
Abstract
Purpose of Review
This review will discuss how the steroid hormones, estrogen and progesterone, as well as treatments that target steroid receptors, can regulate cancer stem cell (CSC) activity. The CSC theory proposes a hierarchical organization in tumors where at its apex lies a subpopulation of cancer cells endowed with self-renewal and differentiation capacity.
Recent Findings
In breast cancer (BC), CSCs have been suggested to play a key role in tumor maintenance, disease progression, and the formation of metastases. In preclinical models of BC, only a few CSCs are required sustain tumor re-growth, especially after conventional anti-endocrine treatments. CSCs include therapy-resistant clones that survive standard of care treatments like chemotherapy, irradiation, and hormonal therapy.
Summary
The relevance of hormones for both normal mammary gland and BC development is well described, but it was only recently that the activities of hormones on CSCs have been investigated, opening new directions for future BC treatments and CSCs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The cancer stem cell (CSC) concept proposes a hierarchical organization of the cells within a tumor, where only a small subset of cells, the CSCs, drives and sustains tumor growth. In preclinical studies using breast cancer models, CSCs are defined as self-renewing cells that can propagate the tumor, which makes them very important in the processes of tumor recurrence, metastasis, and resistance to therapy. These roles make them an important therapeutic target [1].
The first report providing evidence for the presence of breast CSCs (BCSCs) observed that CD44+/CD24low/ESA+/lineage− (named CD44+/CD24−/lo henceforth) cells (Table 1), isolated from human breast tumors by fluorescence activated cell sorting (FACS), were enriched for CSCs that were adequate to seed tumors in immune-deficient mice [14]. CD44+/CD24−/lo cells were serially passaged and gave rise to tumors containing both CSCs (CD44+/CD24−/lo) and non-CSCs, suggesting self-renewal and differentiation, respectively. Breast cancers with high CD44 and low CD24 have been associated with the triple negative subtype (negative for estrogen receptor (ER), progesterone receptor (PR), and HER2 receptor) and with poorer prognosis [15, 16].
Other strategies have also been used to identify BCSC enriched populations. Mammosphere formation, high aldehyde dehydrogenase (ALDH) activity, and CD49f or CD133 expression are properties that have been utilized to isolate CSCs (Table 1). The mammosphere colony assay tests the capacity of BCSCs to survive in non-adherent culture conditions and to form spherical colonies, called mammospheres [17,18,19]. The activity of ALDH1, which retinaldehyde to retinoic acid, is detected by an enzymatic assay (ALDEFLUOR) and flow cytometry [20]. The proportion of cells with ALDH1 expression in breast cancer has been shown to correlate with poor prognosis [20,21,22]. Finally, CD49f and CD133 (Table 1) have recently been shown to enrich for CSCs in chemotherapy resistant triple negative and endocrine-resistant breast cancer, respectively [7••, 23]. The establishment of BCSC markers suitable for all tumors is hindered by intra-tumor and inter-tumor heterogeneity of CSC populations.
At the present time, the most robust enrichment for BCSCs is achieved through the use of CD44+/CD24−/lo and ALDH+. These two cell populations have been demonstrated to mark BCSCs in different states and with gene expression resembling either mesenchymal (CD44+/CD24−/lo cells) or epithelial characteristics (ALDH+ cells) [24]. A small overlapping population of cells which is both CD44+/CD24−/lo and ALDH+ was identified, which suggested that BCSCs possess cellular plasticity and can dynamically switch between mesenchymal and epithelial states. The epithelial–mesenchymal transition and vice-versa (mesenchymal–epithelial transition) can be driven by the tumor microenvironment, with hypoxia or transforming growth factor beta playing key roles in this [25, 26]. It is likely that other signaling factors that have been reported to regulate BCSC activity, such as hormones, will influence this. Herein, we discuss the regulation of BCSC function by the steroid hormones, particularly estrogen and progesterone, and their antagonists [22].
Estrogen and BCSCs
Estrogen promotes mammary epithelial cell proliferation and is therefore critical for normal breast development, but it also stimulates breast tumor growth through the estrogen receptor (ER) [27]. Estrogen binds to its receptors, ERα and ERβ, which are nuclear ligand-activated transcription factors, to modulate the transcription of target genes [28]. The effects of estrogen in the breast epithelium are mainly mediated by ERα, which has a higher affinity to 17β-estradiol, the physiological form of estrogen, than does ERβ [29]. Transcription factors need nuclear receptor co-regulators to mediate their action on target DNA sequences; in this case, ER signaling is dependent on FOXA1 expression, which promotes local DNA unwinding facilitating the access of ER to DNA [30].
Around three out of four breast tumors express ERα. Its expression is associated with luminal differentiation markers and with a more favorable breast cancer prognosis and is the most important breast cancer predictive factor for endocrine responsiveness [31, 32]. Exposure to high levels of estrogen during women’s lifetime is established to be associated with increased risk of postmenopausal breast cancer [33]. However, exogenous estrogen used as hormone replacement therapy may reduce the risk of invasive breast cancer and breast cancer-specific mortality in postmenopausal women [34]. This paradoxal effect of the role of estrogen in breast cancer initiation and progression might be explained by the different impacts of estrogen on different breast cancer cell types. On one hand, the pro-proliferative function of estrogen in ERα-positive breast cancer cells has been well characterized, but on the other hand, literature detailing the effects of estrogen on breast cancer stem cell (BCSCs) remains relatively scarce [35].
These effects are proposed to occur indirectly via paracrine mechanisms since BCSCs (CD44+ CD24−/lo and ALDH+ cells) are mostly ERα-negative [36,37,38]. It has been reported that treatment of CSC-enriched mammosphere population with estrogen decreases the proportion of BCSCs in ERα-positive breast cancer cells as a result of downregulation of embryonic stem cell genes [39]. This observation could in theory explain the better prognosis of ERα-positive tumors [40]. But it also has been shown that ERα-positive breast cancer cells can secrete FGFR and EGFR ligands in response to estrogen, which can act as paracrine mediators to promote CSC activity and expand the fraction of CD44+ CD24−/lo cells [41, 42]. In contrast, Axlund et al. reported that estrogen does not change cancer stem/progenitor cell properties on its own [43]. The reasons why some data show a protective effect of estrogen whereas others show that it can enhance cancer cell growth are not yet clear, but likely are related to other underlying differences in the tumor models used in the studies.
More recently, despite the fact that BCSCs do not express the classical ERα, estrogens have been suggested to act directly on BCSCs through the ERα36 variant and ERβ. ERα36 variant activates mitogenic signaling via the AKT/GSK3β pathway and is essential for the maintenance of CD44+ CD24−/lo cells of two ER-positive breast cancer cell lines [44]. Ma and colleagues identified ERβ expression to be associated with stem cell markers CD44 and ALDH1 and also to be important for mammospheres formation [45]. Interestingly, ERβ gene expression has been reported to be upregulated in FACS sorted human breast stem cells [46••] compared to the total tumor cell population.
The complex implications of estrogen signaling in human breast cancer cells with stem-like characteristics indicate that further studies are needed to fully elucidate the effects of estrogens on BCSCs. Standardization of experimental conditions is warranted since the use of different BCSC markers, models, or culture conditions alters the analysis of CSC activity.
Anti-estrogen Drugs and BCSCs
Anti-estrogen therapies are used for breast cancer treatment of ER-positive tumors in both the adjuvant and metastatic settings. The principal classes of drugs are selective estrogen receptor modulators (SERMs, e.g., tamoxifen) and downregulators (SERDs, e.g., fulvestrant) as well as aromatase inhibitors, that reduce estrogen synthesis [47]. Since BCSCs are mostly ER-negative, they are not targeted by anti-estrogen therapies and several publications have reported that these therapies enrich for cells with BCSC characteristics.
Tumors treated with letrozole (aromatase inhibitor) increased in CD44+CD24−/lo mammosphere forming cells [48]. Piva and colleagues reported that tamoxifen-resistant MCF-7 cells have increased CD44+CD24−/lo and ALDH+ populations and form more mammospheres than the parental cells. In addition, they established that expression of the embryonic stem cell marker SOX2 and consequent activation of WNT signaling pathway was key for BCSCs survival after tamoxifen treatment [49]. Another study showed that ERα36 promotes tamoxifen resistance by increasing the proportion of CD44+CD24−/lo cells and mammosphere-forming cells [44]. Our group has shown that BCSCs (ALDH+ cells) are enriched following anti-estrogen treatment of breast cancer cells both in vitro using patient samples and in vivo using patient-derived xenografts. We also found that ALDH+ cells have high expression of JAG1 ligand and NOTCH4 receptor and that high ALDH1 expression predicts anti-estrogen resistance in women treated with tamoxifen [38]. Recently, two different studies from Sansone and colleagues demonstrated how the transfer of miR-221 or full mitochondrial DNA from cancer-associated fibroblasts to breast cancer cells through circulating extracellular vesicles could promote an exit from dormancy of BCSCs (CD133+) leading to endocrine therapy resistance [50, 51].
Together, these findings suggest that inhibition of estrogen signaling in breast cancer cells may lead to an increase of the proportion of BCSCs. It still needs to be addressed whether this phenomenon occurs through selection and survival of the ER negative BCSCs, through induction of BCSCs characteristics in the ER+ cells, or by both processes. Either way, we hypothesize that BCSCs that survive anti-estrogen treatments can enter a dormant state and eventually re-initiate tumor growth, sometimes several years after the therapy. Further interest in this field has given rise to several clinical trials directly targeting CSCs (via recognized markers like ALDH) or using different signaling pathways linked to CSCs (Table 2).
Progesterone and BCSCs
Progesterone plays a pivotal role in lobuloalveolar development of the mouse mammary gland during pregnancy [52, 53]. In premenopausal women, breast epithelial cell proliferation is highest in the luteal phase of the menstrual cycle during maximum progesterone secretion [54, 55]. Progesterone signaling is mediated by the progesterone receptor (PR), expressed as two isoforms (PRA and PRB) that are only different by a third activation domain on the 5′ end of PRB [56]. Importantly, the ratio of these two isoforms is key in the normal development of the mammary gland [57]. Further evidence from isoform-specific murine mutants demonstrates that mammary gland morphogenesis is linked to PRB, whereas PRA plays a prominent role in the ovarian homeostasis [58, 59]. The gene expression patterns while largely overlapping indicate that PRB can regulate gene expression of more genes in comparison to its counterparts [60].
In normal human breast cells, stimulation with progesterone in matrix-embedded culture increases bi-potent cell numbers [61]. Evidence in mouse models corroborates that progesterone and PR signaling drives mammary gland development by expansion of the mammary stem cell population; this signaling is also appropriated in carcinogen-induced mammary tumor formation [62,63,64]. In established cancer cell lines, progestin administration leads to an increase of progenitor cells and CSC markers [65].
In the normal breast epithelium, the ratio of the PR isoforms remains balanced, but this is disrupted in the cancer setting, favoring the expression of PRA [61, 66]. The increased risk of developing breast cancer has been linked to atypical hyperplasia [67] which often exhibits loss of PRB, equally, altered ER expression, sole PRA expression, and preferential PRB loss is also reported in the normal breast tissue of women with germline BRCA1/2 mutations [68]. Such women demonstrate PR isoform imbalance and double the circulating progesterone levels compared to matched controls; however, the cause and significance of these findings remain obscure [69].
During mammary gland expansion, PR mediates proliferation via paracrine signals, including RANKL (receptor activator of nuclear factor-κB ligand) and Wnt4. These signals are secreted from PR+ sensor cells and act on PR− progenitor cells, expressing RANK and the Wnt receptors Frizzled and LRP5/6 [63, 64]. In multiple rodent models, deletion or inhibition of PR or the RANK/RANKL pathway results in significant reduction in mammary carcinogenesis [62, 63, 70, 71]. More recently, Nolan and colleagues have also shown the potential of RANKL as a therapeutic target in a Brca1-deficient mouse model, while in normal breast tissue of BRCA1-mutation carriers, identifying luminal RANK(+) progenitors that are highly proliferative and bear a molecular signature similar to that of basal-like BC, this indicates RANKL inhibition as a promising strategy in the prevention setting [72•].
One of the mediators of progesterone-induced stem/progenitor cell functions in normal mammary gland is CXCR4/CXCL12 [73]. Signaling by progesterone occurs in a paracrine manner on luminal cells expressing CXCL12 while CXCR4 expression is also induced in both basal and luminal PR− cells. Inhibition of CXCR4-CXCL12 signaling axis can arrest the progesterone induced expansion of mammary stem/progenitor cells. Ginestier and colleagues translated the inhibition of CXCR1 with either a specific blocking antibody or by methanesulfonamide (a CXCR inhibitor known as Reparixin), in which both depleted the CSC population of two BC cell lines in vitro and in vivo [74]. This approach is currently under evaluation in the clinic, using reparixin in both early and advance breast cancer (Table 2). A further effect of progesterone is the secretion of growth hormone (GH) in human breast epithelial cells, driving proliferation of the stem/progenitor breast cells expressing growth hormone receptor (GHR) [75].
Similarly, the increase in CK5+ cell population (linked to tumor-initiating properties and therapy resistance) and CD44hi or CD44+CD24− BCSCs has been linked to progesterone in several ER+PR+ cell lines but particularly in T47D cells, which have high PR levels, through gene amplification, even in the absence of estrogen [65, 76,77,78]. In cell lines where PR expression is still dependent on estrogen, co-stimulation with estrogen and progesterone is required, while estrogen alone was not able to induce BCSCs.
In terms of potential mechanisms, reports have shown that PR signaling inhibits the expression of miR-29 and miR-141, while de-repressing KLF4 and STAT5A, respectively [77, 79]. Both studies showed expansion of the CK5+/CD44+ CSC population with an increase in colony formation and in vivo tumor initiating capacity. KLF4 is a transcription factor required for maintenance of both BCSCs [80] and pluripotency in embryonic stem cells [81] whereas STAT5A is a transcription factor that regulates the mammary luminal progenitor population [82]. The maintenance of leukemic stem cells heavily depends of BCL6 expression while also essential for progesterone-induction of CK5+ cells in luminal breast cancer [83]. This progesterone-induced expression of BCL6 is suppressed by prolactin, further demonstrating the interplay taking place in hormonal signaling in the regulation of BCSCs [84]. The paracrine signaling taking place in the normal mammary gland between PR+ and PR− cells may indeed be acting in the same fashion with PR-BCSCs. Furthermore paracrine signaling of non-endogenous overexpressed RANKL in human breast cell lines increases the CD44+CD24− BCSC pool, promoting tumor initiation and metastasis [85]. However, despite strong preclinical data, clinical trials of denosumab, a monoclonal antibody targeting RANKL, have not translated to any improvement in cancer specific survival despite their valuable role in reducing skeletal complications from bone metastases. Altogether, this large body of evidence indicates that the expansion of both normal and BCSC is largely or in part driven by progesterone, although the exact mechanisms remain to be elucidated. Inhibition of PR directly or its paracrine/downstream mediators could translate to rational drug targets for breast cancer prevention and therapy.
Anti-progesterone Drugs and BCSCs
The Women’s Health Initiative study reports that combination of estrogen with progestin (synthetic progesterone derivative), but not estrogen alone, was associated with an increased breast cancer incidence and mortality [86]. Tumorigenesis in the mammary gland can be attributed to the effects of progesterone signaling expanding the stem cell pool, which may transform to BCSCs and eventually lead to the formation of ER+/PR+ tumors [87]. Recent reports in vitro have shown that natural and synthetic progestins can increase CSC-related markers ALDHhigh and CD44high (Table 1) and that this enrichment of a subpopulation of cancer cells may be of functional significance in the development of BC in vivo [65]. The potential of progestin modulators as anti-tumor agents has recently been addressed using a patient derived xenograft model of breast cancer; investigators showed ulipristal acetate, a selective progesterone receptor modulator translated to significant anti-tumor effect, with reduction in Ki67 and Cyclin D1 [88].
During much of the 1990s, a great investment in anti-progestins as therapeutic agents was seen. Several trials were initiated for BC and other indications, as monotherapies and or in combinations. However, despite much interest, no anti-progestin is currently used as the recommended standard of care in any cancer setting either through lack of activity or tolerability. An example of these was the onapristone phase I trials which showed liver function test abnormalities, halting its clinical development [89, 90]. Recent years have seen a new series of clinical trials (Table 3) using anti-progestin drugs like mifepristone and onapristone (the latter, now administered in a new formulation to avoid previous observed hepatoxicity) in breast cancer and other solid tumors [91,92,93,94,95]. Based on recent research literature, these drugs may target BCSCs in ER+/PR+ tumors; although hypothetical, this merits further investigation. Trials in the prevention setting are also investigating the effects of ulipristal acetate, a selective progesterone receptor modulator, assessing proliferation and CSC markers in normal breast tissue [94]. New evidence on the complex interaction of estrogen and progesterone now elucidates the co-regulation of proliferative signaling under both steroid hormones. Progesterone acting through PR is able to ameliorate the effects of estrogen by reducing it activation of downstream effectors [96••]. This may offer and explanation as to why double positive endocrine tumors (ER+/PR+) are classified as less aggressive than single ER + breast cancers, translating to better prognosis [96••, 97]. A hypothesis currently being evaluated in clinical trials investigates the potential benefits of PR agonists, as single agents in improving and prolonging response or in combination with cholecalciferol or letrozole (Table 3). The renewed interest in PR as potential therapeutic target may hold clinical benefits by either modulating ER co-regulations by progestins or by reducing the progenitor pool via PR signaling. Despite new advances and insights into the effects of PR, more clinical work is need to validate the preclinical data.
Conclusions
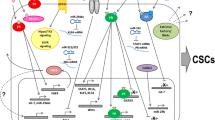
In breast cancer, both estrogen and progesterone signaling have effects on CSC activity. BCSCs are reported to be low or negative for steroid hormone receptors, and therefore, effects must be indirect, mediated through paracrine or juxtacrine cell–cell signaling (Fig. 1). It remains possible that there is a degree of autocrine signaling downstream of hormones that regulates some BCSCs. The effects of estrogen and progesterone have only been partly described in cancer, for progesterone particularly; there is more data from normal mammary epithelium than from cancer. For estrogen, there is evidence that following in vitro treatment of serum-starved breast cancer cells, CSC activity is upregulated and that this is regulated by EGF, FGF, or Notch1 receptors, indicating indirect, paracrine or juxtacrine signaling between cells (Fig. 1). In contrast, anti-estrogens, such as tamoxifen or fulvestrant, block direct estrogenic effects on cell proliferation, and indirect signals to the ER− BCSCs. Surprisingly, however, tamoxifen can actually increase BCSC activity in mammosphere colony culture [38, 39, 98], and more recently, the same has been confirmed for both tamoxifen and fulvestrant in vivo [38]. In breast cancer, both estrogen and progesterone signaling have effects on CSC activity. BCSCs are reported to be low or negative for steroid hormone receptors, and therefore, effects must be indirect, mediated through paracrine or juxtacrine cell–cell signaling (Fig. 1). It remains possible that there is a degree of autocrine signaling downstream of hormones that regulates some BCSCs. The effects of estrogen and progesterone have only been partly described in cancer, for progesterone particularly; there is more data from normal mammary epithelium than from cancer. For estrogen, there is evidence that following in vitro treatment of serum-starved breast cancer cells, CSC activity is upregulated and that this is regulated by EGF, FG,F or Notch1 receptors, indicating indirect, paracrine or juxtacrine signaling between cells (Fig. 1). In contrast, anti-estrogens, such as tamoxifen or fulvestrant, block direct estrogenic effects on cell proliferation and indirect signals to the ER− BCSCs. Nevertheless, as mentioned previously, tamoxifen can actually increase BCSC activity [38].

Representation of juxtacrine and paracrine signals involved in estrogen and progesterone regulation of BCSCs. Estrogen (E2) and progesterone (Pg) bind to their receptors along with nuclear transcription factors, respectively, regulating expression of downstream target genes. Estrogen sensor cells (non-BCSCs) increase transcription of EGF (epidermal growth factor), AREG (amphiregulin), TGFα (transforming growth factor α), and FGF (fibroblast growth factor), which will signal to the BCSCs through the EGFR and FGFR receptors. Non-BCSCs can also signal with BCSCs via Notch signaling. Progesterone sensor cells (non-BCSCs) upregulate the transcription of several key signaling factors. Regulation of BCSCs via Pg may occur via activation of RANK/RANKL, Wnt receptors/Wnt4, CXCR4/CXCL12, and GHR/GH paracrine signaling (dashed lines). Estrogen and progesterone-induced signals can be blocked by anti-estrogens (e.g., tamoxifen and fulvestrant) and anti-progesterone drugs (e.g., mifepristone and onapristone)
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. https://doi.org/10.1038/35102167.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–8. https://doi.org/10.1073/pnas.0530291100.
Tsang JY, Huang YH, Luo MH, Ni YB, Chan SK, Lui PC, et al. Cancer stem cell markers are associated with adverse biomarker profiles and molecular subtypes of breast cancer. Breast Cancer Res Treat. 2012;136(2):407–17. https://doi.org/10.1007/s10549-012-2271-6.
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–67. https://doi.org/10.1016/j.stem.2007.08.014.
Hwang-Verslues WW, Kuo WH, Chang PH, Pan CC, Wang HH, Tsai ST, et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PLoS One. 2009;4(12):e8377. https://doi.org/10.1371/journal.pone.0008377.
Bensimon J, Altmeyer-Morel S, Benjelloun H, Chevillard S, Lebeau J. CD24(−/low) stem-like breast cancer marker defines the radiation-resistant cells involved in memorization and transmission of radiation-induced genomic instability. Oncogene. 2013;32(2):251–8. https://doi.org/10.1038/onc.2012.31.
•• Sansone P, Ceccarelli C, Berishaj M, Chang Q, Rajasekhar VK, Perna F, et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat Commun. 2016;7:10442. https://doi.org/10.1038/ncomms10442. The study demonstrates that the enrichment of CD133hi/ERlo cancer cells is found in clinical samples after neoadjuvant endocrine therapy and in refractory metastatic disease. They represent a therapy-resistant, self-renewing (CD133hi/ERlo/IL6hi) pool of cancer stem cells which are linked to the main cause of relapse.
Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Research : BCR. 2008;10(1):R10. https://doi.org/10.1186/bcr1855.
Liu TJ, Sun BC, Zhao XL, Zhao XM, Sun T, Gu Q, et al. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene. 2013;32(5):544–53. https://doi.org/10.1038/onc.2012.85.
Vaillant F, Asselin-Labat ML, Shackleton M, Forrest NC, Lindeman GJ, Visvader JE. The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. 2008;68(19):7711–7. https://doi.org/10.1158/0008-5472.CAN-08-1949.
Vassilopoulos A, Chisholm C, Lahusen T, Zheng H, Deng CX. A critical role of CD29 and CD49f in mediating metastasis for cancer-initiating cells isolated from a Brca1-associated mouse model of breast cancer. Oncogene. 2014;33(47):5477–82. https://doi.org/10.1038/onc.2013.516.
Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, et al. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439(7079):993–7. https://doi.org/10.1038/nature04496.
Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140(1):62–73. https://doi.org/10.1016/j.cell.2009.12.007.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci. 2003;100(7):3983–8. https://doi.org/10.1073/pnas.0530291100.
Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356(3):217–26. https://doi.org/10.1056/nejmoa063994.
Honeth G, Bendahl P-O, Ringnér M, Saal LH, Gruvberger-Saal SK, Lövgren K et al. The CD44+/CD24-phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008;10(3). doi:https://doi.org/10.1186/bcr2108.
Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17(10):1253–70. https://doi.org/10.1101/gad.1061803.
Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65(13):5506–11. https://doi.org/10.1158/0008-5472.can-05-0626.
Farnie G, Clarke RB, Spence K, Pinnock N, Brennan K, Anderson NG, et al. Novel cell culture technique for primary ductal carcinoma in situ: role of notch and epidermal growth factor receptor signaling pathways. JNCI Journal of the National Cancer Institute. 2007;99(8):616–27. https://doi.org/10.1093/jnci/djk133.
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–67. https://doi.org/10.1016/j.stem.2007.08.014.
Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res. 2010;16(1):45–55. https://doi.org/10.1158/1078-0432.CCR-09-1630.
Simoes BM, Alferez DG, Howell SJ, Clarke RB. The role of steroid hormones in breast cancer stem cells. Endocr Relat Cancer. 2015;22(6):T177–86. https://doi.org/10.1530/ERC-15-0350.
Gomez-Miragaya J, González-Suárez E. Tumor-initiating CD49f cells are a hallmark of chemoresistant triple negative breast cancer. Mol Cell Oncol. 2017;4(4):e1338208. https://doi.org/10.1080/23723556.2017.1338208.
Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. 2014;2(1):78–91. https://doi.org/10.1016/j.stemcr.2013.11.009.
Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–54. https://doi.org/10.1038/nrc822.
Yang M-H, Wu M-Z, Chiou S-H, Chen P-M, Chang S-Y, Liu C-J, et al. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat Cell Biol. 2008;10(3):295–305. https://doi.org/10.1038/ncb1691.
Arendt LM, Kuperwasser C. Form and function: how estrogen and progesterone regulate the mammary epithelial hierarchy. J Mammary Gland Biol Neoplasia. 2015;20(1–2):9–25. https://doi.org/10.1007/s10911-015-9337-0.
Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. 1987;51(6):941–51. https://doi.org/10.1016/0092-8674(87)90581-2.
Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138(3):863–70. https://doi.org/10.1210/endo.138.3.4979.
Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122(1):33–43. https://doi.org/10.1016/j.cell.2005.05.008.
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. https://doi.org/10.1038/35021093.
Osborne CK, Schiff R, Arpino G, Lee AS, Hilsenbeck VG. Endocrine responsiveness: understanding how progesterone receptor can be used to select endocrine therapy. Breast. 2005;14(6):458–65. https://doi.org/10.1016/j.breast.2005.08.024.
Clemons M, Goss P. Estrogen and the risk of breast cancer. N Engl J Med. 2001;344(4):276–85. https://doi.org/10.1056/NEJM200101253440407.
LaCroix AZ, Chlebowski RT, Manson JE, Aragaki AK, Johnson KC, Martin L, et al. Health outcomes after stopping conjugated equine estrogens among postmenopausal women with prior hysterectomy: a randomized controlled trial. JAMA. 2011;305(13):1305–14. https://doi.org/10.1001/jama.2011.382.
Simoes BM, Vivanco MD. Cancer stem cells in the human mammary gland and regulation of their differentiation by estrogen. Future Oncol. 2011;7(8):995–1006. https://doi.org/10.2217/fon.11.80.
Morimoto K, Kim SJ, Tanei T, Shimazu K, Tanji Y, Taguchi T, et al. Stem cell marker aldehyde dehydrogenase 1-positive breast cancers are characterized by negative estrogen receptor, positive human epidermal growth factor receptor type 2, and high Ki67 expression. Cancer Sci. 2009;100(6):1062–8. https://doi.org/10.1111/j.1349-7006.2009.01151.x.
Harrison H, Simões BM, Rogerson L, Howell SJ, Landberg G, Clarke RB. Oestrogen increases the activity of oestrogen receptor negative breast cancer stem cells through paracrine EGFR and Notch signalling. Breast Cancer Res. 2013;15(2). doi:https://doi.org/10.1186/bcr3396.
Simoes BM, O'Brien CS, Eyre R, Silva A, Yu L, Sarmiento-Castro A, et al. Anti-estrogen resistance in human breast tumors is driven by JAG1-NOTCH4-dependent cancer stem cell activity. Cell Rep. 2015;12(12):1968–77. https://doi.org/10.1016/j.celrep.2015.08.050.
Simoes BM, Piva M, Iriondo O, Comaills V, Lopez-Ruiz JA, Zabalza I, et al. Effects of estrogen on the proportion of stem cells in the breast. Breast Cancer Res Treat. 2011;129(1):23–35. https://doi.org/10.1007/s10549-010-1169-4.
Fisher B, Redmond C, Fisher ER, Caplan R. Relative worth of estrogen or progesterone receptor and pathologic characteristics of differentiation as indicators of prognosis in node negative breast cancer patients: findings from National Surgical Adjuvant Breast and Bowel Project Protocol B-06. J Clin Oncol: Off J Am Soc Clin Oncol. 1988;6(7):1076–87. https://doi.org/10.1200/JCO.1988.6.7.1076.
Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U S A. 2010;107(50):21737–42. https://doi.org/10.1073/pnas.1007863107.
Harrison H, Rogerson L, Gregson HJ, Brennan KR, Clarke RB, Landberg G. Contrasting hypoxic effects on breast cancer stem cell hierarchy is dependent on ER-alpha status. Cancer Res. 2013;73(4):1420–33. https://doi.org/10.1158/0008-5472.CAN-12-2505.
Axlund SD, Yoo BH, Rosen RB, Schaack J, Kabos P, Labarbera DV, et al. Progesterone-inducible cytokeratin 5-positive cells in luminal breast cancer exhibit progenitor properties. Hormones Cancer. 2013;4(1):36–49. https://doi.org/10.1007/s12672-012-0127-5.
Deng H, Zhang XT, Wang ML, Zheng HY, Liu LJ, Wang ZY. ER-alpha36-mediated rapid estrogen signaling positively regulates ER-positive breast cancer stem/progenitor cells. PLoS One. 2014;9(2):e88034. https://doi.org/10.1371/journal.pone.0088034.
Ma R, Karthik GM, Lovrot J, Haglund F, Rosin G, Katchy A, et al. Estrogen receptor beta as a therapeutic target in breast cancer stem cells. J Natl Cancer Inst. 2017;109(3):1–14. https://doi.org/10.1093/jnci/djw236.
•• Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526(7571):131–5. https://doi.org/10.1038/nature15260. This paper provides evidence of a hierarchical model for metastasis, in which metastases are initiated by stem-like cells that proliferate and differentiate to produce advanced metastatic disease.
Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62(1):233–47. https://doi.org/10.1146/annurev-med-070909-182917.
Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106(33):13820–5. https://doi.org/10.1073/pnas.0905718106.
Piva M, Domenici G, Iriondo O, Rábano M, Simões BM, Comaills V, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014;6(1):66–79. https://doi.org/10.1002/emmm.201303411.
Sansone P, Berishaj M, Rajasekhar VK, Ceccarelli C, Chang Q, Strillacci A, et al. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res. 2017;77(8):1927–41. https://doi.org/10.1158/0008-5472.CAN-16-2129.
Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci U S A. 2017;114(43):E9066–E75. https://doi.org/10.1073/pnas.1704862114.
Lydon JP, DeMayo FJ, Funk CR, Mani SK, Hughes AR, Montgomery CA Jr, et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9(18):2266–78. https://doi.org/10.1101/gad.9.18.2266.
Brisken C. Progesterone signalling in breast cancer: a neglected hormone coming into the limelight. Nat Rev Cancer. 2013;13(6):385–96. https://doi.org/10.1038/nrc3518.
Potten CS, Watson RJ, Williams GT, Tickle S, Roberts SA, Harris M, et al. The effect of age and menstrual cycle upon proliferative activity of the normal human breast. Br J Cancer. 1988;58(2):163–70. https://doi.org/10.1038/bjc.1988.185.
Navarrete MA, Maier CM, Falzoni R, Quadros LG, Lima GR, Baracat EC, et al. Assessment of the proliferative, apoptotic and cellular renovation indices of the human mammary epithelium during the follicular and luteal phases of the menstrual cycle. Breast Cancer Res: BCR. 2005;7(3):R306–13. https://doi.org/10.1186/bcr994.
Kastner P, Krust A, Turcotte B, Stropp U, Tora L, Gronemeyer H, et al. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990;9(5):1603–14.
Shyamala G, Yang X, Cardiff RD, Dale E. Impact of progesterone receptor on cell-fate decisions during mammary gland development. Proc Natl Acad Sci U S A. 2000;97(7):3044–9. https://doi.org/10.1073/pnas.97.7.3044.
Mulac-Jericevic B, Mullinax RA, DeMayo FJ, Lydon JP, Conneely OM. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform. Science. 2000;289(5485):1751–4. https://doi.org/10.1126/science.289.5485.1751.
Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci U S A. 2003;100(17):9744–9. https://doi.org/10.1073/pnas.1732707100.
Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277(7):5209–18. https://doi.org/10.1074/jbc.M110090200.
Graham JD, Mote PA, Salagame U, van Dijk JH, Balleine RL, Huschtscha LI, et al. DNA replication licensing and progenitor numbers are increased by progesterone in normal human breast. Endocrinology. 2009;150(7):3318–26. https://doi.org/10.1210/en.2008-1630.
Lydon JP, Ge G, Kittrell FS, Medina D, O’Malley BW. Murine mammary gland carcinogenesis is critically dependent on progesterone receptor function. Cancer Res. 1999;59(17):4276–84.
Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, et al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature. 2010;468(7320):103–7. https://doi.org/10.1038/nature09495.
Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, et al. Progesterone induces adult mammary stem cell expansion. Nature. 2010;465(7299):803–7. https://doi.org/10.1038/nature09091.
Goyette S, Liang Y, Mafuvadze B, Cook MT, Munir M, Hyder SM. Natural and synthetic progestins enrich cancer stem cell-like cells in hormone-responsive human breast cancer cell populations in vitro. Breast Cancer (Dove Med Press). 2017;9:347–57. https://doi.org/10.2147/BCTT.S135371.
Graham JD, Yager ML, Hill HD, Byth K, O’Neill GM, Clarke CL. Altered progesterone receptor isoform expression remodels progestin responsiveness of breast cancer cells. Mol Endocrinol. 2005;19(11):2713–35. https://doi.org/10.1210/me.2005-0126.
Hartmann LC, Degnim AC, Dupont WD. Atypical hyperplasia of the breast. N Engl J Med. 2015;372(13):1271–2. https://doi.org/10.1056/NEJMc1501046.
Mote PA, Leary JA, Avery KA, Sandelin K, Chenevix-Trench G, Kirk JA, et al. Germ-line mutations in BRCA1 or BRCA2 in the normal breast are associated with altered expression of estrogen-responsive proteins and the predominance of progesterone receptor A. Genes Chromosomes Cancer. 2004;39(3):236–48. https://doi.org/10.1002/gcc.10321.
Widschwendter M, Rosenthal AN, Philpott S, Rizzuto I, Fraser L, Hayward J, et al. The sex hormone system in carriers of BRCA1/2 mutations: a case-control study. The Lancet Oncology. 2013;14(12):1226–32. https://doi.org/10.1016/S1470-2045(13)70448-0.
Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature. 2010;468(7320):98–102. https://doi.org/10.1038/nature09387.
Poole AJ, Li Y, Kim Y, Lin SC, Lee WH, Lee EY. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314(5804):1467–70. https://doi.org/10.1126/science.1130471.
• Nolan E, Vaillant F, Branstetter D, Pal B, Giner G, Whitehead L, et al. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat Med. 2016;22(8):933–9. https://doi.org/10.1038/nm.4118. This study shows a targetable pathway in the cells of origin for BRCA1 mutation carriers and implicates RANKL inhibition as a promising strategy in the prevention of breast cancer.
Shiah YJ, Tharmapalan P, Casey AE, Joshi PA, McKee TD, Jackson HW, et al. A progesterone-CXCR4 axis controls mammary progenitor cell fate in the adult gland. Stem Cell Reports. 2015;4(3):313–22. https://doi.org/10.1016/j.stemcr.2015.01.011.
Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010;120(2):485–97. https://doi.org/10.1172/JCI39397.
Lombardi S, Honeth G, Ginestier C, Shinomiya I, Marlow R, Buchupalli B, et al. Growth hormone is secreted by normal breast epithelium upon progesterone stimulation and increases proliferation of stem/progenitor cells. Stem cell reports. 2014;2(6):780–93. https://doi.org/10.1016/j.stemcr.2014.05.005.
Kabos P, Haughian JM, Wang X, Dye WW, Finlayson C, Elias A, et al. Cytokeratin 5 positive cells represent a steroid receptor negative and therapy resistant subpopulation in luminal breast cancers. Breast Cancer Res Treat. 2011;128(1):45–55. https://doi.org/10.1007/s10549-010-1078-6.
Finlay-Schultz J, Cittelly DM, Hendricks P, Patel P, Kabos P, Jacobsen BM, et al. Progesterone downregulation of miR-141 contributes to expansion of stem-like breast cancer cells through maintenance of progesterone receptor and Stat5a. Oncogene. 2015;34(28):3676–87. https://doi.org/10.1038/onc.2014.298.
Hilton HN, Santucci N, Silvestri A, Kantimm S, Huschtscha LI, Graham JD, et al. Progesterone stimulates progenitor cells in normal human breast and breast cancer cells. Breast Cancer Res Treat. 2014;143(3):423–33. https://doi.org/10.1007/s10549-013-2817-2.
Cittelly DM, Finlay-Schultz J, Howe EN, Spoelstra NS, Axlund SD, Hendricks P, et al. Progestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene. 2013;32(20):2555–64. https://doi.org/10.1038/onc.2012.275.
Yu F, Li J, Chen H, Fu J, Ray S, Huang S, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30(18):2161–72. https://doi.org/10.1038/onc.2010.591.
Zhang P, Andrianakos R, Yang Y, Liu C, Lu W. Kruppel-like factor 4 (Klf4) prevents embryonic stem (ES) cell differentiation by regulating Nanog gene expression. J Biol Chem. 2010;285(12):9180–9. https://doi.org/10.1074/jbc.M109.077958.
Yamaji D, Na R, Feuermann Y, Pechhold S, Chen W, Robinson GW, et al. Development of mammary luminal progenitor cells is controlled by the transcription factor STAT5A. Genes Dev. 2009;23(20):2382–7. https://doi.org/10.1101/gad.1840109.
Hurtz C, Hatzi K, Cerchietti L, Braig M, Park E, Kim YM, et al. BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J Exp Med. 2011;208(11):2163–74. https://doi.org/10.1084/jem.20110304.
Sato T, Tran TH, Peck AR, Girondo MA, Liu C, Goodman CR, et al. Prolactin suppresses a progestin-induced CK5-positive cell population in luminal breast cancer through inhibition of progestin-driven BCL6 expression. Oncogene. 2014;33(17):2215–24. https://doi.org/10.1038/onc.2013.172.
Palafox M, Ferrer I, Pellegrini P, Vila S, Hernandez-Ortega S, Urruticoechea A, et al. RANK induces epithelial-mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res. 2012;72(11):2879–88. https://doi.org/10.1158/0008-5472.CAN-12-0044.
Chlebowski RT, Anderson GL, Gass M, Lane DS, Aragaki AK, Kuller LH, et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA. 2010;304(15):1684–92. https://doi.org/10.1001/jama.2010.1500.
Narod SA. Hormone replacement therapy and the risk of breast cancer. Nat Rev Clin Oncol. 2011;8(11):669–76. https://doi.org/10.1038/nrclinonc.2011.110.
Esber N, Cherbonnier C, Resche-Rigon M, Hamze A, Alami M, Fagart J, et al. Anti-tumoral effects of anti-progestins in a patient-derived breast cancer xenograft model. Hormones & Cancer. 2016;7(2):137–47. https://doi.org/10.1007/s12672-016-0255-4.
Robertson JFR, Willsher PC, Winterbottom L, Blamey RW, Thorpe S. Onapristone, a progesterone receptor antagonist, as first-line therapy in primary breast cancer. Eur J Cancer. 1999;35(2):214–8. https://doi.org/10.1016/S0959-8049(98)00388-8.
Jonat W, Bachelot T, Ruhstaller T, Kuss I, Reimann U, Robertson JF. Randomized phase II study of lonaprisan as second-line therapy for progesterone receptor-positive breast cancer. Ann Oncol: Off J Eur Soc Med Oncol. 2013;24(10):2543–8. https://doi.org/10.1093/annonc/mdt216.
NCT02046421. Carboplatin, gemcitabine hydrochloride, and mifepristone in treating patients with advanced breast cancer or recurrent or persistent ovarian epithelial, fallopian tube, or primary peritoneal cancer. -. 2014.(https://clinicaltrials.gov/ct2/show/NCT02046421?term=NCT02046421&rank=1). doi:-.
NCT02049190. Phase 1–2 study of onapristone in patients with advanced castration-resistant prostate cancer. United Kingdom. 2014.(https://clinicaltrials.gov/ct2/show/NCT02049190?term=ONAPRISTONE&rank=1). doi:-.
NCT02052128. Phase 1–2 study of onapristone in patients with progesterone receptor expressing cancers. France. 2014.(https://clinicaltrials.gov/ct2/show/NCT02052128?term=ONAPRISTONE&rank=2).
NCT02408770. Breast cancer—Anti-Progestin Prevention Study 1 (BC-APPS1). UK. 2015.(https://clinicaltrials.gov/ct2/show/NCT02408770?term=NCT02408770&rank=1).
NCT02014337. Mifepristone and eribulin in patients with metastatic triple negative breast ancer or other specified solid tumors. 2013.(https://clinicaltrials.gov/ct2/show/NCT02014337?term=NCT02014337&rank=1).
•• Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, et al. Progesterone receptor modulates ERα action in breast cancer. Nature. 2015;523(7560):313–7. https://doi.org/10.1038/nature14583. https://www.nature.com/articles/nature14583#supplementary-information. This paper provides evidence showing PR functions as a modulator of ERα binding, with important implications for prognosis and therapeutic interventions for patients
Bae SY, Kim S, Lee JH, Lee HC, Lee SK, Kil WH, et al. Poor prognosis of single hormone receptor- positive breast cancer: similar outcome as triple-negative breast cancer. BMC Cancer. 2015;15(1):138. https://doi.org/10.1186/s12885-015-1121-4.
Piva M, Domenici G, Iriondo O, Rabano M, Simoes BM, Comaills V, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Molecular Medicine. 2014;6(1):66–79. https://doi.org/10.1002/emmm.201303411.
Acknowledgments
The authors would like to apologize to those authors whose work was not cited due to space limitations.
Funding
The authors’ work is supported by funding from Breast Cancer Now.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Denis G. Alferez, Dr. Bruno M. Simões, Dr. Sacha J. Howell, and Dr. Robert B. Clarke declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Cell:Cell Interactions in Stem Cell Maintenance
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Alferez, D.G., Simões, B.M., Howell, S.J. et al. The Role of Steroid Hormones in Breast and Effects on Cancer Stem Cells. Curr Stem Cell Rep 4, 81–94 (2018). https://doi.org/10.1007/s40778-018-0114-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-018-0114-z