
Abstract
Anorexia nervosa is a disorder associated with serious adverse health outcomes, for which there is currently considerable treatment ineffectiveness. Characterised by restrictive eating behaviours, distorted body image perceptions and excessive physical activity, there is growing recognition anorexia nervosa is associated with underlying dysfunction in excitatory and inhibitory neurometabolite metabolism and signalling. This narrative review critically explores the role of N-methyl-d-aspartate receptor-mediated excitatory and inhibitory neurometabolite dysfunction in anorexia nervosa and its associated biomarkers. The existing magnetic resonance spectroscopy literature in anorexia nervosa is reviewed and we outline the brain region-specific neurometabolite changes that have been reported and their connection to anorexia nervosa psychopathology. Considering the proposed role of dysfunctional neurotransmission in anorexia nervosa, the potential utility of zinc supplementation and sub-anaesthetic doses of ketamine in normalising this is discussed with reference to previous research in anorexia nervosa and other neuropsychiatric conditions. The rationale for future research to investigate the combined use of low-dose ketamine and zinc supplementation to potentially extend the therapeutic benefits in anorexia nervosa is subsequently explored and promising biological markers for assessing and potentially predicting treatment response are outlined.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Zinc deficiency may contribute to the pathogenesis of anorexia nervosa via multiple physiological pathways including glutamatergic and GABAergic signalling, N-methyl-d-aspartate receptor regulation, astrocyte dysfunction and pro-inflammatory signalling. |
A growing body of neurobiological evidence indicates that low-dose ketamine and zinc are independently efficacious in treating neuropsychiatric conditions, including anorexia nervosa. |
Via dual modulation of the N-methyl-d-aspartate receptor, the novel combination of low-dose ketamine with zinc may engage the necessary mechanisms to uncouple dysfunctional neural circuits and normalise those required for addressing impairments in executive control, reward processing and interoceptive awareness. |
1 Introduction
Anorexia nervosa (AN) is an eating disorder sub-type diagnosed in around 1% of the population today [1], which is clinically defined by severe malnutrition, ritualised and restrictive eating behaviours, an overwhelming fear of gaining weight and excessive physical activity [2]. Despite signals indicating homeostatic distress [3,4,5], individuals living with AN often deny being ill and resist treatment [6]. This treatment resistance likely contributes to chronic disease, as whilst on average it takes between 5 and 10 years from AN diagnosis to recovery [7], between 30 and 60% of people experience an enduring illness even at 30 years follow-up [8, 9]. Concerningly, this extended disease burden may lead to increased mortality from medical complications [10] and suicide [11].
Current treatments for AN typically incorporate the use of antidepressants alone, or in combination with psychotherapy and dietary interventions that emphasise weight restoration [12, 13]. Whilst these measures have contributed to improvements in some cases, their efficacy in promoting functional remission in AN is limited [5, 12], and to date no medication or physical therapies have been approved for AN [13,14,15]. Lack of treatment efficacy across existing treatment options for AN is of critical concern [16], as repeated failures not only promote treatment hesitancy, but also lead to disease progression, potentially past the point amenable to appropriate therapeutic intervention. Accordingly, clinical and research specialists have called for a revision of existing treatments, increased innovative research into safe and neurobiologically informed novel treatments for AN, and for the establishment of associated treatment biomarkers to optimise treatment prescription [17, 18].
The glutamatergic hypothesis, which proposes that disturbances in excitatory and inhibitory neuronal signalling are involved in the pathogenesis and maintenance of dysfunctional cognition and behaviour, has previously been applied to psychiatric conditions [19]. Recently, this framework was explored in AN [20], with support from preclinical [21,22,23] and clinical research [24, 25]. The efficacy of sub-anaesthetic doses of ketamine (0.1–3.0 mg/kg); an N-methyl-d-aspartate receptor (NMDAr) non-competitive antagonist, in promoting rapid and significant reductions in behavioural and psychological symptoms of AN is gaining increasing recognition [26, 27]. However, low-dose ketamine is not effective in all cases of AN and therapeutic effects dissipate with time, suggesting that although ketamine is capable of ameliorating neuropsychiatric pathology, the system may tend back towards the pathological wiring.
Recently, Hermens et al. [20] critically discussed the role of zinc deficiency in the pathogenesis of glutamatergic dysfunction in AN, providing a preliminary outline of the rationale for combined low-dose ketamine and zinc supplementation as a treatment for AN. Expanding on this, the current review synthesises relevant evidence to argue that whilst low-dose ketamine may stimulate the synaptic remodelling necessary for therapeutic relief in AN, long-term functional maintenance of these synaptic changes may be supported by zinc supplementation. Specifically, this review addresses: (a) the pathogenesis of excitatory and inhibitory neurotransmitter dysfunction in AN with a focus on the role of zinc deficiency and caloric restriction; (b) relevant biomarkers in AN and the case for investigating central cellular mediators of metabolism and synaptic plasticity; (c) neurometabolites associated with AN; (d) the mechanisms and previous therapeutic uses of zinc and low-dose ketamine in neuropsychiatric conditions and AN; and (e) the neurobiological rationale for exploring their combined use.
This rationale is not exclusive to AN, as the combination of zinc with low-dose ketamine may extend the therapeutic effects of ketamine (and the associated neurobiological changes) previously reported in psychiatric disorders [28]. However, the use of zinc as an adjunct to low-dose ketamine in AN is of particular interest because zinc deficiency has been reported in participants with AN [29,30,31,32]. The extent that the proposed neurobiological rationale may apply to other neuropsychiatric conditions in which altered neurotransmission is similarly implicated is noted, but does not form a major focus of this review. The rationale for the independent use of low-dose ketamine or zinc in normalising aberrant synaptic plasticity and neurotransmission associated with AN and other psychiatric conditions has been previously reviewed [33,34,35]. Whilst the evidence regarding the use of low-dose ketamine and zinc in treating psychiatric conditions is discussed, the reader should refer to these reviews for a more comprehensive analysis.
2 Literature Search
A literature search in the electronic databases PubMed, SCOPUS and Web of Science was performed up to November 2022. An analysis of relevant publications generated with a search strategy including MeSH and free-text keywords such as “ketamine”, “zinc”, “magnetic resonance spectroscopy”, “anorexia nervosa”, “neurotransmitter*”, “gluta*”, “GABA” and “blood biomarker*” was conducted. Consistent with the scope and aims of a narrative review [36, 37], stringent inclusion criteria were not imposed apart from the inclusion of magnetic resonance spectroscopy (MRS) studies conducted in AN populations. No constraint was placed on publication date and English-language studies only were included. Animal and human studies were considered, and no restrictions were placed for the type of article; case-studies, case-series, population-based studies, randomised controlled trials (RCTs), reviews, systematic reviews, meta-analyses, clinical trials and theoretical papers were included in this narrative review. Secondary screening of each publication’s reference lists and snowballing techniques (SCOPUS) were implemented to identify additional eligible articles for inclusion.
3 Glutamatergic Dysfunction in Anorexia Nervosa
To critically discuss the pathogenesis of glutamatergic dysfunction in AN and its implications for cognitive function, it is necessary to briefly review the principles of synaptic plasticity and the critical function of the NMDAr in mediating this.
3.1 NMDAr and Synaptic Plasticity
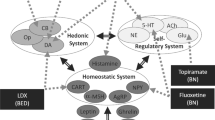
The NMDAr (see Fig. 1) is a heterotetrametric membrane receptor composed of two obligatory GluN1 subunits and different constellations of the GluN2 (A-D) or GluN3 (A-B) subunits [38, 39], although the specific subunit composition varies throughout the cortex [40, 41]. Whilst the GluN1 subunit appears to be a common prerequisite for the expression of functional NMDArs, it is the GluN2/3 subunits that largely determine the bio-physiological properties of the ion channel, such as mean conductance time and sensitivity to Mg2+ block [42]. The NMDAr is expressed both synaptically and extra-synaptically and activation of these spatially distinct populations independently influences synaptic plasticity processes [43]. At the synapse, excitatory (glutamate) and inhibitory (gamma-amino butyric acid [GABA]) signalling exerts a push–pull relationship on the post-synaptic membrane potential [44]. Whilst α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAr) and GABA receptor (GABAr) activation and ion conductance is critical to this process, the NMDAr is necessary for adjusting the synaptic weights within a distributed neural network [45], which ultimately encodes experience, explicit behaviour and memory [46].

N-methyl-d-aspartate (NMDA) receptor and its ligand binding sites. Note: zinc binds to the GluN2/3 subunit allosteric sites, whilst ketamine binds at the deep channel blocker site within the receptor pore. Created on BioRender.com with Publication and Licensing Rights obtained
The ability of the NMDAr to dynamically regulate synaptic plasticity is associated with its unique mode of activation, which requires the binding of glutamate and glycine to the GluN2 subunit and GluN1 subunits, respectively, in addition to the simultaneous depolarisation of the post-synaptic neuron to remove a Mg2+ ion from its channel pore [44, 45, 47]. This dual activation of the NMDAr acts as a coincidence detector [45]; both pre-synaptic signalling and post-synaptic depolarisation are needed for NMDAr activation. Building on the principles of Hebbian learning [48], modern neuroscience has established that plasticity is spike-timing dependent, such that the timing and frequency of pre-post synaptic firing are critical to the subsequent direction of synaptic plasticity, and therefore information encoding and processing [49, 50]. Strong correlated firing between the pre- and post-synapse, with the presynaptic neurons firing within 40 milliseconds or less of the postsynaptic neurons, leads to a NMDAr-mediated surge in intracellular Ca2+, which via a host of second messengers, promotes long-term potentiation (LTP). Conversely, the synapse undergoes long-term depression (LTD) when pre- and post-synaptic neuron firing is not correlated, stimulating a small increase in NMDA-mediated intracellular Ca2+ [44, 49]. This strengthening (LTP) and weakening (LTD) of synaptic connections that subserves neuroplasticity is not a unidimensional process, but includes changes in dendritic spine numbers and numbers of synaptic contacts, neurotransmitter release probability and its auto-regulatory and post-synaptic effects, membrane sensitivity, expression of re-uptake transporters, AMPAr/NMDAr membrane density and AMPAr/NMDAr subunit composition [46, 47].
Increasing evidence indicates NMDAr and AMPAr subunit composition as key regulators of neuroplasticity [47], and changes to the composition (e.g. genetic and/or environmentally induced) may contribute to dysfunctional neurotransmission in AN [22, 51]. A relevant example of this is that NMDAr subunit composition regulates neural metaplasticity; the dynamic threshold for LTP/LTD that varies as a function of prior activation [47]. Increased expression of the GluN2B subunit, which decreases the GluN2A/B ratio, favours LTP, whilst a relative increase in GluN2A expression requires stronger stimulation to induce LTP [47]. Notably, during early mammalian development, the composition of NMDAr gradually tends towards GluN2A dominance with a concurrent GluN2B decrease, which is thought to suppress excessive LTP and help establish mature synapse formation for refined cortical activity [52, 53]. Considering the activation of the NMDAr mediates the degree and direction of synaptic plasticity [46], any modification of its stereochemistry (e.g. chemical modification and/or subunit composition), and thus its biophysical properties, almost certainly would influence neuroplasticity.
3.2 Pathogenesis
3.2.1 Zinc Deficiency
Zinc is an essential dietary trace element within the human body that is involved in a myriad of biological processes, serving a catalytic, structural and regulatory function in metabolism, oxidative stress, gene regulation and the immune system [54, 55]. Within the brain, zinc is primarily bound to proteins; however, a portion (approximately 15%) is found concentrated in synaptic vesicles of glutamatergic neurons in the hippocampus, prefrontal cortex (PFC) and amygdala [34, 56]. Termed zinc-enriched neurons (ZENs), zinc is co-released with glutamate into the synaptic cleft upon stimulation, with measurements of extracellular zinc levels in hippocampal tissue that range from sub-micromolar to greater than 100 micromolar depending on the level of neuronal activity [57, 58].
The role of zinc deficiency in the pathophysiology of major depressive disorder (MDD) and schizophrenia [33, 34, 59, 60], and more generally in perturbed learning, memory and attention has previously been demonstrated [61, 62]. Zinc deficiency in AN has also been reported in studies using peripheral blood samples and urinalysis [29, 31], and dietary intake assessments [30, 32], consistent with the fact that the physiological availability of zinc is exclusively determined by dietary intake [34]. However, the degree to which peripheral zinc measures accurately indicate levels in the brain, and how synaptic zinc signalling is impacted in psychiatric conditions are not well understood. It should be noted that changes in ZEN signalling may occur irrespective of dietary factors, as chronically elevated cortisol levels, which are present in AN [3], cause dendritic and synaptic atrophy of glutamatergic neurons in the PFC and hippocampus, whilst stimulating dendritic arborisation in the nucleus accumbens (NAc) and amygdala [63, 64]. Holding the distribution of ZENs to be valid [34, 56], this could lead to a disproportionate loss of zinc regulation in the frontal regions.
Whilst the implications of zinc deficiency are undoubtedly multiple owing to its diverse physiological role, one consequence of significance to glutamatergic dysfunction is the decrease in homeostatic allosteric inhibition of NMDAr activity [61, 65]. Functionally, decreased zinc-mediated inhibition of the NMDAr leads to greater calcium (Ca2+) conductance, increasing the probability of Ca2+-induced excitotoxicity [66]. In rodents, zinc restriction leads to significantly increased synaptic expression of GluN2A and GluN2B NMDAr subunits in glutamatergic neurons of the hippocampus and PFC [67]. Notably, mice with the greatest vulnerability in activity-based anorexia (ABA) models (e.g. most weight loss, most running) had the highest levels of NMDAr density on the post-synaptic hippocampal pyramidal neurons [68], indicating that zinc deficiency-induced changes in NMDAr expression may contribute to AN pathogenesis.
Changes in glutamatergic NMDAr GluN2A/B subunit expression may be particularly relevant to zinc deficiency-mediated glutamatergic dysfunction in AN, as zinc’s NMDAr inhibition varies according to the specific subunit. Specifically, zinc voltage-independent inhibition of the GluN2A subunit occurs at much lower (5–80 nanomolar) levels (i.e. higher affinity) than the GluN2B subunit (1.6–9.5 micromolar) [69, 70]. Zinc also binds to the amino terminal domain of the GluN3 sub-unit; however, the exact nature of its binding dynamics and subsequent effects on activity are not well established [71]. In fact, compared with GluN1 and GluN2 sub-units, very little is known about the GluN3 sub-unit, albeit growing evidence indicates it has a critical role in shaping early post-natal central nervous system (CNS) development in the thalamus, hypothalamus, brain stem, hippocampal and parahippocampal areas [72].
Zinc deficiency may also contribute to altered inhibitory (GABA) signalling, as zinc normally exerts inhibitory action on GABAergic signalling via the NMDAr [73] and the GABAA receptor [74, 75]. Albeit the exact nature of zinc’s effects are complicated by reports of zinc-mediated potentiation of GABA release [62, 76], a discrepancy likely due in part to the subunit specific regulation of GABAr by zinc [77]. The link between zinc deficiency and altered neurotransmission extends beyond its regulatory effects on the NMDAr and GABAA receptor, as the enzymatic catabolism of glutamate by glutamate dehydrogenase is normally suppressed by zinc [78, 79]. Thus, zinc deficiency may result in greater enzyme-mediated glutamate breakdown (i.e. decreased glutamate). This is consistent with the decreased levels of glutamate neurometabolites reported in some proton (1H)-MRS studies of AN [25, 80, 81].
Considering the differing affinity of zinc for the Glu2A/2B subunits [82] and previous reports that perisynaptic zinc levels reach levels sufficient to occupy high (GluN2A) but not low (GluN2B) affinity NMDAr after bursts of stimulation [57], the degree of activity-mediated inhibition exerted by zinc at glutamatergic neurons may depend on the state of NMDAr subunit expression. In animal models of AN, decreased GluN2A and increased GluN2B subunit expression on glutamatergic neurons are found [22, 83], suggesting decreased zinc-mediated NMDAr inhibition may occur independent of gross changes in zinc levels. However, these perisynaptic zinc measurements were established in vitro [57], with others reporting levels greater than 100 micromolar [58]. Furthermore, the impact of pathological conditions on perisynaptic zinc levels has not been established, therefore such conclusions remain speculative until further research is conducted. Notably, cultured rodent cortical interneurons display an altered subunit ratio, with up to a five-fold higher 2A-2B subunit expression compared with pyramidal neurons [84, 85]. Compared with the aforementioned studies of zinc’s activity in cultured glutamatergic neurons [57, 58], the impact of this differential NMDAr subunit expression on zinc’s NMDAr-mediated regulation of inhibitory neuron activity is not understood and requires investigation.
3.2.2 Caloric Restriction and Hyperactivity
The observation of core features of AN; self-imposed starvation, weight loss and hyperactivity, in response to food restriction and access to a running wheel has provided a valuable preclinical model for investigating the neurobiology of AN (see reviews [23, 86]). Preclinical ABA models have demonstrated these behaviours are associated with a host of physiological changes relevant to glutamatergic function, including increased extracellular glutamate in the NAc [22], and increased expression of GluN2B NMDAr subunit in the hippocampus [68, 87, 88]. Notably, Aoki et al. [88] found the extent of GluN2B levels in dendritic spines positively correlated with weight loss in the ABA group, but not in a group that was subjected to similar food restriction, suggesting excessive activity is a key driver of GluN2B upregulation. Region-specific morphological changes in dendrites of glutamatergic pyramidal neurons in the hippocampal cornu ammonis 1 have also been reported, with branch degeneration in the rostral-dorsal region and increased branching in the ventral region [89]. Furthermore, a recent ABA study found a maladaptive reduction in glutamatergic spine density and a change in AMPAr and NMDAr membrane stability in the medial PFC (mPFC), which was associated with impaired cognitive control [90]. The changes are not limited to excitatory signalling, with changes in GABAergic neuron subunit composition noted within the hippocampus [91, 92] and amygdala [93]. Morphological changes in the GABAergic axons have also been noted [94], with increased contact providing a protective effect against ABA vulnerability. Therefore, the vulnerability to AN, and the specific type and severity of behaviours developed is likely to manifest as a function of both excitatory and inhibitory mechanisms.
The changes in AMPAr and NMDAr composition found across ABA studies [22, 68, 87, 95] provide strong evidence that glutamatergic dysfunction may contribute to the maladaptive symptoms observed in AN. The increased relative expression of the GluN2B subunit is particularly notable, as this subunit is implicated in antidepressant [96] and eating behaviour [51, 97], antidepressant effects of ketamine [98] and ketamine-induced changes in brain connectivity [99]. More broadly, the increase in hippocampal and NAc GluN2B subunit expression noted in animal models of AN may recapitulate a period of heightened neuroplasticity that contributes to dysfunctional circuit formation and susceptibility to psychiatric disorders [53, 100]. However, it must be noted there is healthy speculation of the degree to which the ABA model accurately captures AN, which is founded upon some discordance in psychological, behavioural and pathophysiological features [101]. Despite these limitations, the ABA model is currently the most robust model for AN, replicating many of the behaviours and pathophysiology observed in humans [86].
3.2.3 Inflammation and Astrocyte Dysfunction
Elevated levels of pro-inflammatory cytokines, interleukin-1B, interleukin-6, and tumor-necrosis factor-alpha, have been reported in adolescent and young adult participants living with AN [102, 103], and these may indirectly promote glutamatergic dysfunction. Stress and inflammation are associated with zinc deficiency [79], and decreased zinc within ZENs of the hippocampus promote increased HPA-axis activity [61], potentially creating a self-reinforcing cycle. Furthermore, increased pro-inflammatory cytokines activate the enzyme indoleamine 2,3-dioxygenase, which metabolises tryptophan into quinolinic acid [104]. Not only does the increased metabolism of tryptophan reduce the pool available for serotonin (5-HT) synthesis, which itself is implicated in AN [5, 105], but the increase in quinolinic acid stimulates NMDAr activity [79].
Glutamatergic dysfunction may arise as a consequence of astrocyte damage caused by pro-inflammatory cytokines, leading to impaired extra-synaptic glutamate recycling [106]. Astrocytes regulate extra-synaptic uptake and enzymatic conversion of glutamate to glutamine, before transport of glutamine back to the pre-synaptic bouton for conversion by glutaminase to glutamate [107]. A dehydration-induced AN model found that the glutamate-glutamine homeostasis was disturbed in the orbitofrontal cortex and mPFC (including infralimbic, pre-limbic and anterior cingulate cortex) of young female rats, and this was driven by decreased astrocyte density and glutamate transporter (i.e. GLT-1) expression [21]. This is consistent with previous research showing increased GLT-1 levels in the pre-synaptic membrane of excitatory neurons and astrocytes in the hippocampus dampened the severity of food restriction-evoked running behaviour in an ABA model [95].
The role of astrocytes extends beyond glutamate recycling, with growing research demonstrating they are critical mediators of neuronal energy supply [108]. This is not to mention the release of gliotransmitters (e.g. D-serine and adenosine triphosphate) and neuropeptides (e.g. brain-derived neurotrophic factor [BDNF]) by astrocytes and their influence on neuronal function [109], the complexity of which is only beginning to be understood [110]. No longer considered simply scaffolding cells of the CNS, AN-related disturbances in astrocyte function may participate in the pathogenesis and maintenance of glutamatergic dysfunction. Whilst astrocyte dysfunction has yet to be conclusively demonstrated in participants with AN, evidence suggests that decreased nutrient intake and increased pro-inflammatory cytokines in AN [102, 103] may stimulate astrocytes to transition to a reactive phenotype [111], resulting in disturbed glutamate turn-over and potentially excitotoxicity. Recently, increased glial fibrillary acidic protein expression, an indicator of astrocyte injury, was reported in a sample of acutely underweight participants with AN compared with healthy controls [112], lending further support to this potential mechanism. However, until such findings are replicated in clinical samples of AN and peripheral glial fibrillary acidic protein levels are shown to reflect astrocytic activity in the CNS, strong inferences regarding the contribution of proinflammatory signalling and astrocyte activity to glutamatergic dysfunction in AN, especially when based on peripheral measures, are cautioned.
3.3 Cognitive Deficits and the Glutamatergic System
Anorexia nervosa is characterised by complex psychological, cognitive and behavioural difficulties, including compulsive and inflexible behaviours, emotion dysregulation, distorted perceptions of self particularly around body image, and restrictive eating behaviours despite malnourishment [105, 113,114,115]. Supported by a growing body of neuroimaging findings, it is now understood these maladaptive behaviours are associated with altered processing in the anterior cingulate cortex (ACC), precuneus, thalamus, insula, striatum, NAc and hypothalamus [114,115,116,117,118,119]. Dysfunctional excitatory and inhibitory activity and connectivity within and between these regions is thought to contribute to errors in food-associated reward prediction [120], errors in processing and awareness of motivational, emotional and feeding cues, and hormonal signals [3, 5, 113], and a bias towards negative appraisals of self (particularly body image) [121]. These errors in information processing are thought to drive core cognitive deficits in executive control, reward processing and interoceptive awareness that manifest as cognitive inflexibility [122, 123], detail-focussed processing [124], harm avoidance [125] and impaired emotional regulation [126].
Many of these aspects of AN are replicated in preclinical ABA models that describe changes in the glutamatergic system. Previous reviews of these studies have highlighted changes in the composition of AMPAr, NMDAr and GABAr within the hippocampus, amygdala and NAc are associated with anxiety-like behaviour, impaired inhibitory control and hyperactivity [23, 86]. Although valuable, these models are not capable of replicating the complex cognitive, behavioural and psychological aspects of AN. The clinical literature has provided some insight into the involvement of the glutamatergic system in these aspects of AN. Three 1H-MRS studies have investigated the association between excitatory and inhibitory neurometabolites and AN symptoms [24, 81, 127]. The earliest of these studies by Ohrmann et al. [81] reported decreased levels of the composite neurometabolite measure Glx (glutamate, GABA and glutamine) in the rostral ACC of participants with AN were correlated with increased levels of depression. Maier et al. [24] reported a significant negative correlation between anterior insula levels of Glx and weight and shape concerns in participants with AN as measured by the Eating Disorder Examination Questionnaire. They concluded decreased Glx in this region may contribute to greater negative body image perceptions, a conclusion in line with subsequent work highlighting the insula’s role in interoceptive awareness [113]. Notably, N-acetyl-aspartate (NAA) levels in the insula were also negatively associated with shape concerns, suggesting dysfunctional processing in the insula may be due to neuronal loss. In a recent study, Westwater et al. [127] found reduced glutamate and NAA levels in the right inferior lateral PFC were associated with a higher propensity for automatic routine-like behaviours.
4 Biomarkers of Glutamatergic Dysfunction
4.1 Blood Biomarkers
If the changes in NMDAr composition and neurotransmitter signalling noted from preclinical research similarly contribute to glutamatergic dysfunction in participants AN, the identification of neurotrophic factors and cellular pathways that regulate these mechanisms may serve as valuable biomarkers for evaluating AN severity and treatment response [4, 128]. The expression of BDNF, a neurotrophin key to sustained functional changes in synaptic plasticity [129], may participate in the susceptibility for AN as decreased serum BDNF levels compared with healthy controls have been reported [128, 130, 131]. Decreased BDNF signalling may also occur in the zinc-deficient AN state, as preclinical studies indicate zinc independently activates both the tyrosine receptor kinase-β receptor and the BDNF membrane receptor [132]. Functionally, animal studies have shown reduced BDNF signalling can lead to neuronal shrinkage and dendritic loss [133], which can result in cognitive deficits [134].
Despite the association with AN, the reliability and validity of BDNF as a biomarker is questionable. Currently, there is no consensus regarding the optimal time to measure ketamine-treatment induced increases in BDNF, with preclinical studies indicating the window of detection ranged from 2 to 72 hours post-ketamine [135, 136]. Methodological variations in intra- and inter-individual blood sampling time may also confound results, as BDNF levels display a natural and sexually dimorphic diurnal rhythm [137, 138]. Specifically, male individuals display greater absolute levels of BDNF and greater diurnal variation compared with female individuals. Furthermore, blood processing conditions such as ambient temperature and time spent clotting can influence BDNF measurements [139]. This is not to mention the considerable debate on whether peripheral serum and plasma samples reflect CNS levels, with the evidence en masse indicating only moderate correlations [140]. New methods focused on the analysis of extracellular vesicles enriched for a neuronal origin can provide improvements in the validity of a peripheral biomarker analysis [141]; however, standard enzyme-linked immunosorbent assay methods remain widely used, despite known variations in commercial kits [142]. Accordingly, more central regulators of synaptic plasticity mechanisms may serve as robust biomarker alternatives.
Two such central regulators of relevance to AN are the mammalian target of rapamycin (mTOR) and the silent mating type information regulation 2 mammalian homologue (Sirtuin 1 [SIRT1]), as they are associated with caloric restriction [83, 143, 144], and are known to mediate neurotrophic expression [145, 146]. SIRT1 is a nicotinamide adenine dinucleotide (NAD+) dependent deacetylase that suppresses mTOR activity [147]. Together, mTOR and SIRT1 are responsible for integrating nutrient and cellular stress signals and mediating adaptive metabolic and endocrine responses [83, 144]. In the starved state, limited nutrient availability and growth factors, such as insulin and insulin-like growth factor-1 [148], promote increased SIRT1 activation [143, 149]. Moreover, prolonged fasting increases the biosynthesis of NAD+ [150], fuelling further SIRT1 activation. Animal studies have shown SIRT1 activation produces AN-like behaviours and caloric restriction leads to increased foraging and hyperactivity in rodents, which is blocked by SIRT1 knock-out [151, 152]. SIRT1 activity may be a particularly important mediator of AN pathogenesis, as upregulation of SIRT1 leads to decreased 5-HT levels and increased anxiety-like behaviours in rodents [153]. Furthermore, the NMDAr subunit GluN2A is suppressed by SIRT1 [83], resulting in a decreased GluN2A/GluN2B ratio. Whilst many questions remain to be addressed, the preclinical findings showing SIRT1 activation alone can produce AN-like behaviours [151, 152] and accelerate the progression of ABA models [83] suggest treatments that impinge on this central regulator (i.e. zinc) may target a core regulator of AN pathophysiology.
4.2 MRS
Aberrant excitatory and inhibitory neurotransmission has been reported in preclinical models of depression and AN; however, this has predominantly come from animal studies capable of measuring changes at the cellular scale [86, 154, 155]. Although the sensitivity of MRS measurements (2–4 cm3 voxels) pale in comparison to that achievable with imaging methodologies in preclinical research (nanometers), MRS offers a non-invasive way to measure neurometabolites in defined two-dimensional or three-dimensional cortical volumes, allowing for the identification of disease-specific biomarkers [156], which have previously been shown to correlate with the clinical response of ketamine [157]. The existing MRS literature (including 1H and phosphorous-31 (31P) in AN is summarised (Table 1) and critically evaluated here.
Within the literature, most studies investigated markers of neuronal integrity (e.g. NAA) [158,159,160,161]. Regarding the studies that measured brain neurometabolites involved in excitatory and inhibitory neurotransmission, five investigated glutamate independent of Glx [25, 127, 162,163,164], six quantified the composite Glx [24, 80, 81, 163, 165, 166] and only one study measured GABA [25]. Compared with healthy controls, significantly decreased levels of glutamate in participants with AN were only reported in one study [25]. Conversely, glutamine levels were not significantly changed. Considering that pre-synaptic glutamate replenishment is driven by astrocytic conversion of glutamine to glutamate [107], the findings of Godlewska et al. [25] suggest dysfunctional neurotransmission in AN may be mediated in part by impaired astrocyte function. Whilst this is contradicted by the lack of glutamate change recorded in three studies [162,163,164], these studies were conducted using a 1.5–3 Tesla scanner. Quantification of glutamate at such field strengths is confounded by a glutamine and glutathione chemical overlap (reviewed in [167]).
Six studies associated changes in brain neurometabolites with measures of AN symptoms; four with body mass index (BMI) [80, 161, 164, 168], and two with other eating disorder symptoms [24, 81]. Within the frontal lobe, Castro-Fornieles et al. [80] found improvements in BMI were associated with significant increases in NAA levels, a measure of neuronal integrity. Notably, they also found improvements in BMI were associated with increased insulin-like growth factor-1 and decreased cortisol, suggesting these may serve as treatment biomarkers. In a subsequent study, Castro-Fornieles et al. [165] found total cholineand creatine were increased in AN, and normalised following serotonin-selective re-uptake inhibitor treatment. Roser et al. [164] and Rzanny et al. [161] similarly found markers of neuronal density and membrane integrity in what they termed the ‘frontal white matter’, namely the ratios of NAA/Cho and phosphodiester/phosphate, were associated with BMI, such that decreases in these ratios was associated with lower BMI in participants with AN compared with healthy controls.
Reflecting the high prevalence of AN in female individuals, the imaging literature on brain neurotransmitters has almost exclusively been conducted in female samples, with only two studies including male individuals diagnosed with AN [80, 164], albeit at small numbers (one and two, respectively). The age of participants is similarly skewed, with samples primarily consisting of adolescents and young adults (mean = 20.5 years), although two studies included older adults (41 and 30) [25, 161]. Furthermore, variation in magnetic field strength (1.5-Tesla) and a predominance of studies using 1.5 and 3 Tesla scanners further complicates the findings, as the capacity to accurately discern the spectroscopic signature of chemically similar glutamate, GABA and glutamine from the composite Glx is limited at these field strengths without editing sequences [169]. It should be noted that the only study that found reduced glutamate in AN was conducted at 7 Tesla [25]. At present, the heterogeneity in MRS methodology and findings belies the formation of confident inferences about the degree and locality of dysfunctional neurotransmission in AN, specifically that of glutamate, glutamine and GABA. Future MRS research that (1) is conducted at high (3–7 Tesla) field strengths, (2) uses advanced MRS pulse sequence design and editing techniques to reliably quantify glutamate, GABA, glutathione and glutamine [170] and (3) meets the recently developed minimum MRS design and reporting standards [171] is needed in AN.
5 Zinc and Ketamine
Despite the mixed MRS evidence investigating excitatory and inhibitory neurometabolite profiles, participants living with AN have responded positively to zinc and low-dose ketamine treatment independently [35, 172, 173]. Considering, zinc and low-dose ketamine modulate excitatory and inhibitory signalling through binding to two different sites on the NMDAr (see Fig. 1), an exploration of their independent mechanisms of action and treatment applications is warranted to develop the rationale for their combination.
5.1 Zinc Mechanism
The proposed mechanisms of zinc therapeutic effects have predominantly been inferred from preclinical studies of induced zinc deficiency, which have observed increases in NMDAr expression [67] and hippocampal extracellular glutamate [174], decreased neurogenesis [175] and altered monoaminergic signalling [33]. Furthermore, as reviewed in Sect. 3.3, zinc supplementation may normalise glutamatergic function by re-establishing homeostatic glutamate metabolism, regulation of both the NMDAr and GABAr, and decreasing pro-inflammatory cytokine signalling. Notably, there is some preclinical evidence to indicate zinc’s interaction with the NMDAr is critical to its action, as antagonism of the receptor occluded zinc’s antidepressant effects [176].
Preclinical research has also shown zinc’s antidepressant effects are associated with changes in AMPAr sub-unit and synaptic protein expression in the PFC [177], serotonergic receptor expression in the hippocampus and increased activation of the mTOR [33], BDNF and cAMP-response element binding intracellular signalling pathways [34]. In part, zinc’s up-regulation of BDNF and cAMP-response element binding expression occurs via its interaction with the G-protein-coupled receptor (GPR-39), as decreased GPR-39 signalling, either dietary induced and genetic knockout, reduced cAMP-response element binding and BDNF expression in the frontal cortex [67] and hippocampus [178, 179]. Similar to ketamine, mTOR phosphorylation is critical to zinc’s antidepressant effects, as rapamycin occluded the effects of a single dose of zinc in rodents [177]. Thus, zinc treatment may assist in re-establishing functional neural circuit dynamics by promoting synaptogenesis and regulating excitatory and inhibitory signalling.
5.2 Zinc Treatment
Zinc’s therapeutic treatment outcomes have primarily been observed in preclinical and clinical studies on depression, which have previously been reviewed in great detail [33, 34]. Compared with male rats fed with zinc supplemented diets, male rats fed with a zinc-deficient diet show increased depression-like (anhedonia and anxiety) and anorexia-like (severe weight-loss) behaviour [180], and separate studies have shown zinc supplementation is capable of ameliorating these depressive symptoms [176, 177, 181]. Clinically, zinc supplementation has led to clinical improvements in depressive symptoms both as a monotherapy [182,183,184] and as an adjunct to typical antidepressants [185], even in treatment-resistant cases [186, 187]. One double-blind placebo-controlled RCT assessed the effect of 220 mg of zinc sulphate as an adjunct to 6 mg of risperidone (an antipsychotic) in schizophrenia [188]. The study reported significant improvements in both positive and negative symptomology and a reduction in aggressive behaviour, compared with the risperidone active control. In AN, several RCTs have demonstrated that zinc supplementation results in rapid weight gain in children [31] and adults [173], and decreased depression/anxiety in adolescents living with AN [30].
Hermens et al. [20] recently outlined the zinc deficiency-induced model of glutamatergic dysfunction in AN, proposing that zinc supplementation would play a key role in ameliorating this pathophysiology. However, they suggest stronger NMDAr antagonism, beyond the allosteric modulation of zinc, may be required to produce long-term therapeutic outcomes in the chronic AN state. Low doses of ketamine presents as a promising candidate to fulfil the therapeutic role as an NMDAr antagonist combined with zinc supplementation, especially considering its utility in alleviating glutamatergic dysfunction implicated in other psychiatric conditions [35, 154], as suggested by Petrilli et al. [34].
5.3 Ketamine Mechanism
The mechanisms of action of ketamine are primarily discussed with respect to its role as a non-competitive NMDAr antagonist; however, the reader is referred to previous reviews describing its impact on opioid, serotonergic and dopaminergic systems [189, 190]. Owing to the fast-firing properties of parvalbumin and calbindin/calretinin-binding GABAergic interneurons in the mPFC [191, 192], low-dose ketamine is thought to preferentially block the NMDAr of these neurons. In rodents, this results in increased glutamatergic neurotransmission in the PFC, due to disinhibition of glutamatergic pyramidal neurons [154, 193], as evidenced by increased gamma-band firing [194] and increased extracellular glutamate and glutamate cycling in this region [195, 196].
At glutamatergic synapses, preclinical studies show ketamine-mediated NMDAr inhibition decreases miniature excitatory post-synaptic potentials, and inactivates eukaryotic elongation factor-2, a protein that normally suppress post-synaptic protein synthesis [197]. This leads to increased AMPAr and BDNF expression [135, 146]. Extra-synaptic NMDAr are similarly inhibited by low-dose ketamine, and this too stimulates an increase in the expression of BDNF and vascular endothelial growth factor [145, 146], proteins involved in synaptogenesis via an mTOR-dependent pathway [146]. Importantly, it is the combination of these effects that underpins the antidepressant effects of ketamine [155]. For instance, selective knockdown of GluN2B subunits in NMDAr expressed by GABA interneurons, including somatostatin and parvalbumin sub-types [193], occludes the antidepressant action of ketamine. This was also observed when synaptic protein synthesis or activity was blocked, via mTOR inhibition [146], AMPAr antagonism [135, 136] or in animals lacking a functional BDNF gene [198].
The mechanism of action of ketamine also involves interactions with the monoaminergic systems. Extracellular serotonin levels are increased following a sub-anaesthetic ketamine dose [199], and pharmacological blockade of 5-HT signalling prevent the antidepressant effects of ketamine [200]. Notably, the effect of ketamine on the 5-HT systems is stereoisomer and metabolite specific [201]. In the dopaminergic system, ketamine stimulates increased activity in the ventral tegmental area, leading to an increase in mPFC dopaminergic signalling [196, 202]. The subsequent increase in dopamine-1 (D1) receptor activation in the mPFC upregulates NMDAr and AMPAr expression [40, 203, 204], which, via an increase in neuron excitability, promotes synapse formation and potentiation in the mPFC. Pothula et al. [205] had demonstrated the importance of this mechanism of action of ketamine, as selective knockdown of the D1 receptor in the mPFC completely occludes the antidepressant behavioural effects. This is consistent with the role of the D1 receptor activation, which stimulates increased AMPAr and NMDAr expression and neuron excitability [204].
The effects of ketamine on the dopaminergic system may also be stimulated by changes in AMPAr composition and neuronal firing patterns in brain regions involved in reward learning. Skiteva et al. [206] found one injection of low-dose ketamine led to a removal of GluA2-lacking (calcium permeable) AMPAr from dopaminergic neurons in the ventral tegmental area (VTA). Conversely, GluA2-lacking (calcium permeable) AMPArs were inserted at glutamatergic synapses in the NAc. Such ketamine-induced alterations in AMPA calcium conductance of neurons within the meso-limbic circuit may be of particular relevance to normalising dysfunctional reward-processing in AN, as one study reported changes in neural firing patterns (tonic vs phasic) in this circuit influenced drug-seeking behaviour [207, 208]. The dynamic interplay between these NMDAr-dependent and NMDAr-independent mechanisms engages the physiological parameters required for synaptic plasticity, which is thoroughly covered in previous reviews [154, 155].
Considerable neuroimaging research in humans has also been conducted in an attempt to explain the rapid and profound shifts in cognition, perception and psychological experience associated with low-dose ketamine. In structural magnetic resonance imaging and functional magnetic resonance imaging studies, the anti-depressant effects of low-dose ketamine treatment have been associated with NMDAr-mediated synaptic remodelling in ventral striato-limbic regions, including decreased [209] and increased NAc volume [210], and increased PFC, hippocampus, putamen, caudate, amygdala and thalamus connectivity/volume [145, 209,210,211,212]. At a systems level, the effects of ketamine are observed as less integrated and more segregated brain networks, and there is evidence this ketamine-induced network reconfiguration is conserved across rodents and humans [213]. Considering the overlap in afflicted brain regions shared across the conditions [64, 105, 125, 214,215,216], it may be reasoned that low-dose ketamine may promote similar therapeutic responses in AN.
The effect of low doses of ketamine on brain neurometabolites in clinical studies has also provided some insight into its acute and delayed effects on glutamatergic function. Increases in GABA [157] and Glx [217] levels within the mPFC were found following low-dose (0.1–0.5 mg/kg) intravenous ketamine in clinical samples, which predicted greater clinical response in obsessive compulsive disorder and depression, respectively. In a double-blind RCT of intravenous ketamine (0.5 mg/kg) in healthy individuals, Li et al. [218] reported a delayed effect, with a significant increase in glutamine-to-glutamine ratio in the pregenual ACC at 24 hours post-ketamine treatment compared with saline-injected controls. The implication of the role of pregenual ACC in ketamine’s treatment response was replicated in a sample (n = 17) with MDD [219], with an increased glutamate level at 24 hours following a single dose of ketamine predictive of both greater clinical outcomes and heightened activity in this region. However, several studies have also reported no changes in brain neurometabolite levels following ketamine treatment [220,221,222]. In part, these disparate findings may be explained by the exploration of effects of ketamine in healthy samples and neuropsychiatric conditions, in addition to the significant variability in the brain region of interest and timing of the MRS scan (during administration up to 24 hours post).
5.4 Ketamine Treatment
In the preclinical literature, the rationale for low-dose ketamine treatment in AN has primarily been developed from models of depression [154], with growing preclinical evidence showing low-dose ketamine opposes the pathophysiology and maladaptive behaviours found in ABA and inflammation-induced anorexia models [223]. Notably, one study found a single dose of ketamine (30 mg/kg) conferred resilience to ABA-induced hyperactivity, anxiety and decreased food intake in adolescent female mice [224]. Clinically, the basis for exploring low-dose ketamine in AN has been heavily influenced by the therapeutic effects in neuropsychiatric conditions that commonly co-present with AN, including MDD [225], suicidality [28, 226] and obsessive-compulsive disorder [27]. For instance, several RCTs have reported rapid antidepressant effects of intravenous ketamine in populations that had not responded to conventional treatments [27, 227,228,229]. A recent treatment-resistant depression case-control study found intravenous ketamine (0.5 mg/kg) infused over 40 minutes led to improvements in optimism bias (tendency to update beliefs more after good than bad news) as early as 4 hours after infusion and these improvements mediated reductions in depressive symptoms 1-week post-treatment [230]. However, this finding requires validation and replication, as the study sample included individuals undergoing concurrent anti-depressant treatment.
To date, no RCTs have investigated the use of low-dose ketamine in participants living with AN; however, preliminary reports indicate it can be a safe and clinically promising treatment option. The earliest known study, which included a small (n = 15) treatment-resistant sample of participants with AN, reported that nine participants responded positively following two to nine ketamine infusions (5–21 days), with prolonged remission and over half maintaining healthier eating for 2 years after hospital discharge [231]. The specific dose of ketamine was not clearly specified, with infusion delivered at “20mg per hour for 10 hours”. Three subsequent case reports [232,233,234], and three case series [229, 235, 236] reported similar findings, with improvements in depression, suicidality, as well as obsessive and compulsive restrictive eating behaviours and thoughts. Notably, two of these studies reported remission from chronic AN when a ketogenic diet was combined with low-dose ketamine treatment [233, 236]. The vast majority of these studies demonstrated single and repeated sub-anaesthetic doses (0.4–1.2 mg/kg) were safe and did not produce any serious side effects. However, one case report noted an episode of apnoea in a 13-year-old individual with severe AN following ketamine (1.3 mg/kg) administration [237]. Despite the promising results reported, the generalisation of these findings is limited by small sample sizes, the primarily adult samples, variability in dose frequency, and the use of intramuscular or intravenous injections in all studies.
Setting aside methodological shortcomings of the emerging body of evidence, the profound responses to low doses of ketamine may provide hope to those suffering most severely from AN. In a case series of ketamine-assisted psychotherapy, participants reported “my eating disorder (ED) symptoms were greatly improved during the short treatment …. it was helpful to catch the vision of what life could be like without them”, and “the effect of easing my rigidity toward ED rules was extremely helpful at making me face challenges that were normally too anxiety provoking to face alone ([235], pg. 6). These responses, whilst subjective, suggest low-dose ketamine is capable of altering brain function in such a way that allows the obsessive behaviours and mental inflexibility characteristic of AN to subside, permitting awareness of one’s own behaviour, internal dialogue and physiological state.
Whilst low-dose ketamine administration appears necessary to access this therapeutic window of neuroplasticity and the associated behavioural change, it is not sustained, as treatment response typically fails to persist past 7 days [154]. Multiple ketamine doses have proved effective in prolonging the therapeutic effects, with two case reports of ketamine delivered at 0.5 mg/kg over three and six sessions showing antidepressant effects at 1 and 2 months. However, treatment response is not seen in all individuals and relapse typically occurs within 2 weeks from treatment cessation [238]. This is not to mention the increased risk of adverse physiological (e.g. hepatotoxicity, memory difficulties and urinary tract dysfunction) and addictive consequences associated with repeated ketamine use [35], albeit low-dose ketamine is less addictive than other currently prescribed psychotropics [239]. Accordingly, alternative means by which the synaptic plasticity-induced therapeutic effects of low-dose ketamine can be maintained, without increasing patient risks and minimising barriers to acceptability are required.
6 Zinc as a Treatment Adjunct
The concept of zinc supplementation as an efficacious and safe adjunct to pharmacological treatments has been demonstrated previously in the treatment of mood disorders [183,184,185,186,187] and schizophrenia [188]. Whilst this research indicates that zinc supplementation may be an effective adjuvant treatment for psychiatric conditions, Petrilli et al. [34] speculate that the combination of zinc with an NMDAr antagonist that targets D1 receptor activity may prove more effective in reducing glutamatergic dysfunction compared with typical psychotics, which work by binding to dopamine-2 receptors, particularly within the nigro-striatal pathways [240]. They justify this by citing the more robust response found following clozapine, an antipsychotic with a higher affinity for D1 compared to dopamine-2 receptors, inferring the heightened response may be due to a stronger effect on NMDAr-mediated hyperactivity in the PFC [240]. Considering this in the context of the recent suggestions by Hermens et al. [20], the combination of ketamine with zinc supplementation warrants consideration.
7 Two Sites of the Same Receptor
In light of the individual neurobiological mechanisms and shared target (the NMDAr), the novel combination of low-dose ketamine with zinc may provide a synergistic action capable of extending the therapeutic effects on psychological, behavioural and cognitive symptoms of AN. Mechanistically, this synergistic action may come into effect by low-dose ketamine engaging the molecular and cellular processes necessary for synaptic remodelling; NMDAr non-competitive inhibition, increased BDNF and AMPAr expression, and increased glutamatergic firing, which concurrent zinc supplementation maintaining this through regulating NMDAr and GABAergic function, BDNF signalling and SIRT1 activity (see Fig. 2). Considering zinc allosterically modulates the NMDAr, and that zinc levels may be depleted in AN [29,30,31, 79], the possibility that low-dose ketamine and zinc synergistically induce and sustain therapeutic neuroplasticity in AN is compelling, and has a sound neurobiological basis. Furthermore, considering the implication of glutamatergic dysfunction in both depression and AN, and the similarity in brain regions associated with this pathology, the mechanisms by which ketamine ameliorates of depression-like symptoms may be ‘transferred’ into the amelioration of anorexia-like behaviours and symptoms. Whether this is the case, and what the specific nature of this relationship is remain to be elucidated however.

a Acute anorexia nervosa (AN): a hyperglutamatergic state is promoted due to decreased zinc-mediated N-methyl-d-aspartate receptor (NMDAr) inhibition and pro-inflammatory cytokines induced inhibition of astrocyte-mediated glutamate uptake and recycling, resulting in decreased synaptic cleft clearance. Serotonin (5-HT) and dopamine (DA) signalling are altered in AN, and this may influence pre-synaptic neurotransmitter glutamate release. The resulting hyperglutamatergic state increases calcium conductance into the post-synaptic neuron, contributing to calcium-induced excitotoxicity. Decreased nutrient signals (insulin/insulin-like growth factor-1) and zinc deficiency may promote increased silent mating type information regulation 2 mammalian homologue (SIRT1) activity, suppressing mammalian target of rapamycin (mTOR)-mediated protein synthesis, and promoting dysfunctional NMDAr-mediated synaptic plasticity via downregulation of GluN2A expression. b Chronic AN: decreased zinc-mediated inhibition of GABAergic neurons (via NMDA and GABAA receptors) inhibits glutamate release, and downregulates BDNF and CREB expression (via the GPR39 receptor). Increased SIRT1 expression leads to a higher NMDAr GluN2B subunit ratio, which coupled with decreased zinc-mediated NMDAr inhibition, decreased glutamate clearance, and increased expression of calcium permeable (CP) AMPAr, promotes dysfunctional activity and connectivity in brain regions implicated in AN. (c) Ketamine and zinc: ketamine binds to NMDArs on GABAergic interneurons in the prefrontal cortex, disinhibiting pyramidal glutamatergic neurons and consequently increasing extracellular glutamate. Increased zinc similarly inhibits GABAergic interneurons. At the post-synaptic neuron, ketamine and zinc inhibit NMDAr activity, leading to decreased NMDAr-mediated calcium influx and reduced NMDAr expression. Synaptic and extra-synaptic NMDAr ketamine inhibition promotes protein (AMPA) synthesis and membrane insertion. The combined AMPAr insertion and increased excitatory PFC firing drives synaptic remodelling. Zinc and ketamine increase 5-HT and DA signalling; however, the specific effects on pre-synaptic firing are not known. Increased zinc levels may downregulate SIRT1 and pro-inflammatory cytokine activity, promoting increased synaptic protein synthesis and astrocyte-mediated glutamate clearance, respectively. The resulting changes in pre-synaptic firing, post-synaptic NMDAr and AMPR expression and composition, and synaptic glutamate clearance may stimulate region-specific synaptic remodelling. (d) The healthy state: ketamine and zinc-induced changes in pre-synaptic neurotransmitter release, post-synaptic receptor expression and glutamate clearance may be sustained by zinc’s regulatory action. Sustained zinc-mediated NMDAr inhibition and reduction in pro-inflammatory signalling prevents relapse of the hyperglutamatergic state, and normalises GABAergic and 5-HT activity. AMPAr alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, BDNF brain-derived neurotrophic factor, CREB cAMP-response element binding protein, EAAT excitatory amino acid transporter, GPR-39 G protein-coupled receptor 39, GABA gamma-aminobutyric acid, GABAr GABA receptor, IL interleukin, TNF-α tumor necrosis factor-alpha. Created on BioRender.com with Publication and Licensing Rights obtained
Previous trials investigating the individual use of repeated doses of low-dose ketamine or zinc have demonstrated considerable variability in the duration of response, lasting from days to months post-treatment cessation [26, 154, 238]. The waning treatment effects of low-dose ketamine and zinc supplementation suggest the changes in synaptic plasticity (increased neurotransmitter release, AMPAr and NMDAr expression and density, and number of synaptic contacts) are: (a) not translated into the changes in gene expression needed for long-term consolidation; (b) reversed by the pathological state or (c) a combination of both. The novel combination of low-dose ketamine and zinc may address this.
Theoretically, through modulating glutamatergic and GABAergic neurotransmission, regulators of synaptic plasticity (i.e. BDNF and mTOR), and proinflammatory signalling (depicted in Fig. 2), combined low-dose ketamine and zinc may be able to uncouple dysfunctional neural circuits, and normalise those required for addressing impairments in executive control, reward processing and interoceptive awareness [122,123,124,125,126]. For those living with AN, the possibility of improving their cognitive function may provide a more meaningful reason for exploring novel treatments such as that proposed here, in contrast to the typical focus placed on weight gain and the associated aberrant cognition [35]. Furthermore, normalisation of glutamatergic function within the brain regions associated with altered cognitive control, emotional and reward processing may promote adaptive, and sustainable behavioural changes that translate to long-term functional remission.
8 Conclusions
Effective and sustainable frontline treatment options for people living with AN are lacking, suggesting treatments are failing to address the precipitating and maintaining neurobiological mechanisms. Considering low-dose ketamine and zinc both target the NMDAr, a receptor intimately involved in neurotransmitter signalling dynamics, an exploratory investigation of their combination with blood and neuroimaging biomarker measures may provide useful insight into the safety and tolerability of the novel treatment, and neurobiological correlates of clinical response. It is important to re-iterate the proposed rationale for combining zinc with low doses of ketamine is not exclusive to AN, as their convergence at the NMDAr may provide synergistic therapeutic action in other neuropsychiatric conditions (e.g. MDD) associated with glutamatergic dysfunction. In contrast to other neuropsychiatric conditions, AN provides a robust model for exploring the potential therapeutic role that dual modulation of the NMDAr may have, as zinc deficiency has previously been reported in AN.
As a novel treatment option, the safety, tolerability and feasibility must be rigorously tested. Currently, there are no data on the combined use of low-dose oral ketamine and zinc either in AN or other neuropsychological conditions, nor on the neurobiological factors that moderate treatment response. Additionally, much of the current understanding of the neurobiological mechanisms of low-dose ketamine and zinc and therapeutic outcomes has come from preclinical models focused on the antidepressant effects. Further to this point, the majority of preclinical research has been conducted with normal-weight animals, and hence, lacks generalisability to the malnourished state. Whilst there is a growing body of neuroimaging research exploring these mechanisms in vivo, future preclinical and clinical research examining the mechanisms and therapeutic effects of ketamine and zinc in the context of the complex physiological, behavioural and psychological disturbances associated with AN is required.
Of arguably equal importance is the need to investigate and associate treatment-induced changes in AN symptomatology with a detailed individualised analysis of brain neurometabolites and blood biomarkers. Whilst the existing MRS literature indicates dysfunctional glutamatergic transmission in the frontal lobe, ACC, parieto-occipital lobe and thalamus may contribute to AN, the findings are mixed and several key methodological limitations are present. Considering changes in excitatory and inhibitory neurotransmission do not occur in isolation of each other, future MRS studies that robustly map excitatory and inhibitory neurometabolites are needed. Relatedly, blood biomarkers must be carefully considered, and should align with the treatment’s mechanism of action. In the case of combined low-dose ketamine and zinc for AN, such biomarkers may include zinc, insulin/insulin-like growth factor-1, SIRT1 and mTOR. Specifically, SIRT1 and mTOR present as promising biomarkers for evaluating low-dose ketamine and zinc-induced changes in clinical symptoms and brain function owing to the proposed role they have in mediating treatments effects. In conclusion, the novel treatment combination of low-dose ketamine and zinc supplementation may have therapeutic utility in AN because of their combined influence on glutamatergic function via modulation of the NMDAr.
Change history
27 February 2023
Missing Open Access funding information has been added in the Funding Note.
References
van Eeden AE, van Hoeken D, Hoek HW. Incidence, prevalence and mortality of anorexia nervosa and bulimia nervosa. Curr Opin Psychiatry. 2021;34(6):515–24.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-5™. 5th ed. Arlington: American Psychiatric Publishing, Inc.; 2013. p. xliv (947-xliv).
Berner LA, Brown TA, Lavender JM, Lopez E, Wierenga CE, Kaye WH. Neuroendocrinology of reward in anorexia nervosa and bulimia nervosa: beyond leptin and ghrelin. Mol Cell Endocrinol. 2019;497: 110320.
Stoving RK. Mechanisms in endocrinology: anorexia nervosa and endocrinology: a clinical update. Eur J Endocrinol. 2019;180(1):R9-27.
Gorwood P, Blanchet-Collet C, Chartrel N, Duclos J, Dechelotte P, Hanachi M, et al. New insights in anorexia nervosa. Front Neurosci. 2016;10:256.
Jowik K, Tyszkiewicz-Nwafor M, Slopien A. Anorexia nervosa: what has changed in the state of knowledge about nutritional rehabilitation for patients over the past 10 years? A review of literature. Nutrients. 2021;13(11): 3819.
Morris J, Twaddle S. Anorexia nervosa. BMJ. 2007;334(7599):894–8.
Dobrescu SR, Dinkler L, Gillberg C, Råstam M, Gillberg C, Wentz E. Anorexia nervosa: 30-year outcome. Br J Psychiatry. 2020;216(2):97–104.
Fichter MM, Quadflieg N, Crosby RD, Koch S. Long-term outcome of anorexia nervosa: results from a large clinical longitudinal study. Int J Eat Disord. 2017;50(9):1018–30.
van Hoeken D, Hoek HW. Review of the burden of eating disorders: mortality, disability, costs, quality of life, and family burden. Curr Opin Psychiatry. 2020;33(6):521–7.
Smith AR, Zuromski KL, Dodd DR. Eating disorders and suicidality: what we know, what we don’t know, and suggestions for future research. Curr Opin Psychol. 2018;22:63–7.
Blanchet C, Guillaume S, Bat-Pitault F, Carles M-E, Clarke J, Dodin V, et al. Medication in AN: a multidisciplinary overview of meta-analyses and systematic reviews. J Clin Med. 2019;8(2):278.
Zeeck A, Herpertz-Dahlmann B, Friederich H-C, Brockmeyer T, Resmark G, Hagenah U, et al. Psychotherapeutic treatment for anorexia nervosa: a systematic review and network meta-analysis. Front Psychiatry. 2018;9:158.
Aigner M, Treasure J, Kaye W, Kasper S. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of eating disorders. World J Biol Psychiatry. 2011;12(6):400–43.
Zipfel S, Giel KE, Bulik CM, Hay P, Schmidt U. Anorexia nervosa: aetiology, assessment, and treatment. Lancet Psychiatry. 2015;2(12):1099–111.
Wonderlich SA, Bulik CM, Schmidt U, Steiger H, Hoek HW. Severe and enduring anorexia nervosa: update and observations about the current clinical reality. Int J Eat Disord. 2020;53(8):1303–12.
Zipfel S, Schmidt U, Giel KE. The hidden burden of eating disorders during the COVID-19 pandemic. Lancet Psychiatry. 2022;9(1):9–11.
Touyz S, Hay P. The future of eating disorders research: an editorial. J Eat Disord. 2022;10(1):10.
Sohal VS, Rubenstein JLR. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry. 2019;24(9):1248–57.
Hermens DF, Simcock G, Dutton M, Boucas AP, Can AT, Lilley C, et al. Anorexia nervosa, zinc deficiency and the glutamate system: the ketamine option. Prog Neuropsychopharmacol Biol Psychiatry. 2020;101: 109921.
Reyes-Ortega P, Soria-Ortiz MB, Rodríguez VM, Vázquez-Martínez EO, Díaz-Muñoz M, Reyes-Haro D. Anorexia disrupts glutamate-glutamine homeostasis associated with astroglia in the prefrontal cortex of young female rats. Behav Brain Res. 2022;420: 113715.
Mottarlini F, Bottan G, Tarenzi B, Colciago A, Fumagalli F, Caffino L. Activity-based anorexia dynamically dysregulates the glutamatergic synapse in the nucleus accumbens of female adolescent rats. Nutrients. 2020;12(12):3661.
Schalla MA, Stengel A. Activity based anorexia as an animal model for anorexia nervosa: a systematic review. Front Nutr. 2019;6:69.
Maier S, Nickel K, Perlov E, Kukies A, Zeeck A, van Elst LT, et al. Insular cell integrity markers linked to weight concern in anorexia nervosa: an MR-spectroscopy Study. J Clin Med. 2020;9(5):1292.
Godlewska BR, Pike A, Sharpley AL, Ayton A, Park RJ, Cowen PJ, et al. Brain glutamate in anorexia nervosa: a magnetic resonance spectroscopy case control study at 7 Tesla. Psychopharmacology. 2017;234(3):421–6.
Keeler JL, Treasure J, Juruena MF, Kan CR, Himmerich H. Ketamine as a treatment for anorexia nervosa: a narrative review. Nutrients. 2021;13(11).
Martinotti G, Chiappini S, Pettorruso M, Mosca A, Miuli A, Di Carlo F, et al. Therapeutic potentials of ketamine and esketamine in obsessive-compulsive disorder (OCD), substance use disorders (SUD) and eating disorders (ED): a review of the current literature. Brain Sci. 2021;11(7):856.
Walsh Z, Mollaahmetoglu OM, Rootman J, Golsof S, Keeler J, Marsh B, et al. Ketamine for the treatment of mental health and substance use disorders: comprehensive systematic review. BJPsych Open. 2021;8(1):e19.
Hanachi M, Dicembre M, Rives-Lange C, Ropers J, Bemer P, Zazzo JF, et al. Micronutrients deficiencies in 374 severely malnourished anorexia nervosa inpatients. Nutrients. 2019;11(4):792.
Katz RL, Keen CL, Litt IF, Hurley LS, Kellams-Harrison KM, Glader LJ. Zinc deficiency in anorexia nervosa. J Adolesc Health Care. 1987;8(5):400–6.
Lask B, Fosson A, Rolfe U, Thomas S. Zinc deficiency and childhood-onset anorexia nervosa. J Clin Psychiatry. 1993;54(2):63–6.
Raatz SK, Jahns L, Johnson LK, Crosby R, Mitchell JE, Crow S, et al. Nutritional adequacy of dietary intake in women with anorexia nervosa. Nutrients. 2015;7(5):3652–65.
Doboszewska U, Wlaź P, Nowak G, Radziwoń-Zaleska M, Cui R, Młyniec K. Zinc in the monoaminergic theory of depression: its relationship to neural plasticity. Neural Plasticity. 2017; 2017: 1-18.
Petrilli MA, Kranz TM, Kleinhaus K, Joe P, Getz M, Johnson P, et al. The emerging role for zinc in depression and psychosis. Front Pharmacol. 2017;8:414.
Keeler JL, Treasure J, Juruena MF, Kan C, Himmerich H. Ketamine as a treatment for anorexia nervosa: a narrative review. Nutrients. 2021;13(11):4158.
Greenhalgh T, Thorne S, Malterud K. Time to challenge the spurious hierarchy of systematic over narrative reviews? Eur J Clin Invest. 2018;48(6): e12931.
Ferrari R. Writing narrative style literature reviews. Med Writing. 2015;24(4):230–5.
Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56(1):2–5.
Stroebel D, Paoletti P. Architecture and function of NMDA receptors: an evolutionary perspective. J Physiol. 2021;599(10):2615–38.
Gao C, Wolf ME. Dopamine receptors regulate NMDA receptor surface expression in prefrontal cortex neurons. J Neurochem. 2008;106(6):2489–501.
Zhang XM, Luo JH. GluN2A versus GluN2B: twins, but quite different. Neurosci Bull. 2013;29(6):761–72.
Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, et al. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256(5060):1217–21.
Zanos P, Moaddel R, Morris PJ, Riggs LM, Highland JN, Georgiou P, et al. Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol Rev. 2018;70(3):621–60.
Bear MF, Connors BW, Paradiso MA. Neuroscience: exploring the brain. 4th ed. Wolters Kluwer; 2015.
Hunt DL, Castillo PE. Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr Opin Neurobiol. 2012;22(3):496–508.
Merzenich MM, Van Vleet TM, Nahum M. Brain plasticity-based therapeutics. Front Hum Neurosci. 2014;8:385.
Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14(6):383–400.
Hebb DO. The organization of behavior: a neuropsychological theory. New York: Wiley; 1949. p. 378.
Keysers C, Gazzola V. Hebbian learning and predictive mirror neurons for actions, sensations and emotions. Philos Trans R Soc Lond B Biol Sci. 2014;369(1644):20130175.
Dan Y, Poo M-M. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86(3):1033–48
Koronyo-Hamaoui M, Frisch A, Stein D, Denziger Y, Leor S, Michaelovsky E, et al. Dual contribution of NR2B subunit of NMDA receptor and SK3 Ca(2+)-activated K+ channel to genetic predisposition to anorexia nervosa. J Psychiatr Res. 2007;41(1–2):160–7.
Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12(3):529–40.
Dumas TC. Developmental regulation of cognitive abilities: modified composition of the molecular switch on associative learning. Prog Neurobiol. 2005;76(3):189–211.
King JC, Brown KH, Gibson RS, Krebs NF, Lowe NM, Siekmann JH, et al. Biomarkers of nutrition for development (BOND): zinc review. J Nutr. 2015;146(4):858S-S885.
Stefanidou M, Maravelias C, Dona A, Spiliopoulou C. Zinc: a multipurpose trace element. Arch Toxicol. 2006;80(1):1–9.
Nowak G, Szewczyk B, Sadlik K, Piekoszewski W, Trela F, Florek E, et al. Reduced potency of zinc to interact with NMDA receptors in hippocampal tissue of suicide victims. Pol J Pharmacol. 2003;55(3):455–9.
Vergnano AM, Rebola N, Savtchenko LP, Pinheiro PS, Casado M, Kieffer BL, et al. Zinc dynamics and action at excitatory synapses. Neuron. 2014;82(5):1101–14.
Sensi SL, Paoletti P, Koh JY, Aizenman E, Bush AI, Hershfinkel M. The neurophysiology and pathology of brain zinc. J Neurosci. 2011;31(45):16076–85.
Siwek M, Dudek D, Schlegel-Zawadzka M, Morawska A, Piekoszewski W, Opoka W, et al. Serum zinc level in depressed patients during zinc supplementation of imipramine treatment. J Affect Disord. 2010;126(3):447–52.
Swardfager W, Herrmann N, Mazereeuw G, Goldberger K, Harimoto T, Lanctôt KL. Zinc in depression: a meta-analysis. Biol Psychiatry. 2013;74(12):872–8.
Nowak G. Does interaction between zinc and glutamate system play a significant role in the mechanism of antidepressant action? Acta Pol Pharm. 2001;58(1):73–5.
Howland JG, Wang YT. Chapter 8 Synaptic plasticity in learning and memory: stress effects in the hippocampus. Prog Brain Res. 2008;169:145–58.
Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13(1):22–37.
Martin Monzon B, Henderson LA, Madden S, Macefield VG, Touyz S, Kohn MR, et al. Grey matter volume in adolescents with anorexia nervosa and associated eating disorder symptoms. Eur J Neurosci. 2017;46(7):2297–307.
Marger L, Schubert CR, Bertrand D. Zinc: an underappreciated modulatory factor of brain function. Biochem Pharmacol. 2014;91(4):426–35.
Mlyniec K. Zinc in the glutamatergic theory of depression. Curr Neuropharmacol. 2015;13(4):505–13.
Doboszewska U, Sowa-Kućma M, Młyniec K, Pochwat B, Hołuj M, Ostachowicz B, et al. Zinc deficiency in rats is associated with up-regulation of hippocampal NMDA receptor. Prog Neuropsychopharmacol Biol Psychiatry. 2015;56:254–63.
Chen YW, Actor-Engel H, Sherpa AD, Klingensmith L, Chowdhury TG, Aoki C. NR2A- and NR2B-NMDA receptors and drebrin within postsynaptic spines of the hippocampus correlate with hunger-evoked exercise. Brain Struct Funct. 2017;222(5):2271–94.
Williams K. Separating dual effects of zinc at recombinant N-methyl-D-aspartate receptors. Neurosci Lett. 1996;215(1):9–12.
Chen N, Moshaver A, Raymond LA. Differential sensitivity of recombinant N-methyl-D-aspartate receptor subtypes to zinc inhibition. Mol Pharmacol. 1997;51(6):1015–23.
Madry C, Betz H, Geiger JR, Laube B. Supralinear potentiation of NR1/NR3A excitatory glycine receptors by Zn2+ and NR1 antagonist. Proc Natl Acad Sci U S A. 2008;105(34):12563–8.
Kehoe LA, Bernardinelli Y, Muller D. GluN3A: an NMDA receptor subunit with exquisite properties and functions. Neural Plast. 2013;2013: 145387.
Nakashima AS, Dyck RH. Zinc and cortical plasticity. Brain Res Rev. 2009;59(2):347–73.
Legendre P, Westbrook GL. Noncompetitive inhibition of γ-aminobutyric acid(A) channels by Zn. Mol Pharmacol. 1991;39(3):267–74.
Ruiz A, Walker MC, Fabian-Fine R, Kullmann DM. Endogenous zinc inhibits GABAa receptors in a hippocampal pathway. J Neurophysiol. 2004;91:1091–6.
Takeda A, Minami A, Seki Y, Oku N. Differential effects of zinc on glutamatergic and GABAergic neurotransmitter systems in the hippocampus. J Neurosci Res. 2004;75(2):225–9.
Draguhn A, Verdorn TA, Ewert M, Seeburg PH, Sakmann B. Functional and molecular distinction between recombinant rat GABAA receptor subtypes by Zn2+. Neuron. 1990;5(6):781–8.
Bailey J, Powell L, Sinanan L, Neal J, Li M, Smith T, et al. A novel mechanism of V-type zinc inhibition of glutamate dehydrogenase results from disruption of subunit interactions necessary for efficient catalysis. FEBS J. 2011;278(17):3140–51.
Prakash A, Bharti K, Majeed ABA. Zinc: indications in brain disorders. Fundam Clin Pharmacol. 2015;29(2):131–49.
Castro-Fornieles J, Bargallo N, Lazaro L, Andres S, Falcon C, Plana MT, et al. Adolescent anorexia nervosa: cross-sectional and follow-up frontal gray matter disturbances detected with proton magnetic resonance spectroscopy. J Psychiatr Res. 2007;41(11):952–8.
Ohrmann P, Kersting A, Suslow T, Lalee-Mentzel J, Donges US, Fiebich M, et al. Proton magnetic resonance spectroscopy in anorexia nervosa: correlations with cognition. NeuroReport. 2004;15(3):549–53.
Paoletti P, Ascher P, Neyton J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci. 1997;17(15):5711–25.
Robinette TM, Nicholatos JW, Francisco AB, Brooks KE, Diao RY, Sorbi S, et al. SIRT1 accelerates the progression of activity-based anorexia. Nat Commun. 2020;11(1):2814.
Xi D, Keeler B, Zhang W, Houle JD, Gao WJ. NMDA receptor subunit expression in GABAergic interneurons in the prefrontal cortex: application of laser microdissection technique. J Neurosci Methods. 2009;176(2):172–81.
Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26(5):1604–15.
Spadini S, Ferro M, Lamanna J, Malgaroli A. Activity-based anorexia animal model: a review of the main neurobiological findings. J Eat Disord. 2021;9(1):123.
Mendonca D, Chen Y-W, Varela K, Aoki C. Changes in NR2B-NMDA-receptor expression relate to individual differences in adaptability to food restriction stress. Columbia Undergrad Sci Jour. 2018; 12:1-9.
Aoki C, Chowdhury TG, Wable GS, Chen YW. Synaptic changes in the hippocampus of adolescent female rodents associated with resilience to anxiety and suppression of food restriction-evoked hyperactivity in an animal model for anorexia nervosa. Brain Res. 2017;1654(Pt B):102–15.
Chowdhury TG, Barbarich-Marsteller NC, Chan TE, Aoki C. Activity-based anorexia has differential effects on apical dendritic branching in dorsal and ventral hippocampal CA1. Brain Struct Funct. 2014;219(6):1935–45.
Mottarlini F, Targa G, Bottan G, Tarenzi B, Fumagalli F, Caffino L. Cortical reorganization of the glutamate synapse in the activity-based anorexia rat model: impact on cognition. J Neurochem. 2022;161(4):350–65.
Aoki C, Sabaliauskas N, Chowdhury T, Min JY, Colacino AR, Laurino K, et al. Adolescent female rats exhibiting activity-based anorexia express elevated levels of GABA(A) receptor α4 and δ subunits at the plasma membrane of hippocampal CA1 spines. Synapse. 2012;66(5):391–407.
Aoki C, Wable G, Chowdhury TG, Sabaliauskas NA, Laurino K, Barbarich-Marsteller NC. α4βδ-GABAARs in the hippocampal CA1 as a biomarker for resilience to activity-based anorexia. Neuroscience. 2014;265:108–23.
Wable GS, Barbarich-Marsteller NC, Chowdhury TG, Sabaliauskas NA, Farb CR, Aoki C. Excitatory synapses on dendritic shafts of the caudal basal amygdala exhibit elevated levels of GABAA receptor α4 subunits following the induction of activity-based anorexia. Synapse. 2014;68(1):1–15.
Chen YW, Wable GS, Chowdhury TG, Aoki C. Enlargement of axo-somatic contacts formed by GAD-immunoreactive axon terminals onto layer V pyramidal neurons in the medial prefrontal cortex of adolescent female mice is associated with suppression of food restriction-evoked hyperactivity and resilience to activity-based anorexia. Cereb Cortex. 2016;26(6):2574–89.
Bilash OM, Actor-Engel HS, Sherpa AD, Chen YW, Aoki C. Suppression of food restriction-evoked hyperactivity in activity-based anorexia animal model through glutamate transporters GLT-1 at excitatory synapses in the hippocampus. Synapse. 2021;75(7): e22197.
Zhang C, Li Z, Wu Z, Chen J, Wang Z, Peng D, et al. A study of N-methyl-D-aspartate receptor gene (GRIN2B) variants as predictors of treatment-resistant major depression. Psychopharmacology. 2014;231(4):685–93.
Wu Q, Zheng R, Srisai D, McKnight GS, Palmiter RD. NR2B subunit of the NMDA glutamate receptor regulates appetite in the parabrachial nucleus. Proc Natl Acad Sci U S A. 2013;110(36):14765–70.
Miller OH, Yang L, Wang CC, Hargroder EA, Zhang Y, Delpire E, et al. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elife. 2014;3: e03581.
Gass N, Becker R, Sack M, Schwarz AJ, Reinwald J, Cosa-Linan A, et al. Antagonism at the NR2B subunit of NMDA receptors induces increased connectivity of the prefrontal and subcortical regions regulating reward behavior. Psychopharmacology. 2018;235(4):1055–68.
Monaco SA, Gulchina Y, Gao W-J. NR2B subunit in the prefrontal cortex: a double-edged sword for working memory function and psychiatric disorders. Neurosci Biobehav Rev. 2015;56:127-38.
Scharner S, Stengel A. Animal models for anorexia nervosa: a systematic review. Front Hum Neurosci. 2020;14: 596381.
Dalton B, Bartholdy S, Robinson L, Solmi M, Ibrahim MAA, Breen G, et al. A meta-analysis of cytokine concentrations in eating disorders. J Psychiatr Res. 2018;103:252–64.
Solmi M, Veronese N, Favaro A, Santonastaso P, Manzato E, Sergi G, et al. Inflammatory cytokines and anorexia nervosa: a meta-analysis of cross-sectional and longitudinal studies. Psychoneuroendocrinology. 2015;51:237–52.
Liuzzi JP, Cousins RJ. Mammalian zinc transporters. Annu Rev Nutr. 2004; 24: 151–72.
Kaye WH, Wierenga CE, Bailer UF, Simmons AN, Bischoff-Grethe A. Nothing tastes as good as skinny feels: the neurobiology of anorexia nervosa. Trends Neurosci. 2013;36(2):110–20.
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16(3):675–86.
Schousboe A, Waagepetersen HS. Role of astrocytes in glutamate homeostasis: implications for excitotoxicity. Neurotox Res. 2005;8(3–4):221–5.
Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14(6):724–38.
Taber KA, Hurley RA. Astroglia: not just glue. J Neuropsych Clin Neurosci. 2008;20(2):iv–129.
Harada K, Kamiya T, Tsuboi T. Gliotransmitter release from astrocytes: functional, developmental and pathological implications in the brain. Front Neurosci. 2016;9:499.
Kogel V, Trinh S, Gasterich N, Beyer C, Seitz J. Long-term glucose starvation induces inflammatory responses and phenotype switch in primary cortical rat astrocytes. J Mol Neurosci. 2021;71(11):2368–82.
Hellerhoff I, King JA, Tam FI, Pauligk S, Seidel M, Geisler D, et al. Differential longitudinal changes of neuronal and glial damage markers in anorexia nervosa after partial weight restoration. Transl Psychiatry. 2021;11(1):86.
Jacquemot A, Park R. The role of interoception in the pathogenesis and treatment of anorexia nervosa: a narrative review. Front Psychiatry. 2020;11:281.
Frank GK. Altered brain reward circuits in eating disorders: chicken or egg? Curr Psychiatry Rep. 2013;15(10):396.
Gaudio S, Wiemerslage L, Brooks SJ, Schiöth HB. A systematic review of resting-state functional-MRI studies in anorexia nervosa: evidence for functional connectivity impairment in cognitive control and visuospatial and body-signal integration. Neurosci Biobehav Rev. 2016;71:578–89.
Olivo G, Gaudio S, Schioth HB. Brain and cognitive development in adolescents with anorexia nervosa: a systematic review of fMRI studies. Nutrients. 2019;11(8):1907.
O’Hara CB, Campbell IC, Schmidt U. A reward-centred model of anorexia nervosa: a focussed narrative review of the neurological and psychophysiological literature. Neurosci Biobehav Rev. 2015;52:131–52.
Fladung AK, Schulze UME, Schöll F, Bauer K, Grön G. Role of the ventral striatum in developing anorexia nervosa. Transl Psychiatry. 2013;3(10): e315.
Lawson EA, Holsen LM, Desanti R, Santin M, Meenaghan E, Herzog DB, et al. Increased hypothalamic-pituitary-adrenal drive is associated with decreased appetite and hypoactivation of food-motivation neurocircuitry in anorexia nervosa. Eur J Endocrinol. 2013;169(5):639–47.
Kaye WH, Fudge JL, Paulus M. New insights into symptoms and neurocircuit function of anorexia nervosa. Nat Rev Neurosci. 2009;10(8):573–84.
Gaudio S, Quattrocchi CC. Neural basis of a multidimensional model of body image distortion in anorexia nervosa. Neurosci Biobehav Rev. 2012;36(8):1839–47.
Dann KM, Hay P, Touyz S. Are poor set-shifting and central coherence associated with everyday function in anorexia nervosa? A systematic review. J Eat Disord. 2021;9(1):40.
Wu M, Brockmeyer T, Hartmann M, Skunde M, Herzog W, Friederich HC. Set-shifting ability across the spectrum of eating disorders and in overweight and obesity: a systematic review and meta-analysis. Psychol Med. 2014;44(16):3365–85.
Lang K, Roberts M, Harrison A, Lopez C, Goddard E, Khondoker M, et al. Central coherence in eating disorders: a synthesis of studies using the Rey Osterrieth Complex Figure Test. PLoS ONE. 2016;11(11): e0165467.
Frank GKW, DeGuzman MC, Shott ME, Laudenslager ML, Rossi B, Pryor T. Association of brain reward learning response with harm avoidance, weight gain, and hypothalamic effective connectivity in adolescent anorexia nervosa. JAMA Psychiat. 2018;75(10):1071–80.
Danner UN, Sternheim L, Evers C. The importance of distinguishing between the different eating disorders (sub)types when assessing emotion regulation strategies. Psychiatry Res. 2014;215(3):727–32.
Westwater ML, Murley AG, Diederen KMJ, Carpenter TA, Ziauddeen H, Fletcher PC. Characterizing cerebral metabolite profiles in anorexia and bulimia nervosa and their associations with habitual behavior. Transl Psychiatry. 2022;12(1):103.
Keeler JL, Robinson L, Keeler-Schäffeler R, Dalton B, Treasure J, Himmerich H. Growth factors in anorexia nervosa: a systematic review and meta-analysis of cross-sectional and longitudinal data. World J Biol Psychiatry. 2022; 23(8): 1–19.
Lu Y, Christian K, Lu B. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem. 2008;89(3):312–23.
Nakazato M, Tchanturia K, Schmidt U, Campbell IC, Treasure J, Collier DA, et al. Brain-derived neurotrophic factor (BDNF) and set-shifting in currently ill and recovered anorexia nervosa (AN) patients. Psychol Med. 2009;39(6):1029–35.
Monteleone P, Fabrazzo M, Martiadis V, Serritella C, Pannuto M, Maj M. Circulating brain-derived neurotrophic factor is decreased in women with anorexia and bulimia nervosa but not in women with binge-eating disorder: relationships to co-morbid depression, psychopathology and hormonal variables. Psychol Med. 2005;35(6):897–905.
Huang YZ, Pan E, Xiong ZQ, McNamara JO. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron. 2008;57(4):546–58.
Gorski JA, Zeiler SR, Tamowski S, Jones KR. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci. 2003;23(17):6856–65.
Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, et al. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000;26(1):233–45.
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475(7354):91–5.
Maeng S, Zarate CA Jr, Du J, Schloesser RJ, McCammon J, Chen G, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63(4):349–52.
Cain SW, Chang A-M, Vlasac I, Tare A, Anderson C, Czeisler CA, et al. Circadian rhythms in plasma brain-derived neurotrophic factor differ in men and women. J Biol Rhythms. 2017;32(1):75–82.
Piccinni A, Marazziti D, Del Debbio A, Bianchi C, Roncaglia I, Mannari C, et al. Diurnal variation of plasma brain-derived neurotrophic factor (BDNF) in humans: an analysis of sex differences. Chronobiol Int. 2008;25(5):819–26.
Amadio P, Sandrini L, Ieraci A, Tremoli E, Barbieri SS. Effect of clotting duration and temperature on BDNF measurement in human serum. Int J Mol Sci. 2017;18(9):1987.
Miranda M, Morici JF, Zanoni MB, Bekinschtein P. Brain-derived neurotrophic factor: a key molecule for memory in the healthy and the pathological brain. Front Cell Neurosci. 2019;13:363.
Mustapic M, Eitan E, Werner JK, Berkowitz ST, Lazaropoulos MP, Tran J, et al. Plasma extracellular vesicles enriched for neuronal origin: a potential window into brain pathologic processes. Front Neurosci. 2017;11:278.
Polacchini A, Metelli G, Francavilla R, Baj G, Florean M, Mascaretti LG, et al. A method for reproducible measurements of serum BDNF: comparison of the performance of six commercial assays. Sci Rep. 2015;5(1):17989.
Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305(5682):390–2.
Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35.
Abdallah CG, Sanacora G, Duman RS, Krystal JH. The neurobiology of depression, ketamine and rapid-acting antidepressants: Is it glutamate inhibition or activation? Pharmacol Ther. 2018;190:148–58.
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–64.
Ghosh HS, McBurney M, Robbins PD. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS ONE. 2010;5(2): e9199.
Misra M, Klibanski A. Anorexia nervosa and its associated endocrinopathy in young people. Horm Res Paediatr. 2016;85(3):147–57.
Mariani S, di Giorgio MR, Martini P, Persichetti A, Barbaro G, Basciani S, et al. Inverse association of circulating SIRT1 and adiposity: a study on underweight, normal weight, and obese patients. Front Endocrinol. 2018;9:449.
Cantó C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22(1):31–53.
Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310(5754):1641.
Cohen DE, Supinski AM, Bonkowski MS, Donmez G, Guarente LP. Neuronal SIRT1 regulates endocrine and behavioral responses to calorie restriction. Genes Dev. 2009;23(24):2812–7.
Libert S, Pointer K, Bell EL, Das A, Cohen DE, Asara JM, et al. SIRT1 activates MAO-A in the brain to mediate anxiety and exploratory drive. Cell. 2011;147(7):1459–72.
Zanos P, Gould TD. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry. 2018;23(4):801–11.
Shinohara R, Aghajanian GK, Abdallah CG. Neurobiology of the rapid-acting antidepressant effects of ketamine: impact and opportunities. Biol Psychiatry. 2021;90(2):85–95.
van der Graaf M. In vivo magnetic resonance spectroscopy: basic methodology and clinical applications. Eur Biophys J. 2010;39(4):527–40.
Rodriguez CI, Kegeles LS, Levinson A, Ogden RT, Mao X, Milak MS, et al. In vivo effects of ketamine on glutamate-glutamine and gamma-aminobutyric acid in obsessive-compulsive disorder: proof of concept. Psychiatry Res. 2015;233(2):141–7.
Grzelak P, Gajewicz W, Wyszogrodzka-Kucharska A, Rotkiewicz A, Stefańczyk L, Góraj B, et al. Brain metabolism alterations in patients with anorexia nervosa observed in 1H-MRS. Psychiatr Pol. 2005;39(4):761–71.
Möckel R, Schlemmer HP, Gückel F, Göpel C, Becker G, Köpke J, et al. 1H-MR spectroscopy in anorexia nervosa: reversible cerebral metabolic changes. Rofo. 1999;170(4):371–7.
Hentschel J, Möckel R, Schlemmer HP, Markus A, Göpel C, Gückel F, et al. 1H-MR spectroscopy in anorexia nervosa: the characteristic differences between patients and healthy subjects. Rofo. 1999;170(3):284–9.
Rzanny R, Freesmeyer D, Reichenbach JR, Mentzel HJ, Pfleiderer SO, Klemm S, et al. 31P-MR spectroscopy of the brain in patients with anorexia nervosa: characteristic differences in the spectra between patients and healthy control subjects. Rofo. 2003;175(1):75–82.
Blasel S, Pilatus U, Magerkurth J, von Stauffenberg M, Vronski D, Mueller M, et al. Metabolic gray matter changes of adolescents with anorexia nervosa in combined MR proton and phosphorus spectroscopy. Neuroradiology. 2012;54(7):753–64.
Joos AA, Perlov E, Buchert M, Hartmann A, Saum B, Glauche V, et al. Magnetic resonance spectroscopy of the anterior cingulate cortex in eating disorders. Psychiatry Res. 2011;191(3):196–200.
Roser W, Bubl R, Buergin D, Seelig J, Radue EW, Rost B. Metabolic changes in the brain of patients with anorexia and bulimia nervosa as detected by proton magnetic resonance spectroscopy. Int J Eat Disord. 1999;26(2):119-36.
Castro-Fornieles J, Garcia AI, Lazaro L, Andres-Perpina S, Falcon C, Plana MT, et al. Prefrontal brain metabolites in short-term weight-recovered adolescent anorexia nervosa patients. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(6):1049–53.
Florent V, Baroncini M, Jissendi-Tchofo P, Lopes R, Vanhoutte M, Rasika S, et al. Hypothalamic structural and functional imbalances in anorexia nervosa. Neuroendocrinology. 2020;110(6):552–62.
Peek AL, Rebbeck T, Puts NA, Watson J, Aguila MR, Leaver AM. Brain GABA and glutamate levels across pain conditions: a systematic literature review and meta-analysis of 1H-MRS studies using the MRS-Q quality assessment tool. Neuroimage. 2020;210: 116532.
Schlemmer H-P, Mockel R, Marcus A, Hentschel F, Gopel C, Becker G, et al. Proton magnetic resonance spectroscopy in acute, juvenile anorexia nervosa. Psychiatry Res.1998;82(3):171-9.
Ford TC, Crewther DP. A comprehensive review of the (1)H-MRS metabolite spectrum in autism spectrum disorder. Front Mol Neurosci. 2016;9:14.
Bogner W, Hangel G, Esmaeili M, Andronesi OC. 1D-spectral editing and 2D multispectral in vivo(1)H-MRS and (1)H-MRSI: methods and applications. Anal Biochem. 2017;529:48–64.
Lin A, Andronesi O, Bogner W, Choi I-Y, Coello E, Cudalbu C, et al. Minimum reporting standards for in vivo magnetic resonance spectroscopy (MRSinMRS): experts’ consensus recommendations. NMR Biomed. 2021;34(5):e4484.
Birmingham CL, Gritzner S. How does zinc supplementation benefit anorexia nervosa? Eating and weight disorders: studies on anorexia, bulimia and obesity. 2006;11(4):e109–11.
Birmingham CL, Goldner EM, Bakan R. Controlled trial of zinc supplementation in anorexia nervosa. Int J Eat Disord. 1994;15(3):251–5.
Takeda A, Tamano H, Oku N. Involvement of unusual glutamate release in kainate-induced seizures in zinc-deficient adult rats. Epilepsy Res. 2005;66(1–3):137–43.
Suh SW, Won SJ, Hamby AM, Yoo BH, Fan Y, Sheline CT, et al. Decreased brain zinc availability reduces hippocampal neurogenesis in mice and rats. J Cerebral Blood Flow Metab. 2009;29(9):1579–88.
Szewczyk B, Poleszak E, Sowa-Kućma M, Wróbel A, Słotwiński S, Listos J, et al. The involvement of NMDA and AMPA receptors in the mechanism of antidepressant-like action of zinc in the forced swim test. Amino Acids. 2010;39(1):205–17.
Szewczyk B, Pochwat B, Rafało A, Palucha-Poniewiera A, Domin H, Nowak G. Activation of mTOR dependent signaling pathway is a necessary mechanism of antidepressant-like activity of zinc. Neuropharmacology. 2015;99:517–26.
Młyniec K, Budziszewska B, Holst B, Ostachowicz B, Nowak G. GPR39 (zinc receptor) knockout mice exhibit depression-like behavior and CREB/BDNF down-regulation in the hippocampus. Int J Neuropsychopharmacol. 2014;18(3):pyu002.
Młyniec K, Doboszewska U, Szewczyk B, Sowa-Kućma M, Misztak P, Piekoszewski W, et al. The involvement of the GPR39-Zn(2+)-sensing receptor in the pathophysiology of depression: studies in rodent models and suicide victims. Neuropharmacology. 2014;79:290–7.
Tassabehji NM, Corniola RS, Alshingiti A, Levenson CW. Zinc deficiency induces depression-like symptoms in adult rats. Physiol Behav. 2008;95(3):365–9.
Szewczyk B, Poleszak E, Wlaź P, Wróbel A, Blicharska E, Cichy A, et al. The involvement of serotonergic system in the antidepressant effect of zinc in the forced swim test. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(2):323–9.
Safai-Kutti S. Oral zinc supplementation in anorexia nervosa. Acta Psychiatr Scand Suppl. 1990;361:14–7.
Sawada T, Yokoi K. Effect of zinc supplementation on mood states in young women: a pilot study. Eur J Clin Nutr. 2010;64(3):331–3.
Solati Z, Jazayeri S, Tehrani-Doost M, Mahmoodianfard S, Gohari MR. Zinc monotherapy increases serum brainderived neurotrophic factor (BDNF) levels and decreases depressive symptoms in overweight or obese subjects: a double-blind, randomized, placebo-controlled trial. Nutr Neurosci. 2015;18(4):162–8.
Ranjbar E, Shams J, Sabetkasaei M, M-Shirazi M, Rashidkhani B, Mostafavi A, et al. Effects of zinc supplementation on efficacy of antidepressant therapy, inflammatory cytokines, and brain-derived neurotrophic factor in patients with major depression. Nutr Neurosci. 2014;17(2):65–71.
Siwek M, Dudek D, Paul IA, Sowa-Kućma M, Zieba A, Popik P, et al. Zinc supplementation augments efficacy of imipramine in treatment resistant patients: a double blind, placebo-controlled study. J Affect Disord. 2009;118(1–3):187–95.
Nowak G, Siwek M, Dudek D, Zieba A, Pilc A. Effect of zinc supplementation on antidepressant therapy in unipolar depression: a preliminary placebo-controlled study. Polish J Pharmacol. 2003;55(6):1143–7.
Mortazavi M, Farzin D, Zarhghami M, Hosseini SH, Mansoori P, Nateghi G. Efficacy of zinc sulfate as an add-on therapy to risperidone versus risperidone alone in patients with schizophrenia: a double-blind randomized placebo-controlled trial. Iranian J Psychiatry Behav Sci. 2015;9(3).
Colla M, Scheerer H, Weidt S, Seifritz E, Kronenberg G. Novel insights into the neurobiology of the antidepressant response from ketamine research: a mini review. Front Behav Neurosci. 2021;15: 759466.
Driver C, Jackson TNW, Lagopoulos J, Hermens DF. Molecular mechanisms underlying the N-methyl-D-aspartate receptor antagonists: highlight their potential for transdiagnostic therapeutics. Prog Neuropsychopharmacol Biol Psychiatry. 2022; 119: 1-8.
Seamans J. Losing inhibition with ketamine. Nat Chem Biol. 2008;4(2):91–3.
Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27(43):11496–500.
Gerhard DM, Pothula S, Liu RJ, Wu M, Li XY, Girgenti MJ, et al. GABA interneurons are the cellular trigger for ketamine’s rapid antidepressant actions. J Clin Invest. 2020;130(3):1336–49.
Hong LE, Summerfelt A, Buchanan RW, O’Donnell P, Thaker GK, Weiler MA, et al. Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology. 2010;35(3):632–40.
Chowdhury GM, Zhang J, Thomas M, Banasr M, Ma X, Pittman B, et al. Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Mol Psychiatry. 2017;22(1):120–6.
Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17(8):2921–7.
Taha E, Gildish I, Gal-Ben-Ari S, Rosenblum K. The role of eEF2 pathway in learning and synaptic plasticity. Neurobiol Learn Mem. 2013;105:100–6.
Liu RJ, Lee FS, Li XY, Bambico F, Duman RS, Aghajanian GK. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry. 2012;71(11):996–1005.
Nishitani N, Nagayasu K, Asaoka N, Yamashiro M, Shirakawa H, Nakagawa T, et al. Raphe AMPA receptors and nicotinic acetylcholine receptors mediate ketamine-induced serotonin release in the rat prefrontal cortex. Int J Neuropsychopharmacol. 2014;17(8):1321–6.
Gigliucci V, O’Dowd G, Casey S, Egan D, Gibney S, Harkin A. Ketamine elicits sustained antidepressant-like activity via a serotonin-dependent mechanism. Psychopharmacology. 2013;228(1):157–66.
Ago Y, Tanabe W, Higuchi M, Tsukada S, Tanaka T, Yamaguchi T, et al. (R)-ketamine induces a greater increase in prefrontal 5-HT release than (S)-ketamine and ketamine metabolites via an AMPA receptor-independent mechanism. Int J Neuropsychopharmacol. 2019;22(10):665–74.
Witkin JM, Monn JA, Schoepp DD, Li X, Overshiner C, Mitchell SN, et al. The rapidly acting antidepressant ketamine and the mGlu2/3 receptor antagonist LY341495 rapidly engage dopaminergic mood circuits. J Pharmacol Exp Ther. 2016;358(1):71–82.
Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25(32):7342–51.
Lavin A, Grace AA. Stimulation of D1-type dopamine receptors enhances excitability in prefrontal cortical pyramidal neurons in a state-dependent manner. Neuroscience. 2001;104(2):335–46.
Pothula S, Kato T, Liu RJ, Wu M, Gerhard D, Shinohara R, et al. Cell-type specific modulation of NMDA receptors triggers antidepressant actions. Mol Psychiatry. 2021;26(9):5097–111.
Skiteva O, Yao N, Chergui K. Ketamine induces opposite changes in AMPA receptor calcium permeability in the ventral tegmental area and nucleus accumbens. Transl Psychiatry. 2021;11(1):530.
Budygin EA, Bass CE, Grinevich VP, Deal AL, Bonin KD, Weiner JL. Opposite consequences of tonic and phasic increases in accumbal dopamine on alcohol-seeking behavior. iScience. 2020;23(3):100877.
Bass CE, Grinevich VP, Gioia D, Day-Brown JD, Bonin KD, Stuber GD, et al. Optogenetic stimulation of VTA dopamine neurons reveals that tonic but not phasic patterns of dopamine transmission reduce ethanol self-administration. Front Behav Neurosci. 2013;7:173.
Abdallah CG, Jackowski A, Salas R, Gupta S, Sato JR, Mao X, et al. The nucleus accumbens and ketamine treatment in major depressive disorder. Neuropsychopharmacology. 2017;42(8):1739–46.
Gallay CC, Forsyth G, Can AT, Dutton M, Jamieson D, Jensen E, et al. Six-week oral ketamine treatment for chronic suicidality is associated with increased grey matter volume. Psychiatry Res Neuroimaging. 2021;317: 111369.
Ramakrishnan N, Murphy NRE, Walker CP, Cuellar Leal VA, Soares JC, Cho RYJ, et al. Neurophysiological effect of ketamine on prefrontal cortex in treatment-resistant depression: a combined transcranial magnetic stimulation-electroencephalography study. Chronic Stress (Thousand Oaks). 2019;3:2470547019861417.
Zhou Y-L, Wu F-C, Liu W-J, Zheng W, Wang C-Y, Zhan Y-N, et al. Volumetric changes in subcortical structures following repeated ketamine treatment in patients with major depressive disorder: a longitudinal analysis. Transl Psychiatry. 2020;10(1):264.
Becker R, Braun U, Schwarz AJ, Gass N, Schweiger JI, Weber-Fahr W, et al. Species-conserved reconfigurations of brain network topology induced by ketamine. Transl Psychiatry. 2016;6: e786.
Biezonski D, Cha J, Steinglass J, Posner J. Evidence for thalamocortical circuit abnormalities and associated cognitive dysfunctions in underweight individuals with anorexia nervosa. Neuropsychopharmacology. 2016;41(6):1560–8.
Krug S, Müller T, Kayali Ö, Leichter E, Peschel SKV, Jahn N, et al. Altered functional connectivity in common resting-state networks in patients with major depressive disorder: a resting-state functional connectivity study. J Psychiatr Res. 2022;155:33–41.
Su T, Gong J, Tang G, Qiu S, Chen P, Chen G, et al. Structural and functional brain alterations in anorexia nervosa: a multimodal meta-analysis of neuroimaging studies. Hum Brain Mapping. 2021;42(15):5154–69.
Milak MS, Rashid R, Dong Z, Kegeles LS, Grunebaum MF, Ogden RT, et al. Assessment of relationship of ketamine dose with magnetic resonance spectroscopy of Glx and GABA responses in adults with major depression: a randomized clinical trial. JAMA Netw Open. 2020;3(8): e2013211.
Li M, Demenescu LR, Colic L, Metzger CD, Heinze HJ, Steiner J, et al. Temporal dynamics of antidepressant ketamine effects on glutamine cycling follow regional fingerprints of AMPA and NMDA receptor densities. Neuropsychopharmacology. 2017;42(6):1201–9.
Weigand A, Gartner M, Scheidegger M, Wyss PO, Henning A, Seifritz E, et al. Predicting antidepressant effects of ketamine: the role of the pregenual anterior cingulate cortex as a multimodal neuroimaging biomarker. Int J Neuropsychopharmacol. 2022; 25(12): 1003-13.
Evans JW, Lally N, An L, Li N, Nugent AC, Banerjee D, et al. 7T (1)H-MRS in major depressive disorder: a ketamine treatment study. Neuropsychopharmacology. 2018;43(9):1908–14.
Gärtner M, Weigand A, Scheidegger M, Lehmann M, Wyss PO, Wunder A, et al. Acute effects of ketamine on the pregenual anterior cingulate: linking spontaneous activation, functional connectivity, and glutamate metabolism. Eur Arch Psychiatry Clin Neurosci. 2022;272(4):703–14.
Taylor MJ, Tiangga ER, Mhuircheartaigh RN, Cowen PJ. Lack of effect of ketamine on cortical glutamate and glutamine in healthy volunteers: a proton magnetic resonance spectroscopy study. J Psychopharmacol. 2012;26(5):733–7.
Gautron L, Laye S. Neurobiology of inflammation-associated anorexia. 2010;3.
Chen YW, Sherpa AD, Aoki C. Single injection of ketamine during mid-adolescence promotes long-lasting resilience to activity-based anorexia of female mice by increasing food intake and attenuating hyperactivity as well as anxiety-like behavior. Int J Eating Disord. 2018;51(8):1020–5.
Grunebaum MF, Galfalvy HC, Choo TH, Keilp JG, Moitra VK, Parris MS, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327–35.
Can AT, Hermens DF, Dutton M, Gallay CC, Jensen E, Jones M, et al. Low dose oral ketamine treatment in chronic suicidality: an open-label pilot study. Transl Psychiatry. 2021;11(1):101.
Chen MH, Li CT, Lin WC, Hong CJ, Tu PC, Bai YM, et al. Rapid inflammation modulation and antidepressant efficacy of a low-dose ketamine infusion in treatment-resistant depression: a randomized, double-blind control study. Psychiatry Res. 2018;269:207–11.
Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen P Psychiatry. 2006;63(8):856–64.
Schwartz T, Trunko ME, Feifel D, Lopez E, Peterson D, Frank GKW, et al. A longitudinal case series of IM ketamine for patients with severe and enduring eating disorders and comorbid treatment-resistant depression. Clin Case Rep. 2021;9(5): e03869.
Bottemanne H, Morlaas O, Claret A, Sharot T, Fossati P, Schmidt L. Evaluation of early ketamine effects on belief-updating biases in patients with treatment-resistant depression. JAMA Psychiat. 2022;79(11):1124–32.
Mills IH, Park GR, Manara AR, Merriman RJ. Treatment of compulsive behaviour in eating disorders with intermittent ketamine infusions. QJM. 1998;91(7):493–503.
Dechant E, Boyle B, A Ross R. Ketamine in a patient with comorbid anorexia and MDD. J Womens Health Dev. 2020;03(03): 373-75.
Scolnick B, Zupec-Kania B, Calabrese L, Aoki C, Hildebrandt T. Remission from chronic anorexia nervosa with ketogenic diet and ketamine: case report. Front Psychiatry. 2020;11:763.
Ragnhildstveit A, Jackson LK, Cunningham S, Good L, Tanner Q, Roughan M, et al. Case report: unexpected remission from extreme and enduring bulimia nervosa with repeated ketamine assisted psychotherapy. Front Psychiatry. 2021;12:764112.
Robison R, Lafrance A, Brendle M, Smith M, Moore C, Ahuja S, et al. A case series of group-based ketamine-assisted psychotherapy for patients in residential treatment for eating disorders with comorbid depression and anxiety disorders. J Eat Disord. 2022;10(1):65.
Calabrese L, Scolnick B, Zupec-Kania B, Beckwith C, Costello K, Frank GKW. Ketogenic diet and ketamine infusion treatment to target chronic persistent eating disorder psychopathology in anorexia nervosa: a pilot study. Eat Weight Disord. 2022; 27(8): 3751-57.
Joshi R, Marvin W. Apnea with ketamine sedation in a patient with severe anorexia nervosa: a case report. Eat Weight Disord. 2021; 27: 387-89.
Katalinic N, Lai R, Somogyi A, Mitchell PB, Glue P, Loo CK. Ketamine as a new treatment for depression: a review of its efficacy and adverse effects. Aust N Z J Psychiatry. 2013;47(8):710–27.
Swainson J, Klassen LJ, Brennan S, Chokka P, Katzman MA, Tanguay RL, et al. Non-parenteral ketamine for depression: a practical discussion on addiction potential and recommendations for judicious prescribing. CNS Drugs. 2022; 36(3): 1–13.
Paz RD, Tardito S, Atzori M, Tseng KY. Glutamatergic dysfunction in schizophrenia: from basic neuroscience to clinical psychopharmacology. Eur Neuropsychopharmacol. 2008;18(11):773–86.
Kato T, Shioiri T, Murashita J, Inubushi T. Phosphorus-31 magnetic resonance spectroscopic observations in 4 cases with anorexia nervosa. Prog Neuropsychopharmacol Biol Psychiatry. 1997;21(4):719–24.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This work forms part of the corresponding author’s PhD supported by the Australian Government Research Training Program scholarship. Open access publication was funded by University of the Sunshine Coast.
Conflicts of Interest/Competing Interests
All authors declare that they have no competing interests.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent to Publication
Not applicable.
Availability of Data and Material
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Code Availability
Not applicable.
Author’ Contributions
All authors contributed to the conceptualisation, writing, review and editing of the finalised manuscript. JSM developed the original draft and was responsible for the database search, study identification and development of associated visuals, in addition to the writing, review and editing of the finalised manuscript. All authors have read and approved the final version of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Mitchell, J.S., Hermens, D.F., Bennett, M.R. et al. Ketamine and Zinc: Treatment of Anorexia Nervosa Via Dual NMDA Receptor Modulation. CNS Drugs 37, 159–180 (2023). https://doi.org/10.1007/s40263-022-00984-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-022-00984-4