ISEF intersystem extrapolation factor [190, 201, 227, 278, 279], CL int intrinsic clearance, MPPI microsomal protein per gram intestine, PI per intestine, S9PPI S9 protein per gram intestine, E G extraction ratio in the gut [69], PCTS precision cut tissue slices, CL int,G intrinsic clearance per gram intestine, SIM small intestinal metabolism, TM traditional model, SFM segregated flow model, SSFM segmental segregated flow model
Sophisticated PBPK models have been published, such as the segmental segregated flow model (SSFM), which encompass all salient variables, e.g. absorption, gastrointestinal transit, metabolism, transport and efflux [95, 98]. This allows route-dependent intestinal metabolism and zonal distribution of DMEs and transporters to be considered, as has been exemplified with morphine [98]. This in turn provides insight into the likely interplay between luminal transit, metabolism and active transport [250], and can be used in experimental design and to explore ‘what if’ scenarios [232, 236, 237, 251]. Comprehensive review of these dynamic, integrated modelling approaches have been provided elsewhere [238, 240, 252, 253]. Often, disadvantages perceived with these models relate to their inherent complexity and the level of detail required on parameters used in predictions. For example, transporter kinetic data (K m and V max) are often lacking, and abundance data for individual DMEs and transporters for the in vitro models and intact tissues are limited and hence restrictive to IVIVE [16, 246]. In response, proteomic- and mass spectrometry-based methods for protein quantification and establishment of scaling factors have started to emerge [32, 149, 254–256].
6 Strategy
An integrated DMPK strategy has been proposed to identify the right compound in the right assay at the right time [257]. Assessing the oral absorption potential of a candidate drug (CD) is a key component to this strategy. Improvements made to preclinical models of gut-wall metabolism (Table 3), and quantitative, mechanistic understanding of processes governing the magnitude of F G [132, 246, 250], present a compelling rationale for inclusion of a gut-wall metabolism strategy. The strategy outlined in Fig. 3 can be implemented along the drug discovery value chain. Briefly, this would entail profiling and designing out intestinal metabolic liabilities during lead identification (LI) and LO phases. If the potential for gut-wall metabolism still resided in the CD, then detailed PBPK modelling would be required for scenario setting and risk assessment. A decision tree to address gut-wall metabolism is provided in Fig. 4. Known structural motifs (e.g. metabolic handles for direct phase II metabolism or CYP3A substrates), metabolic instability in relevant in vitro systems, or intestinal loss indicated in animal intravenous/oral PK would trigger further evaluation. Signs of human intestinal metabolic instability would necessitate risk assessment. The potential magnitude of human F G would be evaluated in relevant preclinical models (Table 3) factoring in the compounds physicochemical properties, biopharmaceutical classification and projected dose (panel 1, Fig. 4). A workflow (panel 2, Fig. 4) is proposed for situations whereby significant intestinal loss limits in vivo exposure in safety or efficacy models. Circumstances such as these may require additional studies to identify whether absorption (permeability/solubility), transporters and/or intestinal metabolism are responsible.

Strategy to address the impact of gut-wall metabolism on oral bioavailability during drug discovery phases. LI lead identification, LO lead optimization, CD candidate drug, PK pharmacokinetics

Decision tree for risk assessment of gut-wall metabolism mediated via CYP450s, carboxylesterases, SULTs and UGTs. Note, as F H approaches 1 calculation of F a × F G becomes sensitive to the value used for liver blood flow. Intestinal microsomal CLint is scaled to a CLint,u,G then transformed using a model such as Q gut to an estimated F G. #For CYP3A substrates, physiological scaling factors accounting for relative CYP3A abundance in human liver microsomes and hepatocytes can also be applied to estimate CLint,u,G. *The solubility range required for an acceptable F a is dependent on dose. Solubility is frequently reassessed during drug discovery and early development phases and the F G liability, in the context of F a, can be revisited as understanding of an NCEs’ solubility profile is developed. Initial measurement of thermodynamic solubility, potentially on amorphous material, would be replaced with solubility of crystalline material, and then potentially solubility in biorelevant media (e.g., simulated intestinal fluids). Additionally, the pKa (ion class-dependent) can be assessed on representative compounds to allow simulation of F a using established PBPK models [92, 239, 280–282]. CYP450 cytochrome P450, UGTs uridine diphospho-glucuronosyltransferases, SULTs sulphotransferases, CL int intrinsic clearance, Q gut minimal static model of intestinal metabolism, IV intravenous, PO oral, PK pharmacokinetics, NCEs novel chemical entities, PBPK physiological-based pharmacokinetic, F a Fraction of orally administered drug absorbed into enterocytes, F G fraction of drug escaping first-pass metabolism in the enterocytes, ER efflux ratio, P app apparent permeability, F a × F G intestinal availability, F H fraction of drug escaping first-pass hepatic metabolism and biliary secretion, NCEs new chemical entities, CL int,G intrinsic clearance per gram intestine
Certain preclinical models may better complement the strategy and decision tree outlined above. During the LI and LO phases, scaled animal and human intestinal microsomal data could provide an efficient means to benchmark the risk of achieving low human F G, and also help troubleshoot underlying causes of intestinal loss in PK species. These data can also be readily incorporated into PBPK models for quantitative prediction of F G and simulation of oral PK profiles [258, 259]. Towards CD nomination, more physiological relevant models such as the Ussing chamber may be introduced, facilitating estimation of F a in the context of metabolism/transporter interplay. Recently, this technique has shown value in predicting human absorption for drugs that are substrates for DMEs and/or transporters expressed in the gut wall [69].
7 Challenges and Future Perspectives
When two or more drugs are coadministered, the effects could be additive, synergistic or antagonistic due to DDIs affecting the absorption and/or therapeutic profile of the victim drug. Overlapping substrate specificities for multiple enzymes and transporters might also enhance the complexity of the absorption profile along the gastrointestinal tract. Thus, overall understanding is a result of complex interplay between physiological (e.g. enzymes and transporters, blood flow, region of the intestine, luminal fluid composition) and physicochemical factors (e.g. pKa, solubility, dissolution, lipophilicity, substrate to enzymes and/or transporters) characterizing the drug molecule. In addition, genetic polymorphisms in drug transporter and DMEs, as well as disease states, may be responsible for variability in the profile and adverse events arising from co-medication among patients, which may be different from healthy volunteers [260] and is difficult to predict from preclinical tools.
In this review, we have illustrated the PK complexity associated with oral administration of drugs linked to intestinal regional variation in DMEs/transporters, as well as species and model differences. Assessing whether clinical candidates have the right risk/benefit balance for patients can be challenging given the inherent complexities and difficulties in the early screening phase and translation into clinical use. Because of the complexity, PBPK modelling will be a crucial tool as it enables efficient integration of knowledge on compound behaviour with the dynamics of intestinal physiology in the preclinical models and humans [236]. However, for successful modelling, high-quality data from in vitro and in vivo preclinical tools needs to be generated (see above strategy). New bioanalytical tools for quantitatively analyzing DME [149, 255, 261, 262] and transporter [33, 256, 263, 264] isoform abundances are already available to improve the quantitative translation between preclinical animals and humans, and will benefit understanding [185, 256, 265]. Knowledge of the impact of pharmacogenomics and disease on regional intestinal availability and variability in underlying mechanisms is scarce. Alongside this, reports focused on back translation of clinical outcome that enable evaluation of the successes or failures of predictions, made from preclinical data, will be crucial to advancing understanding and selection of the best tools for future development activities.
References
Bartholow M. Top 200 drugs of 2011. Pharmacy times. Available at: http://www.pharmacytimes.com/publications/issue/2012/July2012/Top-200-Drugs-of-2011. Accessed Jan 2013.
Liu G, Franssen E, Fitch MI, Warner E. Patient preferences for oral versus intravenous palliative chemotherapy. J Clin Oncol. 1997;15(1):110–5.
Rafil F, Franklin W, Heflich RH, Cerniglia CE. Reduction of nitroaromatic compounds by anaerobic bacteria isolated from the human gastrointestinal tract. Appl Environ Microbiol. 1991;57(4):962–8.
Nishimuta H, Houston JB, Galetin A. Hepatic, intestinal, renal, and plasma hydrolysis of prodrugs in human, cynomolgus monkey, dog, and rat: implications for in vitro–in vivo extrapolation of clearance of prodrugs. Drug Metab Dispos. 2014;42(9):1522–31.
Goldin BR. Intestinal microflora: metabolism of drugs and carcinogens. Ann Med. 1990;22(1):43–8.
McCabe M, Sane RS, Keith-Luzzi M, Xu J, King I, Whitcher-Johnstone A, et al. Defining the role of gut bacteria in the metabolism of deleobuvir: in vitro and in vivo studies. Drug Metab Dispos. 2015;43(10):1612–8.
Kaminsky LS, Zhang Q-Y. The small intestine as a xenobiotic-metabolizing organ. Drug Metab Dispos. 2003;31(12):1520–5.
Darwich AS, Aslam U, Ashcroft DM, Rostami-Hodjegan A. Meta-analysis of the turnover of intestinal epithelia in preclinical animal species and humans. Drug Metab Dispos. 2014;42(12):2016–22.
Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. The human intestinal cytochrome P450 “pie”. Drug Metab Dispos. 2006;34(5):880–6.
Yang J, Tucker GT, Rostami Hodjegan A. Cytochrome P450 3A expression and activity in the human small intestine. Clin Pharmacol Ther. 2004;76(4):391.
Kolars JC, Watkins P, Merion RM, Awni W. First-pass metabolism of cyclosporin by the gut. Lancet. 1991;338(8781):1488–90.
Paine MF, Shen DD, Kunze KL, Perkins JD, Marsh CL, McVicar JP, et al. First-pass metabolism of midazolam by the human intestine. Clin Pharmacol Ther. 1996;60(1):14–24.
von Richter O, Greiner B, Fromm MF, Fraser R, Omari T, Barclay ML, et al. Determination of in vivo absorption, metabolism, and transport of drugs by the human intestinal wall and liver with a novel perfusion technique. Clin Pharmacol Ther. 2001;70(3):217–27.
Benet L, Cummins C, Wu C. Unmasking the dynamic interplay between efflux transporters and metabolic enzymes. Int J Pharm. 2004;277(1):3–9.
Benet LZ. The drug transporter−metabolism alliance: uncovering and defining the interplay. Mol Pharm. 2009;6(6):1631–43.
Siissalo S, Heikkinen AT. In vitro methods to study the interplay of drug metabolism and efflux in the intestine. Curr Drug Metab. 2013;14(1):102–11.
Fagerholm U. Prediction of human pharmacokinetics: gut-wall metabolism. J Pharm Pharmacol. 2007;59(10):1335–43.
Hellriegel ET, Bjornsson TD, Hauck WW. Interpatient variability in bioavailability is related to the extent of absorption: implications for bioavailability and bioequivalence studies. Clin Pharmacol Ther. 1996;60(6):601–7.
Fahmi OA, Hurst S, Plowchalk D, Cook J, Guo F, Youdim K, et al. Comparison of different algorithms for predicting clinical drug–drug interactions, based on the use of CYP3A4 in vitro data: predictions of compounds as precipitants of interaction. Drug Metab Dispos. 2009;37(8):1658–66.
Fahmi OA, Maurer TS, Kish M, Cardenas E, Boldt S, Nettleton D. A combined model for predicting CYP3A4 clinical net drug–drug interaction based on CYP3A4 inhibition, inactivation, and induction determined in vitro. Drug Metab Dispos. 2008;36(8):1698–708.
Galetin A, Gertz M, Houston JB. Potential role of intestinal first-pass metabolism in the prediction of drug–drug interactions. Expert Opin Drug Metab Toxicol. 2008;4(7):909–22.
Galetin A, Gertz M, Houston JB. Contribution of intestinal cytochrome P450-mediated metabolism to drug–drug inhibition and induction interactions. Drug Metab Pharmacokinet. 2010;25(1):28–47.
Vieira ML, Kirby B, Ragueneau-Majlessi I, Galetin A, Chien J, Einolf H, et al. Evaluation of various static in vitro–in vivo extrapolation models for risk assessment of the CYP3A inhibition potential of an investigational drug. Clin Pharmacol Ther. 2014;95(2):189–98.
Peters SA, Schroeder PE, Giri N, Dolgos H. Evaluation of the use of static and dynamic models to predict drug–drug interaction and its associated variability: impact on drug discovery and early development. Drug Metab Dispos. 2012;40(8):1495–507.
König J, Müller F, Fromm MF. Transporters and drug–drug interactions: important determinants of drug disposition and effects. Pharmacol Rev. 2013;65(3):944–66.
Müller F, Fromm MF. Transporter-mediated drug–drug interactions. Pharmacogenomics. 2011;12(7):1017–37.
Yoshida K, Maeda K, Sugiyama Y. Hepatic and intestinal drug transporters: prediction of pharmacokinetic effects caused by drug–drug interactions and genetic polymorphisms. Ann Rev Pharmacol Toxicol. 2013;53:581–612.
Zakeri-Milani P, Valizadeh H. Intestinal transporters: enhanced absorption through P-glycoprotein-related drug interactions. Expert Opin Drug Metab Toxicol. 2014;10(6):859–71.
Wu C-Y, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23.
Komura H, Iwaki M. In vitro and in vivo small intestinal metabolism of CYP3A and UGT substrates in preclinical animals species and humans: species differences. Drug Metab Rev. 2011;43(4):476–98.
Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006;2(6):875–94.
Tucker TG, Milne AM, Fournel-Gigleux S, Fenner KS, Coughtrie MW. Absolute immunoquantification of the expression of ABC transporters P-glycoprotein, breast cancer resistance protein and multidrug resistance-associated protein 2 in human liver and duodenum. Biochem Pharmacol. 2012;83(2):279–85.
Gröer C, Brück S, Lai Y, Paulick A, Busemann A, Heidecke C, et al. LC–MS/MS-based quantification of clinically relevant intestinal uptake and efflux transporter proteins. J Pharm Biomed Anal. 2013;85:253–61.
Shi S, Li Y. Interplay of drug-metabolizing enzymes and transporters in drug absorption and disposition. Curr Drug Metab. 2015;15(10):915–41.
Mohri K, Uesawa Y. Enzymatic activities in the microsomes prepared from rat small intestinal epithelial cells by differential procedures. Pharm Res. 2001;18(8):1232–6.
Sharer JE, Shipley LA, Vandenbranden MR, Binkley SN, Wrighton SA. Comparisons of phase I and phase II in vitro hepatic enzyme activities of human, dog, rhesus monkey, and cynomolgus monkey. Drug Metab Dispos. 1995;23(11):1231–41.
Kolars JC, Lown KS, Schmiedlin-Ren P, Ghosh M, Fang C, Wrighton SA, et al. CYP3A gene expression in human gut epithelium. Pharmacogenetics. 1994;4(5):247–59.
Hebert MF, Roberts JP, Prueksaritanont T, Benet LZ. Bioavailability of cyclosporine with concomitant rifampin administration is markedly less than predicted by hepatic enzyme induction. Clin Pharmacol Ther. 1992;52(5):453–7.
Tsunoda SM, Velez RL, Moltke LL, Greenblatt DJ. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect of ketoconazole. Clin Pharmacol Ther. 1999;66(5):461–71.
Thummel KE, O’Shea D, Paine MF, Shen DD, Kunze KL, Perkins JD, et al. Oral first-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin Pharmacol Ther. 1996;59(5):491–502.
Lennernäs H, Ahrenstedt Ö, Hällgren R, Knutson L, Ryde M, Paalzow LK. Regional jejunal perfusion, a new in vivo approach to study oral drug absorption in man. Pharm Res. 1992;9(10):1243–51.
Lennernäs H, Nylander S, Ungell A-L. Jejunal permeability: a comparison between the Ussing Chamber technique and the single-pass perfusion in humans. Pharm Res. 1997;14(5):667–71.
van de Kerkhof EG, de Graaf IA, Groothuis GM. In vitro methods to study intestinal drug metabolism. Curr Drug Metab. 2007;8(7):658–75.
Lin JH, Chiba M, Baillie TA. Is the role of the small intestine in first-pass metabolism overemphasized? Pharmacol Rev. 1999;51(2):135–58.
Cubitt HE, Houston JB, Galetin A. Relative importance of intestinal and hepatic glucuronidation: impact on the prediction of drug clearance. Pharm Res. 2009;26(5):1073–83.
Galetin A, Gertz M, Houston JB. Contribution of intestinal cytochrome p450-mediated metabolism to drug–drug inhibition and induction interactions. Drug Metab Pharmacokinet. 2010;25(1):28–47.
Eeckhoudt S, Horsmans Y, Verbeeck R-K. Differential induction of midazolam metabolism in the small intestine and liver by oral and intravenous dexamethasone pretreatment in rat. Xenobiotica. 2002;32(11):975–84.
Kotegawa T, Laurijssens BE, von Moltke LL, Cotreau MM, Perloff MD, Venkatakrishnan K, et al. In vitro, pharmacokinetic, and pharmacodynamic interactions of ketoconazole and midazolam in the rat. J Pharmacol Exper Ther. 2002;302(3):1228–37.
Kadono K, Akabane T, Tabata K, Gato K, Terashita S, Teramura T. Quantitative prediction of intestinal metabolism in humans from a simplified intestinal availability model and empirical scaling factor. Drug Metab Dispos. 2010;38(7):1230–7.
Kato M, Chiba K, Hisaka A, Ishigami M, Kayama M, Mizuno N, et al. The intestinal first-pass metabolism of substrates of CYP3A4 and P-glycoprotein-quantitative analysis based on information from the literature. Drug Metab Pharmacokinet. 2003;18(6):365–72.
Gertz M, Davis JD, Harrison A, Houston JB, Galetin A. Grapefruit juice-drug interaction studies as a method to assess the extent of intestinal availability: utility and limitations. Curr Drug Metab. 2008;9(8):785–95.
Ducharme MP, Warbasse LH, Edwards DJ. Disposition of intravenous and oral cyclosporine after administration with grapefruit juice. Clin Pharmacol Ther. 1995;57(5):485–91.
Farkas D, Oleson LE, Zhao Y, Harmatz JS, Zinny MA, Court MH, et al. Pomegranate juice does not impair clearance of oral or intravenous midazolam, a probe for cytochrome P450-3A activity: comparison with grapefruit juice. J Clin Pharmacol. 2007;47(3):286–94.
Kharasch ED, Walker A, Hoffer C, Sheffels P. Intravenous and oral alfentanil as in vivo probes for hepatic and first-pass cytochrome P450 3A activity: Noninvasive assessment by use of pupillary miosis. Clin Pharmacol Ther. 2004;76(5):452–66.
Kupferschmidt HH, Ha HR, Ziegler WH, Meier PJ, Krähenbühl S. Interaction between grapefruit juice and midazolam in humans. Clin Pharmacol Ther. 1995;58(1):20–8.
Lundahl J, Regårdh C, Edgar B, Johnsson G. Effects of grapefruit juice ingestion—pharmacokinetics and haemodynamics of intravenously and orally administered felodipine in healthy men. Eur J Clin Pharmacol. 1997;52(2):139–45.
Paine MF, Criss AB, Watkins PB. Two major grapefruit juice components differ in intestinal CYP3A4 inhibition kinetic and binding properties. Drug Metab Dispos. 2004;32(10):1146–53.
Lown KS, Bailey DG, Fontana RJ, Janardan SK, Adair CH, Fortlage LA, et al. Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal CYP3A protein expression. J Clin Invest. 1997;99(10):2545.
Rashid T, Martin U, Clarke H, Waller D, Renwick A, George C. Factors affecting the absolute bioavailability of nifedipine. Br J Clin Pharmacol. 1995;40(1):51–8.
Floren LC, Bekersky I, Benet LZ, Mekki Q, Dressler D, Lee JW, et al. Tacrolimus oral bioavailability doubles with coadministration of ketoconazole. Clin Pharmacol Ther. 1997;62(1):41–9.
Guo L-Q, Fukuda K, Ohta T, Yamazoe Y. Role of furanocoumarin derivatives on grapefruit juice-mediated inhibition of human CYP3A activity. Drug Metab Dispos. 2000;28(7):766–71.
Ameer B, Weintraub RA. Drug interactions with grapefruit juice. Clin Pharmacokinet. 1997;33(2):103–21.
Paine MF, Widmer WW, Hart HL, Pusek SN, Beavers KL, Criss AB, et al. A furanocoumarin-free grapefruit juice establishes furanocoumarins as the mediators of the grapefruit juice: felodipine interaction. Am J Clinl Nutr. 2006;83(5):1097–105.
Paine MF, Oberlies NH. Clinical relevance of the small intestine as an organ of drug elimination: drug-fruit juice interactions. Expert Opin Drug Metab Toxicol. 2007;3(1):67–80.
Paine MF, Criss AB, Watkins PB. Two major grapefruit juice components differ in time to onset of intestinal CYP3A4 inhibition. J Pharmacol Exper Ther. 2005;312(3):1151–60.
Neuhoff S, Ungell A-L, Zamora I, Artursson P. pH-dependent bidirectional transport of weakly basic drugs across Caco-2 monolayers: implications for drug–drug interactions. Pharm Res. 2003;20(8):1141–8.
Ungell A-L. In vitro absorption studies and their relevance to absorption from the GI tract. Drug Dev Ind Pharm. 1997;23(9):879–92.
Neuhoff S, Ungell A-L, Zamora I, Artursson P. pH-Dependent passive and active transport of acidic drugs across Caco-2 cell monolayers. Eur J Pharm Sci. 2005;25(2):211–20.
Sjöberg Å, Lutz M, Tannergren C, Wingolf C, Borde A, Ungell A-L. Comprehensive study on regional human intestinal permeability and prediction of fraction absorbed of drugs using the Ussing chamber technique. Eur J Pharm Sci. 2013;48(1):166–80.
Ungell AL, Nylander S, Bergstrand S, Sjöberg Å, Lennernäs H. Membrane transport of drugs in different regions of the intestinal tract of the rat. J Pharm Sci. 1998;87(3):360–6.
Li LY, Amidon GL, Kim JS, Heimbach T, Kesisoglou F, Topliss JT, et al. Intestinal metabolism promotes regional differences in apical uptake of indinavir: coupled effect of P-glycoprotein and cytochrome P450 3A on indinavir membrane permeability in rat. J Pharmacol Exp Ther. 2002;301(2):586–93.
Estudante M, Morais JG, Soveral G, Benet LZ. Intestinal drug transporters: an overview. Adv Drug Deliv Rev. 2013;65(10):1340–56.
Murakami T, Takano M. Intestinal efflux transporters and drug absorption. Expert Opin Drug Metab Toxicol. 2008;4(7):923–39.
Zimmermann C, Gutmann H, Hruz P, Gutzwiller J-P, Beglinger C, Drewe J. Mapping of multidrug resistance gene 1 and multidrug resistance-associated protein isoform 1 to 5 mRNA expression along the human intestinal tract. Drug Metab Dispos. 2005;33(2):219–24.
Makhey VD, Guo A, Norris DA, Hu P, Yan J, Sinko PJ. Characterization of the regional intestinal kinetics of drug efflux in rat and human intestine and in Caco-2 cells. Pharm Res. 1998;15(8):1160–7.
Stephens R, O’Neill C, Warhurst A, Carlson G, Rowland M, Warhurst G. Kinetic profiling of P-glycoprotein-mediated drug efflux in rat and human intestinal epithelia. J Pharmacol Exp Ther. 2001;296(2):584–91.
Englund G, Rorsman F, Rönnblom A, Karlbom U, Lazorova L, Gråsjö J, et al. Regional levels of drug transporters along the human intestinal tract: co-expression of ABC and SLC transporters and comparison with Caco-2 cells. Eur J Pharm Sci. 2006;29(3):269–77.
Seithel A, Karlsson J, Hilgendorf C, Björquist A, Ungell A-L. Variability in mRNA expression of ABC-and SLC-transporters in human intestinal cells: comparison between human segments and Caco-2 cells. Eur J Pharm Sci. 2006;28(4):291–9.
Miao Q, Liu Q, Wang C, Meng Q, Guo X, Peng J, et al. Inhibitory effect of zinc on the absorption of JBP485 via the gastrointestinal oligopeptide transporter (PEPT1) in rats. Drug Metab Pharmacokinet. 2011;26(5):494–502.
Yamamoto-Furusho JK, Mendivil-Rangel EJ, Villeda-Ramirez MA, Fonseca-Camarillo G, Barreto-Zuniga R. Gene expression of carnitine organic cation transporters 1 and 2 (OCTN) is downregulated in patients with ulcerative colitis. Inflamm Bowel Dis. 2011;17(10):2205–6.
Tapaninen T, Neuvonen PJ, Niemi M. Grapefruit juice greatly reduces the plasma concentrations of the OATP2B1 and CYP3A4 substrate aliskiren. Clin Pharmacol Ther. 2010;88(3):339–42.
Herrera-Ruiz D, Wang Q, Gudmundsson OS, Cook TJ, Smith RL, Faria TN, et al. Spatial expression patterns of peptide transporters in the human and rat gastrointestinal tracts, Caco-2 in vitro cell culture model, and multiple human tissues. AAPS PharmSci. 2001;3(1):E9.
Hilgendorf C, Ahlin G, Seithel A, Artursson P, Ungell A-L, Karlsson J. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab Dispos. 2007;35(8):1333–40.
De Waziers I, Cugnenc P, Yang C, Leroux J, Beaune P. Cytochrome P 450 isoenzymes, epoxide hydrolase and glutathione transferases in rat and human hepatic and extrahepatic tissues. J Pharmacol Exp Ther. 1990;253(1):387–94.
Paine MF, Khalighi M, Fisher JM, Shen DD, Kunze KL, Marsh CL, et al. Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J Pharmacol Exp Ther. 1997;283(3):1552–62.
Zhang Q-Y, Dunbar D, Ostrowska A, Zeisloft S, Yang J, Kaminsky LS. Characterization of human small intestinal cytochromes P-450. Drug Metab Dispos. 1999;27(7):804–9.
Thummel KE, Kunze KL, Shen DD. Enzyme-catalyzed processes of first-pass hepatic and intestinal drug extraction. Adv Drug Deliv Rev. 1997;27(2):99–127.
Gervot L, Carrière V, Costet P, Cugnenc P-H, Berger A, Beaune PH, et al. CYP3A5 is the major cytochrome P450 3A expressed in human colon and colonic cell lines. Environ Toxicol Pharmacol. 1996;2(4):381–8.
Kivistö KT, Griese E-U, Fritz P, Linder A, Hakkola J, Raunio H, et al. Expression of cytochrome P 450 3A enzymes in human lung: a combined RT-PCR and immunohistochemical analysis of normal tissue and lung tumours. Naunyn Schmiedebergs Arch Pharmacol. 1996;353(2):207–12.
van de Kerkhof EG, Ungell AL, Sjoberg AK, de Jager MH, Hilgendorf C, de Graaf IA, et al. Innovative methods to study human intestinal drug metabolism in vitro: precision-cut slices compared with ussing chamber preparations. Drug Metab Dispos. 2006;34(11):1893–902.
Bergheim I, Bode C, Parlesak A. Distribution of cytochrome P450 2C, 2E1, 3A4, and 3A5 in human colon mucosa. BMC Pharmacol Toxicol. 2005;5(1):4.
Darwich AS, Neuhoff S, Jamei M, Rostami-Hodjegan A. Interplay of metabolism and transport in determining oral drug absorption and gut wall metabolism: a simulation assessment using the “Advanced Dissolution, Absorption, Metabolism (ADAM)” model. Curr Drug Metab. 2010;11(9):716–29.
Berggren S, Lennernäs P, Ekelund M, Weström B, Hoogstraate J, Lennernäs H. Regional transport and metabolism of ropivacaine and its CYP3A4 metabolite PPX in human intestine. J Pharm Pharmacol. 2003;55(7):963–72.
Le Ferrec E, Chesne C, Artusson P, Brayden D, Fabre G, Gires P, et al. In vitro models of the intestinal barrier. Altern Lab Anim. 2001;29:649–68.
Pang KS. Modeling of intestinal drug absorption: roles of transporters and metabolic enzymes (for the Gillette Review Series). Drug Metab Dispos. 2003;31(12):1507–19.
Koster AS, Hofman GA, Frankhuijzen-Sierevogel A, Noordhoek J. Presystemic and systemic intestinal metabolism of fenoterol in the conscious rat. Drug Metab Dispos. 1985;13(4):464–70.
Saitoh H, Saikachi Y, Kobayashi M, Yamaguchi M, Oda M, Yuhki Y, et al. Limited interaction between tacrolimus and P-glycoprotein in the rat small intestine. Eur J Pharm Sci. 2006;28(1):34–42.
Doherty MM, Pang KS. Route-dependent metabolism of morphine in the vascularly perfused rat small intestine preparation. Pharm Res. 2000;17(3):291–8.
Ilett KF, Tee LB, Reeves PT, Minchin RF. Metabolism of drugs and other xenobiotics in the gut lumen and wall. Pharmacol Ther. 1990;46(1):67–93.
Hirayama H, Morgado J, Gasinska I, Pang K. Estimations of intestinal and liver extraction in the in vivo rat: studies on gentisamide conjugation. Drug Metab Dispos. 1990;18:580–7.
Hirayama H, Pang K. First-pass metabolism of gentisamide: influence of intestinal metabolism on hepatic formation of conjugates. Studies in the once-through vascularly perfused rat intestine-liver preparation. Drug Metab Dispos. 1990;18(5):580–7.
Xu X, Hirayama H, Pang KS. First-pass metabolism of salicylamide. Studies in the once-through vascularly perfused rat intestine-liver preparation. Drug Metab Dispos. 1989;17(5):556–63.
Mistry M, Houston JB. Quantitation of extrahepatic metabolism. Pulmonary and intestinal conjugation of naphthol. Drug Metab Dispos. 1985;13(6):740–5.
Mistry M, Houston JB. Glucuronidation in vitro and in vivo. Comparison of intestinal and hepatic conjugation of morphine, naloxone, and buprenorphine. Drug Metab Dispos. 1987;15(5):710–7.
Raoof AA, Augustijns PR, Verbeeck RK. In vivo assessment of intestinal, hepatic, and pulmonary first pass metabolism of propofol in the rat. Pharm Res. 1996;13(6):891–5.
Bohets H, Annaert P, Mannens G, Anciaux K, Verboven P, Meuldermans W, et al. Strategies for absorption screening in drug discovery and development. Curr Top Med Chem. 2001;1(5):367–83.
Lennernäs H, Renberg L, Hoffmann K-J, Regårdh C. Presystemic elimination of the beta-blocker pafenolol in the rat after oral and intraperitoneal administration and identification of a main metabolite in both rats and humans. Drug Metab Dispos. 1993;21(3):435–40.
Aoki M, Okudaira K, Haga M, Nishigaki R, Hayashi M. Contribution of rat pulmonary metabolism to the elimination of lidocaine, midazolam, and nifedipine. Drug Metab Dispos. 2010;38(7):1183–8.
Cui Z, He P, Luo M, Xia S, Wu M. Phenacetin-O-deethylation in extrahepatic tissues of rats. Eur J Drug Metab Pharmacokinet. 2002;27(2):107–11.
Li X, Xia S, Lv Y, He P, Han J, Wu M. Conjugation metabolism of acetaminophen and bilirubin in extrahepatic tissues of rats. Life Sci. 2004;74(10):1307–15.
Karlsson FH, Bouchene S, Hilgendorf C, Dolgos H, Peters SA. Utility of in vitro systems and preclinical data for the prediction of human intestinal first-pass metabolism during drug discovery and preclinical development. Drug Metab Dispos. 2013;41(12):2033–46.
Musther H, Olivares-Morales A, Hatley OJ, Liu B, Hodjegan AR. Animal versus human oral drug bioavailability: do they correlate? Eur J Pharm Sci. 2014;57:280–91.
Nishimuta H, Sato K, Mizuki Y, Yabuki M, Komuro S. Species differences in intestinal metabolic activities of cytochrome P450 isoforms between cynomolgus monkeys and humans. Drug Metab Pharmacokinet. 2011;26(3):300–6.
Mudra DR, Desino KE, Desai PV. In silico, in vitro and in situ models to assess interplay between CYP3A and P-gp. Curr Drug Metab. 2011;12(8):750–73.
Barr WH, Riegelman S. Intestinal drug absorption and metabolism I: comparison of methods and models to study physiological factors of in vitro and in vivo intestinal absorption. J Pharm Sci. 1970;59(2):154–63.
Gugler R, Lain P, Azarnoff DL. Effect of portacaval shunt on the disposition of drugs with and without first-pass effect. J Pharmacol Exp Ther. 1975;195(3):416–23.
Lo M-W, Pond SM, Effeney DJ, Silber BM, Riegelman S, Tozer TN. Nonlinear formation of propranolol metabolites in dogs after portacaval transpositions. J Pharmacokinet Biopharm. 1984;12(4):401–12.
Doherty MM, Pang KS. First-pass effect: significance of the intestine for absorption and metabolism. Drug Chem Toxicol. 1997;20(4):329–44.
Castle S, Tucker G, Woods H, Underwood J, Nicholson C, Havler M, et al. Assessment of an in situ rat intestine preparation with perfused vascular bed for studying the absorption and first-pass metabolism of drugs. J Pharmacol Methods. 1985;14(4):255–74.
Matsuda Y, Konno Y, Satsukawa M, Kobayashi T, Takimoto Y, Morisaki K, et al. Assessment of intestinal availability of various drugs in the oral absorption process using portal vein-cannulated rats. Drug Metab Dispos. 2012;40(12):2231–8.
Kadono K, Koakutsu A, Naritomi Y, Terashita S, Tabata K, Teramura T. Comparison of intestinal metabolism of CYP3A substrates between rats and humans: application of portal-systemic concentration difference method. Xenobiotica. 2013;44(6):511–21.
Pang K, Yuen V, Fayz S, Te Koppele J, Mulder G. Absorption and metabolism of acetaminophen by the in situ perfused rat small intestine preparation. Drug Metab Dispos. 1986;14(1):102–11.
Pang KS, Cherry W, Ulm E. Disposition of enalapril in the perfused rat intestine-liver preparation: absorption, metabolism and first-pass effect. J Pharmacol Exp Ther. 1985;233(3):788–95.
Uhing MR, Kimura RE. The effect of surgical bowel manipulation and anesthesia on intestinal glucose absorption in rats. J Clin Invest. 1995;95(6):2790.
Yuasa H, Matsuda K, Watanabe J. Influence of anesthetic regimens on intestinal absorption in rats. Pharm Res. 1993;10(6):884–8.
Barthe L, Woodley J, Houin G. Gastrointestinal absorption of drugs: methods and studies. Fundam Clin Pharmacol. 1999;13(2):154–68.
Stappaerts J, Brouwers J, Annaert P, Augustijns P. In situ perfusion in rodents to explore intestinal drug absorption: challenges and opportunities. Int J Pharm. 2015;478(2):665–81.
Griffiths R, Lewis A, Jeffrey P. Models of drug absorption in situ and in conscious animals. In: Borchardt RT, Smith PL, Wilson, editors. Models for assessing drug absorption and metabolism. Springer: Berlin; 1996. p. 67–84.
Jeong EJ, Liu Y, Lin H, Hu M. In situ single-pass perfused rat intestinal model for absorption and metabolism. In: Caldwell GW, Zhengyin Y, editors. Optimization in drug discovery. Springer: Berlin; 2004. p. 65–76.
Schurgers N, Bijdendijk J, Tukker JJ, Crommelin DJ. Comparison of four experimental techniques for studying drug absorption kinetics in the anesthetized rat in situ. J Pharm Sci. 1986;75(2):117–9.
Cummins CL, Salphati L, Reid MJ, Benet LZ. In vivo modulation of intestinal CYP3A metabolism by P-glycoprotein: studies using the rat single-pass intestinal perfusion model. J Pharmacol Exp Ther. 2003;305(1):306–14.
Abuasal BS, Bolger MB, Walker DK, Kaddoumi A. In silico modeling for the nonlinear absorption kinetics of UK-343,664: a P-gp and CYP3A4 substrate. Mol Pharm. 2012;9(3):492–504.
Hu M, Sinko P, Johnson D, Amidon G. Membrane permeability parameters for some amino acids and β-lactam antibiotics: application of the boundary layer approach. J Theor Biol. 1988;131(1):107–14.
Balimane PV, Chong S, Morrison RA. Current methodologies used for evaluation of intestinal permeability and absorption. J Pharmacol Toxicol Methods. 2000;44(1):301–12.
Chiou WL, Barve A. Linear correlation of the fraction of oral dose absorbed of 64 drugs between humans and rats. Pharm Res. 1998;15(11):1792–5.
Chiou WL, Buehler PW. Comparison of oral absorption and bioavailability of drugs between monkey and human. Pharm Res. 2002;19(6):868–74.
Chiou WL, Jeong HY, Chung SM, Wu TC. Evaluation of using dog as an animal model to study the fraction of oral dose absorbed of 43 drugs in humans. Pharm Res. 2000;17(2):135–40.
Ward K, Nagilla R, Jolivette L. Comparative evaluation of oral systemic exposure of 56 xenobiotics in rat, dog, monkey and human. Xenobiotica. 2005;35(2):191–210.
Zhao YH, Abraham MH, Le J, Hersey A, Luscombe CN, Beck G, et al. Evaluation of rat intestinal absorption data and correlation with human intestinal absorption. Eur J Med Chem. 2003;38(3):233–43.
Riches Z, Stanley EL, Bloomer JC, Coughtrie MW. Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT “pie”. Drug Metab Dispos. 2009;37(11):2255–61.
Nishimuta H, Nakagawa T, Nomura N, Yabuki M. Species differences in hepatic and intestinal metabolic activities for 43 human cytochrome P450 substrates between humans and rats or dogs. Xenobiotica. 2013;43(11):948–55.
Furukawa T, Naritomi Y, Tetsuka K, Nakamori F, Moriguchi H, Yamano K, et al. Species differences in intestinal glucuronidation activities between humans, rats, dogs and monkeys. Xenobiotica. 2013;44(3):205–16.
Akabane T, Tabata K, Kadono K, Sakuda S, Terashita S, Teramura T. A comparison of pharmacokinetics between humans and monkeys. Drug Metab Dispos. 2010;38(2):308–16.
Takahashi M, Washio T, Suzuki N, Igeta K, Yamashita S. Investigation of the intestinal permeability and first-pass metabolism of drugs in cynomolgus monkeys using single-pass intestinal perfusion. Biol Pharm Bull. 2009;33(1):111–6.
Bueters T, Juric S, Sohlenius-Sternbeck A-K, Hu Y, Bylund J. Rat poorly predicts the combined non-absorbed and presystemically metabolized fractions in the human. Xenobiotica. 2012;43(7):607–16.
Komura H, Iwaki M. Species differences in in vitro and in vivo small intestinal metabolism of CYP3A substrates. J Pharm Sci. 2008;97(5):1775–800.
Jeong EJ, Liu Y, Lin H, Hu M. Species-and disposition model-dependent metabolism of raloxifene in gut and liver: role of UGT1A10. Drug Metab Dispos. 2005;33(6):785–94.
Teubner W, Meinl W, Florian S, Kretzschmar M, Glatt H. Identification and localization of soluble sulfotransferases in the human gastrointestinal tract. Biochem J. 2007;404:207–15.
Harbourt DE, Fallon JK, Ito S, Baba T, Ritter JK, Glish GL, et al. Quantification of human uridine-diphosphate glucuronosyl transferase 1A isoforms in liver, intestine, and kidney using nanobore liquid chromatography–tandem mass spectrometry. Anal Chem. 2011;84(1):98–105.
Berry LM, Wollenberg L, Zhao Z. Esterase activities in the blood, liver and intestine of several preclinical species and humans. Drug Metab Lett. 2009;3(2):70–7.
Williams ET, Bacon JA, Bender DM, Lowinger JJ, Guo W-K, Ehsani ME, et al. Characterization of the expression and activity of carboxylesterases 1 and 2 from the beagle dog, cynomolgus monkey, and human. Drug Metab Dispos. 2011;39(12):2305–13.
Takara K, Ohnishi N, Horibe S, Yokoyama T. Expression profiles of drug-metabolizing enzyme CYP3A and drug efflux transporter multidrug resistance 1 subfamily mRNAs in rat small intestine. Drug Metab Dispos. 2003;31(10):1235–9.
Fasco MJ, Silkworth J, Dunbar DA, Kaminsky LS. Rat small intestinal cytochromes P450 probed by warfarin metabolism. Mol Pharmacol. 1993;43(2):226–33.
Mitschke D, Reichel A, Fricker G, Moenning U. Characterization of cytochrome P450 protein expression along the entire length of the intestine of male and female rats. Drug Metab Dispos. 2008;36(6):1039–45.
Windmill KF, McKinnon RA, Zhu X, Gaedigk A, Grant DM, McManus ME. The role of xenobiotic metabolizing enzymes in arylamine toxicity and carcinogenesis: functional and localization studies. Mut Res. 1997;376(1):153–60.
Zhang Q-Y, Wikoff J, Dunbar D, Kaminsky L. Characterization of rat small intestinal cytochrome P450 composition and inducibility. Drug Metab Dispos. 1996;24(3):322–8.
van de Kerkhof EG, de Graaf IA, de Jager MH, Meijer DK, Groothuis GM. Characterization of rat small intestinal and colon precision-cut slices as an in vitro system for drug metabolism and induction studies. Drug Metab Dispos. 2005;33(11):1613–20.
Mouly S, Paine MF. P-glycoprotein increases from proximal to distal regions of human small intestine. Pharm Res. 2003;20(10):1595–9.
Haller S, Schuler F, Lazic SE, Bachir-Cherif D, Krämer SD, Parrott NJ, et al. Expression profiles of metabolic enzymes and drug transporters in the liver and along the intestine of beagle dogs. Drug Metab Dispos. 2012;40(8):1603–11.
Rost D, Mahner S, Sugiyama Y, Stremmel W. Expression and localization of the multidrug resistance-associated protein 3 in rat small and large intestine. Am J Physiol Gastrointest Liver Physiol. 2002;282(4):G720–6.
Mutch DM, Anderle P, Fiaux M, Mansourian R, Vidal K, Wahli W, et al. Regional variations in ABC transporter expression along the mouse intestinal tract. Physiol Genomics. 2004;17(1):11–20.
Enokizono J, Kusuhara H, Sugiyama Y. Regional expression and activity of breast cancer resistance protein (Bcrp/Abcg2) in mouse intestine: overlapping distribution with sulfotransferases. Drug Metab Dispos. 2007;35(6):922–8.
van Waterschoot RA, Schinkel AH. A critical analysis of the interplay between cytochrome P450 3A and P-glycoprotein: recent insights from knockout and transgenic mice. Pharmacolog Rev. 2011;63(2):390–410.
Choo EF, Woolsey S, DeMent K, Ly J, Messick K, Qin A, et al. Use of transgenic mouse models to understand the oral disposition and drug–drug interaction potential of cobimetinib, a MEK inhibitor. Drug Metab Dispos. 2015;43(6):864–9.
Shen H-W, Jiang X-L, Gonzalez FJ, Yu A-M. Humanized transgenic mouse models for drug metabolism and pharmacokinetic research. Curr Drug Metab. 2011;12(10):997–1006.
Tang SC, Hendrikx JJ, Beijnen JH, Schinkel AH. Genetically modified mouse models for oral drug absorption and disposition. Curr Opin Pharmacol. 2013;13(6):853–8.
Cheung C, Gonzalez FJ. Humanized mouse lines and their application for prediction of human drug metabolism and toxicological risk assessment. J Pharmacol Exp Ther. 2008;327(2):288–99.
Scheer N, Wolf CR. Genetically humanized mouse models of drug metabolizing enzymes and transporters and their applications. Xenobiotica. 2014;44(2):96–108.
Salyers KL, Xu Y. Animal models for studying drug metabolizing enzymes and transporters. In: Zhang D, Surapaneni S, editors. ADME-enabling technologies in drug design and development. Wiley: London; 2012. p. 253.
Lin JH. Applications and limitations of genetically modified mouse models in drug discovery and development. Curr Drug Metab. 2008;9(5):419–38.
Mols R, Brouwers J, Schinkel AH, Annaert P, Augustijns P. Intestinal perfusion with mesenteric blood sampling in wild-type and knockout mice evaluation of a novel tool in biopharmaceutical drug profiling. Drug Metab Dispos. 2009;37(6):1334–7.
Muruganandan S, Sinal C. Mice as clinically relevant models for the study of cytochrome P450-dependent metabolism. Clin PharmacolTher. 2008;83(6):818–28.
Gonzalez FJ, Fang Z-Z, Ma X. Transgenic mice and metabolomics for study of hepatic xenobiotic metabolism and toxicity. Expert Opin Drug Metab Toxicol. 2015;11(6):869–81.
van Herwaarden AE, Wagenaar E, van der Kruijssen CM, van Waterschoot RA, Smit JW, Song JY, et al. Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. J Clin Invest. 2007;117(11):3583–92.
van Waterschoot RA, ter Heine R, Wagenaar E, van der Kruijssen CM, Rooswinkel RW, Huitema AD, et al. Effects of cytochrome P450 3A (CYP3A) and the drug transporters P-glycoprotein (MDR1/ABCB1) and MRP2 (ABCC2) on the pharmacokinetics of lopinavir. Br J Pharmacol. 2010;160(5):1224–33.
van Waterschoot RA, Rooswinkel RW, Sparidans RW, van Herwaarden AE, Beijnen JH, Schinkel AH. Inhibition and stimulation of intestinal and hepatic CYP3A activity: studies in humanized CYP3A4 transgenic mice using triazolam. Drug Metab Dispos. 2009;37(12):2305–13.
Scheer N, Kapelyukh Y, McEwan J, Beuger V, Stanley LA, Rode A, et al. Modeling human cytochrome P450 2D6 metabolism and drug–drug interaction by a novel panel of knockout and humanized mouse lines. Mol Pharmacol. 2012;81(1):63–72.
Scheer N, Kapelyukh Y, Chatham L, Rode A, Buechel S, Wolf CR. Generation and characterization of novel cytochrome P450 Cyp2c gene cluster knockout and CYP2C9 humanized mouse lines. Mol Pharmacol. 2012;82(6):1022–9.
van Waterschoot RA, van Herwaarden AE, Lagas JS, Sparidans RW, Wagenaar E, van der Kruijssen CM, et al. Midazolam metabolism in cytochrome P450 3A knockout mice can be attributed to up-regulated CYP2C enzymes. Mol Pharmacol. 2008;73(3):1029–36.
Scheer N, McLaughlin LA, Rode A, MacLeod AK, Henderson CJ, Wolf CR. Deletion of 30 murine cytochrome p450 genes results in viable mice with compromised drug metabolism. Drug Metab Dispos. 2014;42(6):1022–30.
Jamei M, Dickinson GL, Rostami-Hodjegan A. A framework for assessing inter-individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of’bottom-up’vs’ top-down’recognition of covariates. Drug Metab Pharmacokinet. 2009;24(1):53–75.
Tsamandouras N, Rostami-Hodjegan A, Aarons L. Combining the ‘bottom up’and ‘top down’approaches in pharmacokinetic modelling: fitting PBPK models to observed clinical data. Br J Clin Pharmacol. 2015;79(1):48–55.
Kostewicz ES, Abrahamsson B, Brewster M, Brouwers J, Butler J, Carlert S, et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2014;57:342–66.
Jones H, Chen Y, Gibson C, Heimbach T, Parrott N, Peters S, et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97(3):247–62.
Heikkinen AT, Fowler S, Gray L, Li J, Peng Y, Yadava P, et al. In vitro to in vivo extrapolation and physiologically based modeling of cytochrome P450 mediated metabolism in beagle dog gut wall and liver. Mol Pharm. 2013;10(4):1388–99.
Klees TM, Sheffels P, Dale O, Kharasch ED. Metabolism of alfentanil by cytochrome p4503a (cyp3a) enzymes. Drug Metab Dispos. 2005;33(3):303–11.
Lalovic B, Phillips B, Risler LL, Howald W, Shen DD. Quantitative contribution of CYP2D6 and CYP3A to oxycodone metabolism in human liver and intestinal microsomes. Drug Metab Dispos. 2004;32(4):447–54.
Baranczewski P, Stanczak A, Sundberg K, Svensson R, Wallin A, Jansson J, et al. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol Rep. 2006;58(4):453–72.
Crespi CL. Xenobiotic-metabolizing Human Cells as Tools for Pharmacological and Toxocological Research. Adv Drug Res. 1995;26:180–237.
Proctor N, Tucker G, Rostami-Hodjegan A. Predicting drug clearance from recombinantly expressed CYPs: intersystem extrapolation factors. Xenobiotica. 2004;34(2):151–78.
Bonkovsky HL, Hauri H-P, Marti U, Gasser R, Meyer UA. Cytochrome P450 of small intestinal epithelial cells. Immunochemical characterization of the increase in cytochrome P450 caused by phenobarbital. Gastroenterology. 1985;88(2):458–67.
Bruyère A, Declevès X, Bouzom F, Proust L, Martinet M, Walther B, et al. Development of an optimized procedure for the preparation of rat intestinal microsomes: comparison of hepatic and intestinal microsomal cytochrome P450 enzyme activities in two rat strains. Xenobiotica. 2009;39(1):22–32.
Cotreau MM, von Moltke LL, Beinfeld MC, Greenblatt DJ. Methodologies to study the induction of rat hepatic and intestinal cytochrome P450 3A at the mRNA, protein, and catalytic activity level. J Pharmacol Toxicol Methods. 2000;43(1):41–54.
Dawson JR, Bridges JW. Intestinal microsomal drug metabolism: A comparison of rat and guinea-pig enzymes, and of rat crypt and villous tip cell enzymes. Biochem Pharmacol. 1981;30(17):2415–20.
Galetin A, Houston JB. Intestinal and hepatic metabolic activity of five cytochrome P450 enzymes: impact on prediction of first-pass metabolism. J Pharmacol Exp Ther. 2006;318(3):1220–9.
Klippert P, Borm P, Noordhoek J. Prediction of intestinal first-pass effect of phenacetin in the rat from enzyme kinetic data: correlation with in in vivo data using mucosal blood flow. Biochem Pharmacol. 1982;31(15):2545–8.
Koster AS, Noordhoek J. Glucuronidation in the rat intestinal wall: Comparison of isolated mucosal cells, latent microsomes and activated microsomes. Biochem Pharm. 1983;32(5):895–900.
Richter O, Burk O, Fromm MF, Thon KP, Eichelbaum M, Kivistö KT. Cytochrome P450 3A4 and P-glycoprotein expression in human small intestinal enterocytes and hepatocytes: a comparative analysis in paired tissue specimens. Clin Pharmacol Ther. 2004;75(3):172–83.
Watkins PB, Wrighton S, Schuetz E, Molowa D, Guzelian P. Identification of glucocorticoid-inducible cytochromes P-450 in the intestinal mucosa of rats and man. J Clin Invest. 1987;80(4):1029.
Weiser MM. Intestinal epithelial cell surface membrane glycoprotein synthesis I. An indicator of cellular differentiation. J Biol Chem. 1973;248(7):2536–41.
Zhang Z, Li Y, Shou M, Zhang Y, Ngui J, Stearns R, et al. Influence of different recombinant systems on the cooperativity exhibited by cytochrome P4503A4. Xenobiotica. 2004;34(5):473–86.
de Graaf IA, de Kanter R, de Jager MH, Camacho R, Langenkamp E, van de Kerkhof EG, et al. Empirical validation of a rat in vitro organ slice model as a tool for in vivo clearance prediction. Drug Metab Dispos. 2006;34(4):591–9.
de Graaf IA, Olinga P, de Jager MH, Merema MT, de Kanter R, van de Kerkhof EG, et al. Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat Protoc. 2010;5(9):1540–51.
de Kanter R, Monshouwer M, Draaisma A, De Jager M, De Graaf I, Proost J, et al. Prediction of whole-body metabolic clearance of drugs through the combined use of slices from rat liver, lung, kidney, small intestine and colon. Xenobiotica. 2004;34(3):229–41.
de Kanter R, Tuin A, van de Kerkhof E, Martignoni M, Draaisma AL, de Jager MH, et al. A new technique for preparing precision-cut slices from small intestine and colon for drug biotransformation studies. J Pharmacol Toxicol Methods. 2005;51(1):65–72.
Groothuis GM, de Graaf IA. Precision-cut intestinal slices as in vitro tool for studies on drug metabolism. Curr Drug Metab. 2013;14(1):112–9.
Nejdfors P, Ekelund M, Jeppsson B, Weström B. Mucosal in vitro permeability in the intestinal tract of the pig, the rat, and man: species-and region-related differences. Scand J Gastroenterol. 2000;35(5):501–7.
Mariappan T, Singh S. Evidence of efflux-mediated and saturable absorption of rifampicin in rat intestine using the ligated loop and everted gut sac techniques. Mol Pharm. 2004;1(5):363–7.
Richter E, Strugala G. An all-glass perfusator for investigation of the intestinal transport and metabolism of foreign compounds in vitro. J Pharmacolog Methods. 1985;14(4):297–304.
Emoto C, Yamazaki H, Yamasaki S, Shimada N, Nakajima M, Yokoi T. Use of everted sacs of mouse small intestine as enzyme sources for the study of drug oxidation activities in vitro. Xenobiotica. 2000;30(10):971–82.
Arellano C, Philibert C, Vachoux C, Woodley J, Houin G. The metabolism of midazolam and comparison with other CYP enzyme substrates during intestinal absorption: in vitro studies with rat everted gut sacs. J Pharm Pharmaceut Sci. 2007;10(1):26–36.
Takemoto K, Yamazaki H, Tanaka Y, Nakajima M, Yokoi T. Catalytic activities of cytochrome P450 enzymes and UDP-glucuronosyltransferases involved in drug metabolism in rat everted sacs and intestinal microsomes. Xenobiotica. 2003;33(1):43–55.
Fisher R, Parsons D. A preparation of surviving rat small intestine for the study of absorption. The J Physiol. 1949;110(1–2):36–46.
Andlauer W, Kolb J, Fürst P. Isoflavones from tofu are absorbed and metabolized in the isolated rat small intestine. J Nutr. 2000;130(12):3021–7.
Andlauer W, Kolb J, Stehle P, Fürst P. Absorption and metabolism of genistein in isolated rat small intestine. J Nutr. 2000;130(4):843–6.
De Vries M, Hofman G, Koster A, Noordhoek J. Systemic intestinal metabolism of 1-naphthol. A study in the isolated vascularly perfused rat small intestine. Drug Metab Dispos. 1989;17(5):573–8.
Kavin H, Levin NW, Stanley MM. Isolated perfused rat small bowel: technic, studies of viability, glucose absorption. J Appl Physiol. 1967;22(3):604–11.
Larsson J, Pantzar N, Permert J, Olaison G. Integrity and metabolism of human ileal mucosa in vitro in the Ussing chamber. Acta Physiol Scand. 1998;162:47–56.
Gotoh Y, Kamada N, Momose D. The advantages of the Ussing Chamber in drug absorption studies. J Biomol Screen. 2005;10(5):517–23.
Polentarutti BI, Peterson AL, Sjöberg ÅK, Anderberg EKI, Utter LM, Ungell A-LB. Evaluation of viability of excised rat intestinal segments in the Ussing chamber: investigation of morphology, electrical parameters, and permeability characteristics. Pharm Res. 1999;16(3):446–54.
Lampen A, Christians U, Guengerich FP, Watkins PB, Kolars JC, Bader A, et al. Metabolism of the immunosuppressant tacrolimus in the small intestine: cytochrome P450, drug interactions, and interindividual variability. Drug Metab Dispos. 1995;23(12):1315–24.
Rogers SM, Back D, Orme M. Intestinal metabolism of ethinyloestradiol and paracetamol in vitro: studies using Ussing chambers. Br J Clin Pharmacol. 1987;23(6):727–34.
Lennernäs H. Animal data: the contributions of the Ussing Chamber and perfusion systems to predicting human oral drug delivery in vivo. Adv Drug Deliv Rev. 2007;59(11):1103–20.
Martignoni M, de Kanter R, Grossi P, Mahnke A, Saturno G, Monshouwer M. An in vivo and in vitro comparison of CYP induction in rat liver and intestine using slices and quantitative RT-PCR. Chem Biol Interact. 2004;151(1):1–11.
Martignoni M, Groothuis G, de Kanter R. Comparison of mouse and rat cytochrome P450-mediated metabolism in liver and intestine. Drug Metab Dispos. 2006;34(6):1047–54.
Worboys PD, Bradbury A, Houston JB. Kinetics of drug metabolism in rat liver slices III. Relationship between metabolic clearance and slice uptake rate. Drug Metab Dispos. 1997;25(4):460–7.
Yang J, Jamei M, Yeo KR, Tucker GT, Rostami-Hodjegan A. Prediction of intestinal first-pass drug metabolism. Curr Drug Metab. 2007;8(7):676–84.
Gertz M, Harrison A, Houston JB, Galetin A. Prediction of human intestinal first-pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab Dispos. 2010;38(7):1147–58.
Bruyere A, Decleves X, Bouzom F, Ball K, Marques C, Treton X, et al. Effect of variations in the amounts of P-glycoprotein (ABCB1), BCRP (ABCG2) and CYP3A4 along the human small intestine on PBPK models for predicting intestinal first pass. Mol Pharm. 2010;7(5):1596–607.
Nishimuta H, Nakagawa T, Nomura N, Yabuki M. Significance of reductive metabolism in human intestine and quantitative prediction of intestinal first-pass metabolism by cytosolic reductive enzymes. Drug Metab Dispos. 2013;41(5):1104–11.
Naritomi Y, Nakamori F, Furukawa T, Tabata K. Prediction of hepatic and intestinal glucuronidation using in vitro–in vivo extrapolation. Drug Metab Pharmacokinet. 2015;30(1):21–9.
Wu B, Dong D, Hu M, Zhang S. Quantitative prediction of glucuronidation in humans using the in vitro-in vivo extrapolation approach. Curr Top Med Chem. 2013;13(11):1343–52.
Hatley O, Jones C, Galetin A, Rostami-Hodjegan A. The rat as a model for screening intestinal metabolism potential. Poster presentation at the 2012 AAPS annual meeting and exposition; october 14–17 2012; chicago. poster m1264; 2012.
Hatley OJD. Mechanistic prediction of intestinal first-pass metabolism using in vitro data in preclinical species and in man [PhD thesis]. Manchester: The University of Manchester; 2014.
Sohlenius-Sternbeck A-K, Orzechowski A. Characterization of the rates of testosterone metabolism to various products and of glutathione transferase and sulfotransferase activities in rat intestine and comparison to the corresponding hepatic and renal drug-metabolizing enzymes. Chem Biol Interact. 2004;148(1):49–56.
Peters SA. Identification of intestinal loss of a drug through physiologically based pharmacokinetic simulation of plasma concentration-time profiles. Clin Pharmacokinet. 2008;47(4):245–59.
Peters SA. Evaluation of a generic physiologically based pharmacokinetic model for lineshape analysis. Clin Pharmacokinet. 2008;47(4):261–75.
Chow EC, Pang SK. Why we need proper PBPK models to examine intestine and liver oral drug absorption. Curr Drug Metab. 2013;14(1):57–79.
Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50:S41–67.
Dokoumetzidis A, Kalantzi L, Fotaki N. Predictive models for oral drug absorption: from in silico methods to integrated dynamical models. Expert Opin Drug Metab Toxicol. 2007;3(4):491–505.
Ando H, Hisaka A, Suzuki H. A new physiologically based pharmacokinetic model for the prediction of gastrointestinal drug absorption: translocation model. Drug Metab Dispos. 2015;43(4):590–602.
Furukawa T, Nakamori F, Tetsuka K, Naritomi Y, Moriguchi H, Yamano K, et al. Quantitative prediction of intestinal glucuronidation of drugs in rats using in vitro metabolic clearance data. Drug Metab Pharmacokinet. 2012;27(2):171–80.
Furukawa T, Yamano K, Naritomi Y, Tanaka K, Terashita S, Teramura T. Method for predicting human intestinal first-pass metabolism of UGT substrate compounds. Xenobiotica. 2012;42(10):980–8.
Nakamori F, Naritomi Y, K-i Hosoya, Moriguchi H, Tetsuka K, Furukawa T, et al. Quantitative prediction of human intestinal glucuronidation effects on intestinal availability of UDP-glucuronosyltransferase substrates using in vitro data. Drug Metab Dispos. 2012;40(9):1771–7.
Nishimuta H, Sato K, Yabuki M, Komuro S. Prediction of the intestinal first-pass metabolism of CYP3A and UGT substrates in humans from in vitro data. Drug Metab Pharmacokinet. 2011;26(6):592–601.
Gertz M, Houston JB, Galetin A. Physiologically based pharmacokinetic modeling of intestinal first-pass metabolism of CYP3A substrates with high intestinal extraction. Drug Metab Dispos. 2011;39(9):1633–42.
Sohlenius-Sternbeck AK, Afzelius L, Prusis P, Neelissen J, Hoogstraate J, Johansson J, et al. Evaluation of the human prediction of clearance from hepatocyte and microsome intrinsic clearance for 52 drug compounds. Xenobiotica. 2010;40(9):637–49.
Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27(11):1350–9.
Jones HM, Houston JB. Substrate depletion approach for determining in vitro metabolic clearance: time dependencies in hepatocyte and microsomal incubations. Drug Metab Dispos. 2004;32(9):973–82.
Tam D, Tirona RG, Pang SK. Segmental intestinal transporters and metabolic enzymes on intestinal drug absorption. Drug Metab Dispos. 2003;31(4):373–83.
Sun H, Pang SK. Physiological modeling to understand the impact of enzymes and transporters on drug and metabolite data and bioavailability estimates. Pharm Res. 2010;27(7):1237–54.
Pang KS, Chow EC. Commentary: theoretical predictions of flow effects on intestinal and systemic availability in physiologically based pharmacokinetic intestine models: the traditional model, segregated flow model, and QGut model. Drug Metab Dispos. 2012;40(10):1869–77.
Fan J, Chen S, Chow EC, Pang SK. PBPK modeling of intestinal and liver enzymes and transporters in drug absorption and sequential metabolism. Curr Drug Metab. 2010;11(9):743–61.
Heikkinen AT, Friedlein A, Lamerz J, Jakob P, Cutler P, Fowler S, et al. Mass spectrometry-based quantification of CYP enzymes to establish in vitro/in vivo scaling factors for intestinal and hepatic metabolism in beagle dog. Pharm Res. 2012;29(7):1832–42.
Heikkinen AT, Friedlein A, Matondo M, Hatley OJ, Petsalo A, Juvonen R, et al. Quantitative ADME proteomics–CYP and UGT enzymes in the beagle dog liver and intestine. Pharm Res. 2015;32(1):74–90.
Uchida Y, Ohtsuki S, Kamiie J, Terasaki T. Blood-brain barrier (BBB) pharmacoproteomics: reconstruction of in vivo brain distribution of 11 P-glycoprotein substrates based on the BBB transporter protein concentration, in vitro intrinsic transport activity, and unbound fraction in plasma and brain in mice. J Pharmacol Exp Ther. 2011;339(2):579–88.
Ballard P, Brassil P, Bui KH, Dolgos H, Petersson C, Tunek A, et al. The right compound in the right assay at the right time: an integrated discovery DMPK strategy. Drug Metab Rev. 2012;44(3):224–52.
Emoto C, Fukuda T, Cox S, Christians U, Vinks A. Development of a physiologically-based pharmacokinetic model for sirolimus: predicting bioavailability based on intestinal CYP3A content. CPT Pharmacometrics Syst Pharmacol. 2013;2(7):1–9.
Heikkinen AT, Baneyx G, Caruso A, Parrott N. Application of PBPK modeling to predict human intestinal metabolism of CYP3A substrates: an evaluation and case study using GastroPlus™. Eur J Pharm Sci. 2012;47(2):375–86.
Pal D, Mitra AK. MDR-and CYP3A4-mediated drug–drug interactions. J Neuroimmune Pharmacol. 2006;1(3):323–39.
Achour B, Russell MR, Barber J, Rostami-Hodjegan A. Simultaneous quantification of the abundance of several cytochrome P450 and uridine 5′-diphospho-glucuronosyltransferase enzymes in human liver microsomes using multiplexed targeted proteomics. Drug Metab Dispos. 2014;42(4):500–10.
Sato Y, Nagata M, Tetsuka K, Tamura K, Miyashita A, Kawamura A, et al. Optimized methods for targeted peptide-based quantification of human uridine 5′-diphosphate-glucuronosyltransferases in biological specimens using liquid chromatography-tandem mass spectrometry. Drug Metab Dispos. 2014;42(5):885–9.
Oswald S, Gröer C, Drozdzik M, Siegmund W. Mass spectrometry-based targeted proteomics as a tool to elucidate the expression and function of intestinal drug transporters. AAPS J. 2013;15(4):1128–40.
Wang L, Prasad B, Salphati L, Chu X, Gupta A, Hop CE, et al. Interspecies variability in expression of hepatobiliary transporters across human, dog, monkey, and rat as determined by quantitative proteomics. Drug Metab Dispos. 2015;43(3):367–74.
Harwood M, Neuhoff S, Carlson G, Warhurst G, Rostami-Hodjegan A. Absolute abundance and function of intestinal drug transporters: a prerequisite for fully mechanistic in vitro–in vivo extrapolation of oral drug absorption. Biopharm Drug Dispos. 2013;34(1):2–28.
Thelen K, Dressman JB. Cytochrome P450-mediated metabolism in the human gut wall. J Pharm Pharmacol. 2009;61(5):541–58.
Thörn M, Finnström N, Lundgren S, Rane A, Lööf L. Cytochromes P450 and MDR1 mRNA expression along the human gastrointestinal tract. Br J Clin Pharmacol. 2005;60(1):54–60.
Uehara S, Murayama N, Nakanishi Y, Nakamura C, Hashizume T, Zeldin DC, et al. Immunochemical detection of cytochrome P450 enzymes in small intestine microsomes of male and female untreated juvenile cynomolgus monkeys. Xenobiotica. 2014;44(9):769–74.
Wang MZ, Wu JQ, Bridges AS, Zeldin DC, Kornbluth S, Tidwell RR, et al. Human enteric microsomal CYP4F enzymes O-demethylate the antiparasitic prodrug pafuramidine. Drug Metab Dispos. 2007;35(11):2067–75.
Borm PJ, Koster AS, Frankhuijzen-Sierevogel A, Noordhoek J. Comparison of two cell isolation procedures to study in vitro intestinal wall biotransformation in control and 3-methyl-cholanthrene pretreated rats. Cell Biochem Funct. 1983;1(3):161–7.
Behera D, Damre A, Varghese A, Addepalli V. In vitro evaluation of hepatic and extra-hepatic metabolism of coumarins using rat subcellular fractions: correlation of in vitro clearance with in vivo data. Drug Metab Drug Interact. 2008;23(3–4):329–50.
Pacifici G, Franchi M, Bencini C, Repetti F, Di Lascio N, Muraro G. Tissue distribution of drug-metabolizing enzymes in humans. Xenobiotica. 1988;18(7):849–56.
Gibbs JP, Yang J-S, Slattery JT. Comparison of human liver and small intestinal glutathione S-transferase-catalyzed busulfan conjugation in vitro. Drug Metab Dispos. 1998;26(1):52–5.
Shirkey R, Chakraborty J, Bridges J. An improved method for preparing rat small intestine microsomal fractions for studying drug metabolism. Anal Biochem. 1979;93:73–81.
Lawrence XY. An integrated model for determining causes of poor oral drug absorption. Pharm Res. 1999;16(12):1883–7.
Sawamoto T, Haruta S, Kurosaki Y, Higaki K, Kimura T. Prediction of the plasma concentration profiles of orally administered drugs in rats on the basis of gastrointestinal transit kinetics and absorbability. J Pharm Pharmacol. 1997;49(4):450–7.
Kimura T, Iwasaki N, Yokoe J-I, Haruta S, Yokoo Y, Ogawara K-I, et al. Analysis and prediction of absorption profile including hepatic first-pass metabolism of N-methyltyramine, a potent stimulant of gastrin release present in beer, after oral ingestion in rats by gastrointestinal-transit-absorption model. Drug Metab Dispos. 2000;28(5):577–81.
Remmel RP, Burchell B. Validation and use of cloned, expressed human drug-metabolizing enzymes in heterologous cells for analysis of drug metabolism and drug–drug interactions. Biochem Pharmacol. 1993;46(4):559–66.
Venkatakrishnan K, von Moltke LL, Harmatz JS, Crespi CL, Greenblatt DJ. Comparison between cytochrome P450 (CYP) content and relative activity approaches to scaling from cDNA-expressed CYPs to human liver microsomes: ratios of accessory proteins as sources of discrepancies between the approaches. Drug Metab Dispos. 2000;28(12):1493–504.
Sjögren E, Westergren J, Grant I, Hanisch G, Lindfors L, Lennernäs H, et al. In silico predictions of gastrointestinal drug absorption in pharmaceutical product development: application of the mechanistic absorption model GI-Sim. Eur J Pharm Sci. 2013;49(4):679–98.
Thelen K, Coboeken K, Willmann S, Burghaus R, Dressman JB, Lippert J. Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part 1: oral solutions. J Pharm Sci. 2011;100(12):5324–45.
Parrott N, Lave T. Applications of physiologically based absorption models in drug discovery and development. Mol Pharm. 2008;5(5):760–75.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the preparation of this manuscript.
Conflicts of interest
Sheila Annie Peters, Christopher R. Jones, Anna-Lena Ungell and Oliver Hatley have no conflicts of interest to declare.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Peters, S.A., Jones, C.R., Ungell, AL. et al. Predicting Drug Extraction in the Human Gut Wall: Assessing Contributions from Drug Metabolizing Enzymes and Transporter Proteins using Preclinical Models. Clin Pharmacokinet 55, 673–696 (2016). https://doi.org/10.1007/s40262-015-0351-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-015-0351-6