
Abstract
Precision medicine is an old concept, but it is not widely applied across human health conditions as yet. Numerous attempts have been made to apply precision medicine in epilepsy, this has been based on a better understanding of aetiological mechanisms and deconstructing disease into multiple biological subsets. The scope of precision medicine is to provide effective strategies for treating individual patients with specific agent(s) that are likely to work best based on the causal biological make-up. We provide an overview of the main applications of precision medicine in epilepsy, including the current limitations and pitfalls, and propose potential strategies for implementation and to achieve a higher rate of success in patient care. Such strategies include establishing a definition of precision medicine and its outcomes; learning from past experiences, from failures and from other fields (e.g. oncology); using appropriate precision medicine strategies (e.g. drug repurposing versus traditional drug discovery process); and using adequate methods to assess efficacy (e.g. randomised controlled trials versus alternative trial designs). Although the progress of diagnostic techniques now allows comprehensive characterisation of each individual epilepsy condition from a molecular, biological, structural and clinical perspective, there remain challenges in the integration of individual data in clinical practice to achieve effective applications of precision medicine in this domain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Precision medicine in epilepsy is based on the understanding of causal mechanisms. |
The scope of precision medicine is to provide effective strategies for treating individual patients with specific agent(s) that are likely to work best based on the causal biological make-up. |
Strategies to improve precision medicine in epilepsy include establishing a unanimous definition of its concept and outcomes; learning from past experiences, from failures and from other fields; using appropriate precision medicine strategies; and using adequate methods to assess efficacy not limited to seizure control. |
1 Introduction
In the current era of genomic medicine, advances in various sequencing strategies, including whole genome sequencing (WGS), exome sequencing (ES) and targeted resequencing have led to major progress in many areas of medicine, including management and development and application of targeted treatments.
The central principle of precision medicine (PM) was already recognised thousands of years ago when Hippocrates fostered a rational and targeted therapeutic strategy since human beings are innately different from one another, and this individuality affects both their predisposition to disease and their response to therapeutics [1]. From traditional Chinese and Islamic medicine to European humoral theory, physicians have long attempted to tailor treatment recommendations to patients’ specific characteristics and particular manifestations of disease.
Various definitions of PM have been proposed, including PM as a holistic approach that uses a person’s genetics, environment, and lifestyle to help determine the best initiatives to prevent or treat disease [2], and PM as an approach ‘to improve stratification and timing of health care by utilising biological information and biomarkers at the level of molecular disease pathways, genetics, proteomics as well as metabolomics’ [3]. According to most PM definitions, the underlying genetic architecture can affect treatment response in each disease, but we do not have systematic evidence for this assumption. Furthermore, the PM concept is referred to with several other names such as genomic, personalised, targeted, stratified or differentiated medicine, although they are not all strictly equivalent. Another used term is ‘P4 Medicine’, which refers to the wider assemblage of making medicine predictive, preventive, personalised and participatory [4]. P4 medicine requires a systems approach to medicine which includes the use of deep phenotyping to characterise the complexities of disease, the use of network biology to understand the mechanistic underpinnings of various types of diseases and identify biomarkers and candidate drug targets, and the creation and integration of new technologies and computational platforms [5, 6]. Pharmacogenetics is one of the applications of PM and involves the study of how the underlying genomic architecture affects the response to drugs and susceptibility to adverse reactions [7]. Although genomics has helped identifying the causes of many diseases, there is still a huge gap between identifying genes associated with a disease and developing a successful targeted treatment for it. We provide an overview of the state of the art of PM in epilepsy and discuss strategies to improve the current approaches.
2 PM in Epilepsy: Current Applications and Limitations
Epilepsy affects around 50 million people worldwide, and accounts for 0.5% of the global burden of disease [8]. The epilepsies are a large heterogeneous group of diseases characterised by many different aetiologies, seizure types and severity, and are often manifested as syndromes. A high level of interindividual variability in the clinical presentation is observed, even within the same aetiology [9, 10].
Despite these complexities, identification of a monogenic aetiology for epilepsy syndromes can sometimes lead to a treatment strategy that targets the underlying pathophysiology and in turn corrects or improves at least some phenotypic features [11]. There are multiple examples of successful PM applications in epilepsy, including metabolic conditions, channelopathies and diseases caused by variants in the mechanistic target of rapamycin (mTOR) pathway genes (reviewed in [10, 12]). We provide some examples for each category below and in Table 1.
Inborn errors of metabolism include epilepsy as one of the commonest symptoms. They present usually in the neonatal period or infancy but can manifest at any time, even in adulthood. In a few metabolic disorders, epilepsy responds to specific treatments based on diet or supplementation of cofactors (vitamin-responsive epilepsies). For example, pyridoxine (vitamin B6)-dependent epilepsy is caused by biallelic variants in the ALDH7A1 gene, which encodes antiquitin. Deficiency of antiquitin causes seizures because of accumulating 1-piperideine-6-carboxylate condenses with pyridoxal 5′-phosphate (PLP) and inactivating this latter enzyme cofactor, which is essential for normal metabolism of neurotransmitters. Seizures do not respond to antiseizure medications (ASMs) but are responsive clinically and electrographically to large daily supplements of pyridoxine (vitamin B6) [13]. B6-responsive seizures may also be due to variants in the pyridox(am)ine 5′-phosphate oxidase (PNPO) gene. In ‘classic’ PNPO deficiency, seizures (including status epilepticus) often begin on the first day of life and typically before the age of 2 weeks, but a later onset is possible. Independent of age of onset, seizures respond to life-long treatment with either PLP or pyridoxine [14]. In GLUT1-deficiency syndrome, caused by heterozygous variants in SLC2A1, there is high phenotypic variability including epilepsy and movement disorders. SLC2A1 variants have been identified in up to 10% of individuals with early-onset absence epilepsy and in about 1% of idiopathic generalised epilepsies (IGEs) overall [15, 16]. In this condition, ketogenic diet improves seizure control and abnormal movements, although its benefit on neurodevelopment remains controversial [17, 18]. Given the IGE-like presentation, there is a great risk of misdiagnosis or underdiagnosis, reducing the chance of successful early PM application.
Ion channels constitute a broad category of transmembrane proteins that share the physiological properties of regulating ion flux across cell membranes and involvement in a diverse range of cellular processes. The clinical features and age of presentation of channelopathies are dependent on the physiological role of the ion channel in question, as well as the tissue- and age-specific expression of its gene(s). Many different mechanisms may lead to ion channel dysfunction, with a complex interplay between genetic, epigenetic and environmental factors, often with unclear genotype–phenotype associations [19]. KCNT1 gain-of-function variants cause early onset epileptic encephalopathies including epilepsy of infancy with migrating focal seizures [20]. KCNT1 encodes a sodium-dependent potassium channel (KNa1.1) and is activated by increased intracellular chloride and sodium concentrations; it is responsible for the slow hyperpolarisation of the transmembrane potential during action potentials. In vitro testing has indicated that the electrophysiological defect of at least some of these variants may be reversed by quinidine, an antiarrhythmic drug, which is a partial blocker of KNa1.1 [21]. Quinidine administration resulted in decreased seizure frequency or freedom from seizures and improved psychomotor development in a few patients with epilepsy of infancy with migrating focal seizures due to KCNT1 variants [22]. However, in an observational study of 43 individuals with KCNT1-related epilepsies, treatment with quinidine resulted in significant seizure reduction in only 20% of individuals, an effect that was not significantly different to non-precision treatments [23]. In a clinical trial of quinidine in six individuals with sleep-related hypermotor epilepsy, another KCNT1-related phenotype, treatment with quinidine was limited by cardiac side effects and did not result in a significant seizure reduction [24]. Such contradictory results of quinidine efficacy in epilepsies caused by KCNT1 gain‐of‐function variants illustrate the complexity of the transition from models to patients. The reasons for its failure may include not achieving therapeutic doses due to side effects, late treatment initiation, type of epilepsy syndrome, type and location of the variant and other factors that have not been fully elucidated yet. Genetic sodium channelopathies may also result in complex neurodevelopmental disorders including epilepsy, developmental delay, brain malformations, autism and movement disorders. Defining the functional effects of a genetic variant is gold standard for proving pathogenicity and requires in vitro and/or in vivo modelling. The most used models involve transfecting cell lines with the variant being investigated, and then using the patch clamp technique to measure the properties of ion currents compared with wild-type cells. Based on such functional characterisations, ion channel variants are often referred to as either ‘loss-of-function’ or ‘gain-of-function’, though in reality such terms oversimplify what are often complex alterations in ion channel function [19, 25, 26].
The mTOR signalling cascade is a key homeostatic regulatory pathway involved in cell growth and replication. Tuberous sclerosis complex (TSC) is an autosomal-dominant, multiorgan disease with widely variable clinical expression, caused by heterozygous germ-line variants in the tumour-suppressor genes TSC1 and TSC2 which encode hamartin and tuberin. These proteins form a heterodimer (TSC1–TSC2 complex) that inhibits the mTOR signalling cascade. The clinical manifestations of TSC are distinctive and include hamartomatous lesions of the brain, skin, heart, lungs and kidneys; epilepsy; autism; and intellectual disability. Everolimus, an mTOR inhibitor, proved effective at reducing focal seizures, in a large-scale phase 3, randomised, double-blind, placebo-controlled trial in TSC [27]. Pathogenic variation in other genes encoding regulators of the mTOR cascade cause epilepsy, malformations of cortical development (MCD) and neurodevelopmental disorders [28]. Brain somatic variants in mTOR pathway genes are a common cause of focal cortical dysplasia (FCD) and hemimegaloencephaly (HME) [29]. Pathogenic variants in genes encoding the GTPase-activating protein (GAP) activity towards Rags 1 complex (GATOR1) cause non-lesional focal epilepsies and FCD-related epilepsies [30]. The anti-seizure effects of mTOR inhibition may represent a PM strategy also in other mTORopathies, as excessive activation of the mTOR pathway appears to be an essential pathomechanism for epileptogenesis in all these disorders, but adequately powered randomised clinical trials are needed. To date, the only evidence for disease modification in monogenic epilepsies in humans is the use of vigabatrin in TSC, where the EPISTOP clinical trial showed that vigabatrin taken as preventive antiseizure treatment reduces the risk of infantile spasms and drug-resistant epilepsy compared with starting treatment after seizure onset [31]. An ongoing phase 2b trial is assessing whether early identification of electroencephalography A(EEG) biomarkers and early versus delayed treatment with vigabatrin in infants with TSC will also have a positive impact on developmental outcomes at 24 months of age (https://clinicaltrials.gov/ct2/show/study/NCT02849457). However, since the mechanisms underlying the antiseizure action of vigabatrin in TSC remain unknown, this drug cannot be strictly considered a PM.
A systematic literature review of the diagnostic yield of various types of genetic testing in epilepsy also assessed the reported impact of a genetic diagnosis at the clinical and individual level [32]. Across 24 studies, involving heterogeneous patient cohorts (e.g. early-onset epilepsies, developmental and epileptic encephalopathies, focal epilepsies, drug-resistant epilepsies) treatment changes were reported in 12–80% of patients with a genetic diagnosis, including avoiding, stopping or initiating specific ASMs or ketogenic diet and halting a plan for surgery after a specific genetic diagnosis. Reported impacts of changes in clinical management focused primarily on seizure control and ASM use. Most non-ASM management changes were reported without detailed examples or anecdotally for representative patients and did not describe outcomes. Additional outcomes from genetic testing included clinical trial eligibility [29, 33], initiation of palliative care for genetic conditions with uniformly poor outcomes [34, 35], reduction in hospitalisations [36], end of the diagnostic odyssey and of additional testing and procedures [34, 35, 37], prognostic information [38] and impact on recurrence risk estimation/family planning [34, 36].
However, in a systematic survey of patients with epilepsy of all ages and with a molecular genetic diagnosis, across six tertiary epilepsy centres, we found a high variability of clinical outcomes after a genetic cause was identified, with a rational precision medicine treatment available only for a minority of patients and its effectiveness being obvious in an even lower proportion [39].
Unfortunately, most current PM applications in the epilepsies are not straightforward, i.e. the pathway from a molecular diagnosis through identification and administration of a pathophysiology-based PM treatment is not linear [40]. This is likely due to several factors, including the complexity of the genome (overall variation beyond a single variant with large effect size, fine dynamic regulation, gene expression programs), the timing of the diagnosis (the later a PM treatment is tried, the less likely its impact), the lack of information on the functional impact of the genetic variant or the gap between its expected functional effects as established in vitro and translation in the human condition [11].
We discuss below a series of strategies that could be potentially implemented to reduce the current limitations of PM framework in epilepsy.
3 Steps to Implement PM in Epilepsy
3.1 Establishing the Definition
There are different levels for considering ‘precision’ in epilepsy, including the aetiologies, the underlying pathophysiology leading to epileptogenesis or the clinical and EEG characteristics of the epilepsy syndromes including comorbidities.
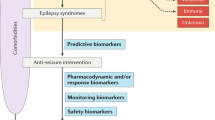
Aetiology-level PM Although most reported PM applications in epilepsy address the neurobiological consequences of genetic aetiologies, there are PM strategies available also for structural (e.g. surgery), metabolic (e.g. correction of metabolic defect), immune (e.g. immunotherapy) and infectious (e.g. antibiotic, antiviral or anthelminthic treatment) causes. Understanding the aetiology and subsequently choosing the most appropriate aetiology-specific treatment improves prognosis, including the possibility of curing epilepsy with surgical treatment in some patients with structural lesions [41].
Pathophysiology-level PM An even more ‘precise’ approach focuses on the specific neurobiological mechanisms leading to disease, aiming for an enhanced understanding of the molecular pathways and network disruption, including a dynamic multilevel assessment [42]. Such a ‘high-definition’ PM approach requires deeper investigation of the underlying aetiologies, and may provide further information for a more pathophysiology-driven treatment even within the same aetiology: for example, genetic characterisation of structural brain lesions may affect surgical management [10] and multilevel genomic analysis including contribution of rare and common variants may help elucidate clinical variability in classical ‘monogenic’ conditions [43, 44].
Syndrome-level PM There are syndrome-specific treatment strategies such as fenfluramine and stiripentol for Dravet syndrome, caused by SCN1A loss-of-function variants, where treatment efficacy is not directly related to the known pathophysiological mechanisms, but they are still erroneously considered PM [45,46,47].
In this context, there is a need to be more ‘precise’ in the PM definition to align assessment and treatment approaches in epilepsy. Next-generation sequencing (NGS) and decision tools by artificial intelligence approaches are certainly already well developed to tackle the complexity underlying epilepsy conditions and advance PM [48]. A more ‘precise’ PM concept should indeed include data-driven approaches that leverage patient heterogeneity and in turn improve treatment decisions so that the right treatment is given to the right patient at the right time.
Here we will employ the pathophysiology-level PM definition, implying that the treatment is tailored to reversing or antagonising the functional alteration caused by the underlying aetiology.
3.2 Defining Outcomes
So far, most reported ‘successful’ PM outcomes have been anecdotal, short-term and mainly focusing on seizures. A more holistic approach including cognition, behaviour and other comorbidities should be instead considered to fully embrace the PM paradigm [11].
In an ideal world, a successful PM therapy would not only reduce the burden of seizures but also improve the patient’s comorbidities and quality of life. Natural history studies of rare diseases are essential to assess PM success. Natural history studies require highly detailed and longitudinal clinical phenotyping that provide information on disease modification operated by PM treatments. Outcome tools are fundamental to assess the level of modification achieved and can in turn be developed and validated through natural history studies.
Registry frameworks promoting multicentre high-quality data collection represent one of the ways forward to obtain natural history information, especially for rare genetic epilepsies, where obtaining homogenous information on large series is challenging. Such frameworks already exist for some rare epilepsy syndromes [49], and should be promoted to address the various challenges of PM implementation, including small populations for clinical studies, difficulty in determining relevant outcome measures and endpoints, and poorly understood natural history, clinical study design and patient recruitment [50].
3.3 Learning from the Past, from Failures and from Other Fields
A relevant step to improve how PM is implemented includes the search for precision explanation for failure of PM treatments. Unsuccessful PM strategies should be systematically documented in the literature as PM successes should include knowledge of follow-up duration. Despite advances in genetic diagnosis, we struggle to figure out the complex interaction of the various biological mechanisms generating the ultimate clinical phenotype, including gene regulation, genomic background modifiers, cell-specific biology and network-level integration. Conventional experimental in vitro or in vivo models of epilepsies such as mouse and rat models or heterologous expression systems have variable throughput and cannot reflect the full complexity of the human phenotype. Novel advanced models of genetic epilepsies, especially the use of induced pluripotent stem cells (iPSCs) to create cultured patient-derived neuronal cultures and organoids, can provide further insights in epilepsy and represent new systems for high-throughput preclinical drug screening. Ideally, conventional and new models should be integrated and complemented with clinical information extrapolated by deep phenotyping and natural history studies to define the appropriate endpoints to use for the preclinical models [51]. Deep phenotyping should be pursued through multimodal techniques [52, 53] which may help bridge the gap between standard clinical phenotype (i.e. clinical information derived from routine practice) and preclinical information provided by in vitro or in vivo model systems.
We should also learn from oncology, where PM is most advanced. Despite thousands of clinical trials of potential gene therapies (http://www.genetherapynet.com/clinical-trials.html), only a few gene therapy products are approved, most being for cancer management and none for epilepsy treatment. The rapid advance of precision oncology with the increasing use of genomic profiling for diagnosis and therapy guidance in many tumour types has led to a growing number of molecularly guided treatment options having received regulatory approval. However, this progress has brought significant challenges for healthcare systems to adapt their infrastructure, methodologies and reimbursement policies to enable wide access to these drugs. These advanced therapies can be costly, and healthcare systems must develop reimbursement policies that address the pricing and reimbursement challenges associated with these novel treatments. It is important to strike a balance between ensuring access to innovative therapies and maintaining the sustainability of healthcare systems. In addition, as PM approaches evolve and more molecularly guided treatments become available, generating robust evidence becomes crucial. Clinical trials and real-world data collection are essential for understanding the clinical utility, effectiveness and long-term outcomes of these treatments. Healthcare systems need to establish mechanisms to facilitate data collection, analysis and sharing to generate evidence and inform treatment decisions. The successful implementation of PM relies on healthcare professionals’ knowledge and expertise. By learning from the experiences and lessons in precision oncology, healthcare systems can work towards overcoming these challenges and optimising the delivery of molecularly advanced diagnostics and treatments to patients in a responsible and accessible manner. In epilepsy, we should foresee similar challenges once more and more genomic diagnostics and treatments based on molecular biomarkers become available.
3.4 What PM Strategies?
Drug repositioning (or repurposing) is an approach where existing medicines are redirected based on a valid target molecule for new therapeutic use(s). This strategy is highly efficient, time saving and low-cost and bears minimum risk of failure. It maximises the therapeutic value of a drug and consequently increases the success rate. Drug repositioning utilises the combined efforts of activity-based or experimental and in silico-based or computational approaches to develop/identify the new uses of drug molecules on a rational basis. Examples of drug repurposing in epilepsy include the use of fluoxetine in KCNC1-related developmental and epileptic encephalopathy [54] or quinidine in KCNT1-related epilepsy [24]. There are a growing number of novel approaches to identify repurposed drugs for specific monogenic epilepsies [55, 56]. However, the mechanisms through which these molecules work are not necessarily understood. For instance, we do not know the exact mechanisms of fenfluramine in controlling seizures in Dravet syndrome nor why it is not effective in all patients with the syndrome.
Alternatively, new molecular entities can be identified by the traditional drug discovery process, which can be a lengthy and expensive venture, but allows specific drug development based on specific molecular targets. Tools that selectively modify the expression of target genes, such as antisense oligonucleotides (ASOs) or RNA interference (RNAi) represent some of the currently closest strategies to the ideal of PM, as they aim to correct a well-defined genetic mechanism. However, altering the expression levels of genes causing developmental and epileptic encephalopathies often requires careful dosing because of the essential roles those genes play in nervous system development. The issue of mosaicism further complicates cellular targeting for many monogenic disorders caused by X‐linked genes or imprinted alleles. Genome editing is a promising alternative to gene supplementation to correct the pathophysiological mechanisms underlying monogenic epilepsy syndromes by restoring the genome to the normal state. The CRISPR/Cas gene editing system has become increasingly prominent in the field of gene therapy in recent years as a programmable form of genome editing [57]. This approach works off the endogenous genes and allows recapitulation of the complex mRNA transcripts; however, it still requires careful dosing and has the risk of off‐target editing from long‐term expression and the potential immunogenic response to a bacterial protein [58]. Gene therapy approaches have proved to be effective in some animal models of monogenic developmental and epileptic encephalopathies. In these disorders, the timing of intervention is crucial as later diagnosis and treatment may not result in complete rescue of the pathology due to developmental changes that have already occurred, and it might be preferable to intervene at or even before the onset of symptoms. However, we are not always able to predict disease evolution after identifying a monogenic cause for a given epilepsy syndrome. For example, would gene therapy be justified in an infant presenting with a prolonged febrile seizure after a pathogenic SCN1A variant is found but evolution towards a genetic epilepsy with febrile seizures plus (GEFS+) or Dravet syndrome remains uncertain?
In conditions caused by ion channel gene variants, such as potassium channels, the affected genes can be delivered directly. For larger genes, such as sodium channels, supplementation in viruses is more challenging; however, canine adenovirus type 2 (CAV-2)-mediated delivery of a codon-modified SCN1A open reading frame into the brain of adolescent Dravet syndrome mice recently provided a proof of concept for the potential of neuronal delivery of an expression cassette encoding SCN1A as a therapeutic approach for Dravet syndrome [59]. In addition, a high proportion of variants affecting ion channels results in toxic gain of function effects, where supplementation is not appropriate. But not all gene therapy strategies being developed are precise with respect to the underlying genetic mechanism [60]. Several virally delivered treatments show in fact great promise in vitro and increasingly in vivo in models of refractory focal epilepsies, aiming to control seizures where there is no established underlying monogenic cause. Such treatments provide very long-term expression of a therapeutic transgene from adeno-associated virus (AAV) vectors, therefore offering long-term benefit from a single intervention [61]. Viral vectors have a relatively restricted spatial spread, permitting more precise targeting of the epileptogenic zone in focal epilepsies, and sparing distinct areas which are not implicated in epileptogenesis. Conversely, this does not represent an advantage in monogenic epilepsies caused by constitutional variants which affect the entire brain.
Current gene therapy approaches in epilepsy still face several limitations including, way of administration (e.g. absence of a vector that can be administered via systemic approaches), timing of intervention, cell type specificity, dominant-negative variants, packaging limit (< 4.7 Kb) of AAV vectors which excludes the delivery of many epilepsy genes such as channels, lack of safety data, unknown outcomes in terms of cognition and other neurological aspects beyond seizures, poor information regarding reaction of the rest of the genome, risk of permanent irreversible effects and other unknown aspects related to the heterogeneity of the epilepsies even within the same ‘monogenic’ aetiology [62]. By addressing these challenges and fostering collaboration among various stakeholders, including researchers, healthcare providers, regulators and policymakers, it may prove possible to facilitate the successful integration of gene therapy into PM for epilepsy. Continued research, innovation and a patient-centred approach are key to advancing the field and maximising the potential benefits of gene therapy in the management of epilepsy.
3.5 How to Measure PM Efficacy
Randomised controlled trials might not be the ideal evaluation paradigm for PM interventions that are only applicable to small numbers of patients. When searching the clinicaltrials.gov website (https://clinicaltrials.gov/), there are 45 studies registered for treatment of monogenic epilepsy syndromes, most of them without a clear PM mechanism. Of these 45 studies, only 13 (29%) have been completed and have results available, the majority of which (8 of 13, 62%) mostly focus on Dravet syndrome. The ‘lumping versus splitting’ attitude has long been a dilemma in design of clinical trials for epilepsy. Whilst a lumping approach is required for drug development when patients cannot be included into a specific aetiology group or syndromic classification, which is still the case for many individuals with epilepsy, the splitting paradigm is most useful to identify potential responders by type of epilepsy, permits reduction in the number of patients included in trials and is closer to the PM paradigm [63]. Pharmaceutical industry plays a big role in the design of clinical trials. The adoption of a PM-based splitting approach implies capacity and intention to switch from medications treating seizures at large to medications treating specific conditions manifesting seizures as one of the symptoms. This paradigm shift comes with a need for both translation and precision [64] and certainly has considerable marketing implications.
Conventional large-scale, placebo-controlled clinical trials may prove challenging in the context of rare diseases due to the small patient populations available for enrolment. In such circumstances, it can be difficult to achieve statistical power and generate meaningful results, and new approaches are required to evaluate short- and long-term safety, efficacy and durability of PM treatments. In rare genetic epilepsies, there is need for functional characterisation of the causative variant and stratification of severity of the clinical phenotype; furthermore, endpoint definition and integration of multimodal data (e.g. combining clinical data, genetic information, neuroimaging, electrophysiology and other relevant data sources) are often challenging. On this basis, adequately powered randomised controlled trials are difficult to design for many genetic epilepsies. Alternative trial designs can overcome issues of low sample size and high interindividual variability. Examples include small crossover trials and prospective, rigorously designed N-of-1 trials that assess the result of an intervention in a single study participant, who undergoes several blocks of crossovers between the intervention and control condition, and the relative effect in each block is established [65, 66]. A rigorously designed multi-crossover N-of-1 randomised controlled trial requires appropriate selection of interventions, a priori specification of treatment doses, durations and placebo washouts of each treatment, blinding, repeated blocks, randomised sequences of treatments in each block and systematic application of outcome measures. Some consider N-of-1 trials to provide Level 1 evidence [67]. Combining data of N-of-1 trials that use the same intervention, trial design and outcome measures in multiple subjects can provide an estimate of response to the intervention in the condition more generally. Such studies can have higher statistical power than parallel group randomised controlled trials, as some sources of bias such as clinical heterogeneity and presence of comorbidities are automatically controlled when subjects serve as their own controls. Despite these advantages, a recent systematic review of N-of-1 trials for rare genetic neurodevelopmental disorders found that only 12 studies complied with the fundamental N-of-1 criteria of a controlled multiple crossover trial, showing how N-of-1 studies are only sporadically reported and their findings often cannot be generalised due to limited use of validated and generalisable outcome measures [68]. However, improving methodological and statistical quality, generalisability, feasibility and personalisation is at reach [68]. Ethics, regulation and equity also pose challenges: development of a therapy for one individual, rather than a population, makes the boundaries less clear-cut between research and medical treatment, and there is the possibility of conflicts of interest and inadequately informed consent when having a single subject as study participant. To advance PM in epilepsy, robust methods for efficacy assessment are required to provide a solid basis for evidence-based interventions for vulnerable patients with rare and complex epilepsies who may have limited capacity to consent to such treatments.
With the advance of PM, also the regulatory agencies need to switch from a population-based to an individual-based approach, to take decisions on benefit–risk assessments and marketing authorisation [69]. Regulatory approval can be very lengthy, especially for cell and gene therapies. By addressing the challenges related to scaling production of complex biological molecules, managing the transportation of living cells, and standardising manufacturing across different facilities, the integration of PM options into clinical practice could be facilitated.
4 Towards the Future
In our current PM paradigm, we require rigorous preclinical testing so as to fully understand which pathophysiological mechanisms to target and how. For genetic aetiologies, we have newer technologies for NGS including long-read sequencing, or third-generation sequencing, which offers several advantages over short-read sequencing. These sequencing technologies, based on the sequencing of a circularised single-molecule DNA (PacBio, Pacific Biosciences) or of a single-molecule DNA through a protein nanopore (Oxford Nanopore Technologies) generate long-reads > 10 Kb. De novo assembly, mapping certainty, transcript isoform identification and detection of structural variants can improve with the use of long reads. Long-read sequencing of both DNA and RNA also eliminates amplification bias while preserving base modifications [70]. On the other hand, long-read sequencing technologies provide lower per read accuracy than short-read sequencing, and error correction remains an important step in long-read analysis pipelines [71]. This limitation could be overcome with the spreading of the HiFi long-read sequencing that provides exceptional read lengths without compromising accuracy [72]. Data from multi-modal experiments including genomics, proteomics, transcriptomics, metabolomics and imaging techniques can be analysed through high-dimensional multivariate statistics. Deep learning techniques analyse available multimodal genomics data. However, there are fields that need further development such as single-cell sequencing across organs; across developmental time points, including all stages of embryonic and foetal development. Furthermore, these multi-modal experimental approaches, although having the power to reach a molecular diagnosis in most patients, are not yet feasible on a large-scale basis due to high costs and complexity. Accurate machine learning methods to predict functional effects of missense and non-coding variants require characterisation of genomic non-coding regions and repetitive regions, and high-throughput systems for saturation mutagenesis and functional readouts across major classes of genes and regulatory sequences [73]. Accurate computational methods to predict the impact of genetic variants in other similar genes without directly assessing them require technology to visualise dynamic changes in protein structure and protein-protein interactions within cells along with the functional assays [74]. Single-cell multi-omic technologies allow to sequence DNA, to obtain the RNA transcriptome and detect epigenomic events simultaneously within the same cell at low cost and high throughput. Single-cell proteomics and metabolomics profiling are possible too, although a concurrent multi-omic profiling combining these omics with DNA or RNA related omics is not yet available [75]. However, single-cell multi-omic technologies need development in epilepsy to understand genotype–phenotype interactions in single cells. Spatial omics technologies enable a deeper understanding of cellular organisations and interactions within a tissue of interest by measuring unbiased DNA, RNA or epigenomic information at low cost and across large spatial regions. However, these techniques also need further development to improve spatial resolution, multiplexing capability, scale/throughput and coverage [76]. The continuous expansion of omics and biological datasets should go in parallel with new computational approaches to drive their collection and guide prediction of new therapeutic targets. Genetic testing should also become more widely available, so to reduce the current disparities in PM application, inclusion in clinical trials and in natural history studies.
PM is not just about genomics. With the advent of high-resolution magnetic resonance imaging (MRI) epilepsy surgery has significantly grown, and the increasing sensitivity of MRI allows detection of more and more subtle structural lesions and increases precision of surgical treatment. Structural 7T MRI is now available, with optimised acquisition and post-acquisition image processing, thus significantly increasing the diagnostic yield [77]. Functional MRI (fMRI) can be used to map eloquent cortical areas and predict the impact of surgical treatment on higher cortical functions. Neuroimaging techniques assessing functional connectivity have markedly improved our understanding of disease-specific effects on brain networks. Diffusion MRI (dMRI) and its derivatives of network analysis and tractography allow assessing the structural basis of connectivity, and guiding optimal resections in individual patients, so mitigating the risk of post-surgical deficits [75]. Identification of autoantibodies and new infectious causes has also notably improved our knowledge of immune and infectious epilepsy aetiologies and related targeted treatments [78, 79].
Progress of diagnostics techniques in epilepsy now often allows for the full characterisation of each individual epilepsy condition from a molecular, biological, structural and clinical perspective. However, not all data from a single individual are routinely integrated in clinical practice and access to advanced diagnostics highly varies worldwide. Major steps to improve PM in epilepsy are required to bridge the gap between identifying the aetiology and choosing the right successful treatment (Table 2).
References
Sykiotis GP, Kalliolias GD, Papavassiliou AG. Pharmacogenetic principles in the hippocratic writings. J Clin Pharmacol [Internet]. 2005 [cited 2023 Jul 9];45:1218–20. http://doi.wiley.com/10.1177/0091270005281091.
National Research Council. Towards precision medicine: building a knowledge network for biomedical research and a new taxonomy of disease. [Internet]. Washington, DC: National Academies Press; 2011. Available from: https://www.nap.edu/catalog/13284/toward-precision-medicine-building-a-knowledge-network-for-biomedical-research.
Schleidgen S, Klingler C, Bertram T, Rogowski WH, Marckmann G. What is personalized medicine: sharpening a vague term based on a systematic literature review. BMC Med Ethics [Internet]. 2013 [cited 2023 Jul 9];14:55. Available from: https://bmcmedethics.biomedcentral.com/articles/10.1186/1472-6939-14-55.
Hood L, Flores M. A personal view on systems medicine and the emergence of proactive P4 medicine: predictive, preventive, personalized and participatory. N Biotechnol. 2012;29:613–24.
Auffray C, Chen Z, Hood L. Systems medicine: the future of medical genomics and healthcare. Genome Med. 2009;1:2.
Hood L. How technology, big data, and systems approaches are transforming medicine. Res Technol Manag [Internet]. 2019 [cited 2023 Jul 9];62:24–30. Available from: https://www.tandfonline.com/doi/full/10.1080/08956308.2019.1661077.
Jones DS. How personalized medicine became genetic, and racial: werner kalow and the formations of pharmacogenetics. Journal Hist Med Allied Sci [Internet]. 2013 [cited 2023 Jul 9];68:1–48. Available from: https://academic.oup.com/jhmas/article-lookup/doi/10.1093/jhmas/jrr046.
GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18:459–80.
Balestrini S, Arzimanoglou A, Blümcke I, Scheffer IE, Wiebe S, Zelano J, et al. The aetiologies of epilepsy. Epileptic Disord. 2021;23:1–16.
Guerrini R, Balestrini S, Wirrell EC, Walker MC. Monogenic epilepsies: disease mechanisms, clinical phenotypes, and targeted therapies. Neurology [Internet]. 2021 [cited 2022 Mar 27];97:817–31. Available from: https://www.neurology.org/lookup/doi/10.1212/WNL.0000000000012744.
Sisodiya SM. Precision medicine and therapies of the future. Epilepsia. 2021;62 Suppl 2(Suppl 2):S90–105. https://doi.org/10.1111/epi.16539.
Balestrini S, Sisodiya SM. Personalized treatment in the epilepsies: challenges and opportunities. Expert Rev Precis Med Drug Dev [Internet]. 2018 [cited 2023 Jul 9];3:237–47. Available from: https://www.tandfonline.com/doi/full/10.1080/23808993.2018.1486189.
Gospe SM. Pyridoxine-dependent epilepsy—ALDH7A1. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993 [cited 2023 Jul 9]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1486/.
Plecko B, Mills P. PNPO deficiency. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993 [cited 2023 Jul 9]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK581452/.
Arsov T, Mullen SA, Damiano JA, Lawrence KM, Huh LL, Nolan M, et al. Early onset absence epilepsy: 1 in 10 cases is caused by GLUT1 deficiency. Epilepsia. 2012;53:e204-207.
Arsov T, Mullen SA, Rogers S, Phillips AM, Lawrence KM, Damiano JA, et al. Glucose transporter 1 deficiency in the idiopathic generalized epilepsies. Ann Neurol. 2012;72:807–15.
Seidner G, Alvarez MG, Yeh JI, O’Driscoll KR, Klepper J, Stump TS, et al. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood–brain barrier hexose carrier. Nat Genet. 1998;18:188–91.
Alter AS, Engelstad K, Hinton VJ, Montes J, Pearson TS, Akman CI, et al. Long-term clinical course of Glut1 deficiency syndrome. J Child Neurol. 2015;30:160–9.
Symonds JD, Zuberi SM. Genetics update: Monogenetics, polygene disorders and the quest for modifying genes. Neuropharmacology. 2018;132:3–19.
McTague A, Nair U, Malhotra S, Meyer E, Trump N, Gazina EV, et al. Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy. Neurology. 2018;90:e55-66.
Milligan CJ, Li M, Gazina EV, Heron SE, Nair U, Trager C, et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine: KCNT1 and Human Epilepsy. Ann Neurol [Internet]. 2014 [cited 2023 Jul 9];75:581–90. Available from: https://onlinelibrary.wiley.com/doi/10.1002/ana.24128.
Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann Neurol. 2014;76:457–61.
Fitzgerald MP, Fiannacca M, Smith DM, Gertler TS, Gunning B, Syrbe S, et al. Treatment responsiveness in KCNT1-related epilepsy. Neurotherapeutics. 2019;16:848–57.
Mullen SA, Carney PW, Roten A, Ching M, Lightfoot PA, Churilov L, et al. Precision therapy for epilepsy due to KCNT1 mutations: a randomized trial of oral quinidine. Neurology. 2018;90:e67-72.
Swanger SA, Chen W, Wells G, Burger PB, Tankovic A, Bhattacharya S, et al. Mechanistic insight into NMDA receptor dysregulation by rare variants in the GluN2A and GluN2B agonist binding domains. Am J Hum Genet. 2016;99:1261–80.
Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017;140:1316–36.
French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet [Internet]. 2016 [cited 2022 Nov 1];388:2153–63. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0140673616314192.
Crino PB. The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol. 2016;12:379–92.
Baldassari S, Ribierre T, Marsan E, Adle-Biassette H, Ferrand-Sorbets S, Bulteau C, et al. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol. 2019;138:885–900.
Baldassari S, Picard F, Verbeek NE, van Kempen M, Brilstra EH, Lesca G, et al. The landscape of epilepsy-related GATOR1 variants. Genet Med. 2019;21:398–408.
Kotulska K, Kwiatkowski DJ, Curatolo P, Weschke B, Riney K, Jansen F, et al. Prevention of epilepsy in infants with tuberous sclerosis complex in the EPISTOP Trial. Ann Neurol. 2021;89(2):304–14. https://doi.org/10.1002/ana.25956. Epub 2020 Nov 27.
Sheidley BR, Malinowski J, Bergner AL, Bier L, Gloss DS, Mu W, et al. Genetic testing for the epilepsies: a systematic review. Epilepsia. 2022;63:375–87.
Hildebrand MS, Myers CT, Carvill GL, Regan BM, Damiano JA, Mullen SA, et al. A targeted resequencing gene panel for focal epilepsy. Neurology. 2016;86:1605–12.
Palmer EE, Schofield D, Shrestha R, Kandula T, Macintosh R, Lawson JA, et al. Integrating exome sequencing into a diagnostic pathway for epileptic encephalopathy: evidence of clinical utility and cost effectiveness. Mol Genet Genomic Med [Internet]. 2018 [cited 2023 Jul 9];6:186–99. Available from: https://onlinelibrary.wiley.com/doi/10.1002/mgg3.355.
Demos M, Guella I, DeGuzman C, McKenzie MB, Buerki SE, Evans DM, et al. Diagnostic yield and treatment impact of targeted exome sequencing in early-onset epilepsy. Front Neurol. 2019;10:434.
Papuc SM, Abela L, Steindl K, Begemann A, Simmons TL, Schmitt B, et al. The role of recessive inheritance in early-onset epileptic encephalopathies: a combined whole-exome sequencing and copy number study. Eur J Hum Genet [Internet]. 2019 [cited 2023 Jul 9];27:408–21. Available from: https://www.nature.com/articles/s41431-018-0299-8.
Lee S, Karp N, Zapata-Aldana E, Sadikovic B, Yang P, Balci TB, et al. Genetic testing in children with epilepsy: report of a single-center experience. Can J Neurol Sci. 2021;48:233–44.
Yuskaitis CJ, Ruzhnikov MRZ, Howell KB, Allen IE, Kapur K, Dlugos DJ, et al. Infantile spasms of unknown cause: predictors of outcome and genotype-phenotype correlation. Pediatr Neurol. 2018;87:48–56.
Balestrini S, Chiarello D, Gogou M, Silvennoinen K, Puvirajasinghe C, Jones WD, et al. Real-life survey of pitfalls and successes of precision medicine in genetic epilepsies. J Neurol Neurosurg Psychiatry [Internet]. 2021 [cited 2021 May 23];jnnp-2020-325932. Available from: https://jnnp.bmj.com/lookup/doi/10.1136/jnnp-2020-325932.
Sisodiya SM. Epilepsy genetics and the precision medicine matrix. Lancet Neurol. 2020;19:29–30.
Guerrini R, Conti V, Mantegazza M, Balestrini S, Galanopoulou AS, Benfenati F. Developmental and epileptic encephalopathies: from genetic heterogeneity to phenotypic continuum. Physiol Rev. 2023;103:433–513.
Torkamani A, Andersen KG, Steinhubl SR, Topol EJ. High-definition medicine. Cell [Internet]. 2017 [cited 2023 Jul 9];170:828–43. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867417309327.
Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–24.
Martins Custodio H, Clayton LM, Bellampalli R, Pagni S, Silvennoinen K, Caswell R, et al. Widespread genomic influences on phenotype in Dravet syndrome, a ‘monogenic’ condition. Brain [Internet]. 2023 [cited 2023 Jul 9];awad111. Available from: https://academic.oup.com/brain/advance-article/doi/10.1093/brain/awad111/7099704.
Chiron C, Marchand M, Tran A, Rey E, d’Athis P, Vincent J, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. Lancet [Internet]. 2000 [cited 2021 Sep 4];356:1638–42. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0140673600031573.
Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet. 2019;394:2243–54.
Nabbout R, Mistry A, Zuberi S, Villeneuve N, Gil-Nagel A, Sanchez-Carpintero R, et al. Fenfluramine for treatment-resistant seizures in patients with dravet syndrome receiving stiripentol-inclusive regimens: a randomized clinical trial. JAMA Neurol [Internet]. 2020 [cited 2021 Jan 17];77:300. Available from: https://jamanetwork.com/journals/jamaneurology/fullarticle/2756124.
Faulkner E, Holtorf A-P, Walton S, Liu CY, Lin H, Biltaj E, et al. Being precise about precision medicine: what should value frameworks incorporate to address precision medicine? a report of the personalized precision medicine special interest group. Value Health. 2020;23:529–39.
Balestrini S, Doccini V, Giometto S, Lucenteforte E, De Masi S, Giarola E, et al. A registry for Dravet syndrome: the Italian experience. Epilepsia Open. 2023;8:517–34.
Mahmood SS, Levy D, Vasan RS, Wang TJ. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet. 2014;383:999–1008.
Demarest ST, Brooks-Kayal A. From molecules to medicines: the dawn of targeted therapies for genetic epilepsies. Nat Rev Neurol [Internet]. 2018 [cited 2023 Jul 9];14:735–45. Available from: https://www.nature.com/articles/s41582-018-0099-3.
Silvennoinen K, Balestrini S, Rothwell JC, Sisodiya SM. Transcranial magnetic stimulation as a tool to understand genetic conditions associated with epilepsy. Epilepsia. 2020;61:1818–39.
Balestrini S, Lopez SM, Chinthapalli K, Sargsyan N, Demurtas R, Vos S, et al. Increased facial asymmetry in focal epilepsies associated with unilateral lesions. Brain Commun. 2021;3:fcab068.
Ambrosino P, Ragona F, Mosca I, Vannicola C, Canafoglia L, Solazzi R, et al. A novel KCNC1 gain-of-function variant causing developmental and epileptic encephalopathy: “Precision medicine” approach with fluoxetine. Epilepsia. 2023;64(7):e148–55. https://doi.org/10.1111/epi.17656. Epub 2023 Jun 2.
Mirza N, Sills GJ, Pirmohamed M, Marson AG. Identifying new antiepileptic drugs through genomics-based drug repurposing. Hum Mol Genet. 2017;26:527–37.
Atkin TA, Maher CM, Gerlach AC, Gay BC, Antonio BM, Santos SC, et al. A comprehensive approach to identifying repurposed drugs to treat SCN8A epilepsy. Epilepsia. 2018;59:802–13.
Doudna JA. The promise and challenge of therapeutic genome editing. Nature [Internet]. 2020 [cited 2023 Jul 9];578:229–36. Available from: https://www.nature.com/articles/s41586-020-1978-5.
Wang D, Mou H, Li S, Li Y, Hough S, Tran K, et al. Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum Gene Ther. 2015;26:432–42.
Fadila S, Beucher B, Dopeso-Reyes IG, Mavashov A, Brusel M, Anderson K, et al. Viral vector-mediated expression of NaV1.1, after seizure onset, reduces epilepsy in mice with Dravet syndrome. J Clin Invest. 2023;133:e159316.
Turner TJ, Zourray C, Schorge S, Lignani G. Recent advances in gene therapy for neurodevelopmental disorders with epilepsy. J Neurochem. 2021;157(2):229–62. https://doi.org/10.1111/jnc.15168. Epub 2020 Sep 28.
Morris G, Schorge S. Gene therapy for neurological disease: state of the art and opportunities for next-generation approaches. Neuroscience. 2022;490:309–14.
Knowles JK, Helbig I, Metcalf CS, Lubbers LS, Isom LL, Demarest S, et al. Precision medicine for genetic epilepsy on the horizon: recent advances, present challenges, and suggestions for continued progress. Epilepsia [Internet]. 2022 [cited 2023 Jul 9];63:2461–75. Available from: https://onlinelibrary.wiley.com/doi/10.1111/epi.17332.
Dulac O, Guerrini R. Seizure types and syndromes: lumping or splitting. Epilepsy Res. 2001;45:37–40 (discussion 41).
Hartl D, de Luca V, Kostikova A, Laramie J, Kennedy S, Ferrero E, et al. Translational precision medicine: an industry perspective. J Transl Med. 2021;19:245.
Gordon K, Bawden H, Camfield P, Mann S, Orlik P. Valproic acid treatment of learning disorder and severely epileptiform EEG without clinical seizures. J Child Neurol. 1996;11:41–3.
Vohra S, Shamseer L, Sampson M, Bukutu C, Schmid CH, Tate R, et al. CONSORT extension for reporting N-of-1 trials (CENT) 2015 Statement. J Clin Epidemiol. 2016;76:9–17.
Howick J, Chalmers I, Glasziou P, Greenhalgh T, Heneghan C, Liberati A, Moschetti I, Phillips B, Thornton H. The 2011 Oxford CEBM levels of evidence (introductory document). [Internet]. Available from: https://www.cebm.ox.ac.uk/resources/levels-of-evidence/ocebm-levels-of-evidence.
Müller AR, Brands MMMG, van de Ven PM, Roes KCB, Cornel MC, van Karnebeek CDM, et al. Systematic review of N-of-1 studies in rare genetic neurodevelopmental disorders: the power of 1. Neurology. 2021;96:529–40.
Breckenridge A, Eichler H-G, Jarow JP. Precision medicine and the changing role of regulatory agencies. Nat Rev Drug Discov. 2016;15:805–6.
Conlin LK, Aref‐Eshghi E, McEldrew DA, Luo M, Rajagopalan R. Long‐read sequencing for molecular diagnostics in constitutional genetic disorders. Hum Mutat [Internet]. 2022 [cited 2023 Jul 9];43:1531–44. Available from: https://onlinelibrary.wiley.com/doi/10.1002/humu.24465.
Watson M, Warr A. Errors in long-read assemblies can critically affect protein prediction. Nat Biotechnol [Internet]. 2019 [cited 2023 Aug 25];37:124–6. Available from: https://www.nature.com/articles/s41587-018-0004-z.
Kucuk E, van der Sanden BPGH, O’Gorman L, Kwint M, Derks R, Wenger AM, et al. Comprehensive de novo mutation discovery with HiFi long-read sequencing. Genome Med. 2023;15:34.
Spielmann M, Kircher M. Computational and experimental methods for classifying variants of unknown clinical significance. Cold Spring Harb Mol Case Stud. 2022;8: a006196.
Høie MH, Cagiada M, Beck Frederiksen AH, Stein A, Lindorff-Larsen K. Predicting and interpreting large-scale mutagenesis data using analyses of protein stability and conservation. Cell Rep. 2022;38: 110207.
Kong S, Li R, Tian Y, Zhang Y, Lu Y, Ou Q, et al. Single-cell omics: a new direction for functional genetic research in human diseases and animal models. Front Genet. 2022;13:1100016.
Park J, Kim J, Lewy T, Rice CM, Elemento O, Rendeiro AF, et al. Spatial omics technologies at multimodal and single cell/subcellular level. Genome Biol. 2022;23:256.
Opheim G, van der Kolk A, Markenroth Bloch K, Colon AJ, Davis KA, Henry TR, et al. 7T Epilepsy Task Force Consensus recommendations on the use of 7T MRI in clinical practice. Neurology. 2021;96:327–41.
Liu X, Guo K, Lin J, Gong X, Li A, Zhou D, et al. Long-term seizure outcomes in patients with autoimmune encephalitis: a prospective observational registry study update. Epilepsia. 2022;63:1812–21.
Yacubian EMT, Kakooza-Mwesige A, Singh G, Carpio A, de Figueiredo NV, Lutzky Saute R, et al. Common infectious and parasitic diseases as a cause of seizures: geographic distribution and contribution to the burden of epilepsy. Epileptic Disord. 2022;24:994–1019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open access funding provided by Università degli Studi di Firenze within the CRUI-CARE Agreement.
Conflict of interest
SB received speaker and consultant honoraria from Angelini, Biocodex and Jazz Pharmaceutics. DM has no competing interests to declare. SMS has received honoraria for educational events from Angelini Pharma, Eisai, Zogenix/UCB and institutional contributions for advisory boards, educational events or consultancy work from Eisai, Jazz Pharma, Stoke Therapeutics, UCB and Zogenix. RG received fees for Advisory Boards from UCB, Zogenix, Biocodex, GW-Jazz Pharmaceutics, Angelini, Takeda and Rapport Therapeutics.
Author contributions
RG and SB contributed to study conceptualisation, SB and DM performed the literature search, SB drafted the manuscript and RG and SMS critically revised the work.
Ethics approval
Not applicable.
Consent (participation and publication)
Not applicable.
Data availability statement
Not applicable.
Code availability
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Balestrini, S., Mei, D., Sisodiya, S.M. et al. Steps to Improve Precision Medicine in Epilepsy. Mol Diagn Ther 27, 661–672 (2023). https://doi.org/10.1007/s40291-023-00676-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-023-00676-9