
Abstract
Background
Knowledge of genetic markers that are correlated to stress tolerance may improve spatial mapping of reef vulnerability and can inform restoration efforts, including the choice of genotypes for breeding and reseeding. In this manuscript we present two methods for screening transcriptome data for candidate genetic markers in two reef building corals, Acropora millepora and Pocillopora damicornis (types α and β). In A. millepora, Single Nucleotide Polymorphisms (SNPs) were pre-selected by targeting genes believed to be involved in the coral thermal stress responses. In P. damicornis (type α and β), SNPs showing varying allele frequencies between two populations from distinct environments were pre-selected. Allele frequencies at nine, five and eight of the pre-selected SNP loci were correlated against gradients of water clarity and temperature in a large number of populations along the Great Barrier Reef.
Results
A significant correlation between environmental category and SNP allele frequency was detected in up to 55% of the tested loci, which is an exceptional success rate for these types of tests. In P. damicornis, SNP allele frequencies of β-hexosaminidase and Elongation factor 1-α were significantly correlated to temperature in type α and to temperature and/or water clarity respectively in type β. Type α also showed a correlation between water clarity and SNP allele frequency in a gene of unknown function. In A. millepora, allele frequencies at five (β-gamma crystallin, Galaxin, Ubiquitin, Ligand of Numb X2 and Thioredoxin) SNP loci showed significant correlations.
Conclusions
After validation of these candidate loci through laboratory or field assessment of relative stress tolerance of colonies harbouring different alleles, it is anticipated that a proportion of these markers may represent the first coral candidate Quantitative Trait Loci for environmental stress tolerance and provide an important genetic tool that can be incorporated into spatial management decisions and restoration efforts of coral reefs. One pertinent example would be to combine spatial data of tolerant populations with genetic connectivity and thus identify high priority conservation reefs and implement targeted coral husbandry and active restoration efforts that use locally- and stress-adapted genotypes.
Similar content being viewed by others

Background
Current models of the Earth’s climate over the coming decades predict rapid changes in a range of parameters including temperature, rainfall, and ocean pH [1]. Given the known and forecasted impacts of climate change on coral reef ecosystems [2–6], a priority area in conservation initiatives should be the determination of the amount of adaptive genetic variation, i.e. variation at loci that have an effect on fitness [7], in key habit forming organisms such as corals. Particularly, information on genetic markers that are correlated to stress tolerance will contribute to the accuracy of spatial mapping of reef vulnerability and can be incorporated into controlled restoration efforts.
There are numerous approaches to detect adaptive genetic variation and signatures of local adaptation (reviewed in [8]). In non-model species, population genomics has become the most commonly used approach for identifying genetic loci that are influenced by selection. Population genomics is a genome-wide application of population genetics, whereby a large number of loci are analysed in multiple populations, allowing for the discrimination between locus-specific (selection) and genome-wide (drift and migration) effects [9, 10] or for site specific adaptation in panmictic species [11]. Some of the more commonly reported methods include the detection of non-random population divergence at specific loci (FST outliers) [12–14], elucidating the genetic architecture of specific phenotypes [15], and associating the frequency distribution of alleles to one or more ecological variables (genetic-environment associations, GEAs) [16, 17]. To date, evidence for genetic variation that is significantly associated to environmental heterogeneity, fitness and phenotype remains scarce [18–20].
Adaptive genetic variation and its link to phenotypic traits has been widely studied in forestry (reviewed in [21]. However, in many non-model organisms, the discovery of phenotype-linked genetic markers has often been serendipitous. For instance, a correlation between allozyme variation and variable thermal stress tolerance occurs in the fish Fundulus heteroclitus, where the gene Lactate dehydrogenase-B shows differences in allele frequencies between populations from distinct thermal environments [22]. This variation is responsible for differences in physiological function and survival at high temperatures. Other examples of genotype to phenotype associations include the link between polymorphism in the Dopamine receptor D4 and personality in great tits (Parus major) [23], and the correlation between melanism and variation in the Melancortin-1 receptor gene (Mc1r) across several species, including moth, mice and humans [18]. In the panmictic American eel, correlations between environmental variables and SNP allele frequencies have been attributed to post dispersal localised selection [11]. The discovery of links such as those described above remains a challenge. However, the increased accessibility of genomic tools and data from non-model organisms make this a rapidly expanding research area.
The Great Barrier Reef (GBR) is the largest coral reef ecosystem in the world. It spans over 2300 km along the East Coast of Australia and encompasses almost 3000 reefs [24]. Like most of the world’s coral reefs, the GBR is under the influence of a range of direct and indirect anthropogenic stressors. Corals derive a large proportion of their energetic needs from the photosynthetic products of an endosymbiotic dinoflagellate (genus Symbiodinium) [25] and thrive in clear, nutrient poor tropical waters, often near their upper thermal tolerance. Not surprisingly, current research shows that two of the main threats to the long term health of the GBR are climate change [5, 26] and declining water quality (in particular, water clarity) due to coastal development and catchment runoff [27, 28].
The most commonly reported thermal stress response in corals is coral bleaching, which is the dissociation between the Symbiodinium and the coral host tissue [2, 29–34]. Variation in bleaching resistance within and among coral species can occur as a result of many factors associated with both the coral host and its Symbiodinium. In the coral Acropora millepora, thermal stress tolerance has been shown to be correlated with ambient water temperatures in three thermally distinct populations [35]. While this is at least partly due to the Symbiodinium type harboured [35], patterns of genetic differentiation at coral host allozyme loci are correlated with latitudinal gradients of sea water temperature [36], suggesting past adaptation of the host genome to local environments. Similarly, in the coral Pocillopora damicornis, laboratory studies have shown a latitudinal difference in bleaching sensitivity that was not correlated to the type of Symbiodinium harboured, indicative of localised adaptation of the coral host to varying thermal regimes [37].
The first studies of gene expression in corals emerged around the turn of the century [38]. Since then, a large number of expressed sequence tags (ESTs) generated through the reverse transcription of gene transcripts (mRNA) expressed during stress, have been made publically available [39–44]. An overlapping subset of stress response genes have been identified in multiple coral species, which allows the identification of key genes and proteins to target for studies of the coral stress response [45]. Acropora millepora has emerged as one of the model species for coral genetics and genomics. The species has been the target of multiple experiments on transcriptome effects of environmental stress [41, 43, 46–50], thus providing comprehensive sequence information for a range of stress response genes. Also, an increasing amount of coral genome and transcriptome data is being made available in public databases [51, 52], including the recent release of the first complete coral genome sequences for Acropora digitifera[53] and A. millepora (http://coralbase.org/). However, the concept of identifying loci, which can be used as markers for relative stress tolerance, remains untested in corals.
This paper presents the first putative genetic markers for environmental stress tolerance in corals. The genetic markers were identified using two novel transcriptome-based strategies. Both strategies make use of next generation sequencing and large scale Single Nucleotide Polymorphism (SNP) genotyping across environmental gradients and build on the concept of identifying GEAs [16]. These strategies increase the likelihood of finding GEAs compared to traditional methods through targeted, population level approaches of transcriptome sequencing and genotyping.
Methods
Two strategies were followed to identify SNPs that show a significant correlation with environmental gradients, which differ in their use of newly generated versus already available transcriptome data. The strategies are not compared with each other for efficiency, rather presented as two alternative approaches to generate a similar outcome.
The first strategy involved three steps: (1) generate population-tagged transcriptome data from a small number of different environmental zones; (2) mine transcriptome data for SNPs that show a distinct difference in allele frequency between the targeted populations; (3) test the selected SNPs on population samples from a more extensive range of the environmental variable. In the second strategy, step (1) was excluded and step (2) was modified to mining already published transcriptome data for SNPs in pre-selected genes; step (3) was the same in both strategies. Both strategies generated genotypic data across a large number of populations from which allele frequencies were calculated. Correlative methods were used to detect significant genome environment associations (GEAs) as per below.
A targeted Next Generation sequencing effort in Pocillopora damicornis
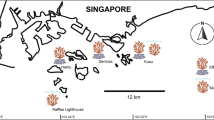
Two nubbins from each of 20 colonies of the common reef coral Pocillopora damicornis were collected from Miall Island in the southern GBR (Keppel Islands), Pelorus Island in the central GBR (Palm Islands), and two sites in the far northern GBR, Wallace Islet and Wilke Reef (Figure 1), spanning a large gradient of temperature and water clarity (Figures 2 and 3), thereby targeting natural gradients of two major stressors on the Great Barrier Reef (GBR), namely elevated sea surface temperatures and declining water quality [4, 54, 55]. The nubbins were snap frozen in liquid nitrogen immediately after collection and stored at −80°C until RNA extractions took place.

Map of sampling locations for P. damicornis (type α) (blue), P. damicornis (type β) (yellow) and A. millepora (red).

Map showing allele frequency patterns of three SNP loci ( β-gamma crystallin right, Galaxin middle and Ubiquitin left) in A. millepora with a significant correlation to temperature categories, plotted on spatial data for average sea surface temperatures between the years 2000 – 2006. Map courtesy of the e-Atlas (http://e-atlas.org.au).

Map showing allele frequency patterns of two SNP loci ( Thioredoxin right, Ligand of numb X2 left) in A. millepora with a significant correlation to water clarity, plotted on spatial data for secchi depths collected between 1992 and 2006. Map courtesy of the e-Atlas (http://e-atlas.org.au).
Recent research has shown that P. damicornis comprises several mtDNA lineages [56, 57], and on the GBR two major distinct lineages (type α & β) have been identified [58]. Therefore, all samples were genotyped using genetic markers that have been developed to separate these two cryptic, sympatric lineages (Torda et al. un-published material). The results revealed that a majority of the colonies that were collected from Wallace Islet and Miall Island reefs were type α, while the Wilke and Pelorus Island reef samples comprised more colonies of type β. As a result, the collections were divided according to their specific lineage.
Messenger RNA was isolated using a modified protocol of the Invitrogen™ Dynabeads® Oligo(dT)25[43]. Two extractions were made from each colony, with a yield of approximately 20 ng/μL per extraction. RNA quality and quantity were verified using the mRNA Pico series kit on the Agilent© 2100 Bioanalyzer. To ensure good quality transcriptome sequences, only samples with low rRNA contamination (<10%) and good quality mRNA were included in the study. As a result, five and six samples of P. damicornis (type β) from Wilke and Pelorus Island reefs were used, respectively. These samples were pooled into two population specific samples. For P. damicornis (type α), mRNA from three Wallace Islet and two Miall Island samples were pooled into a third sample and included in the 454 analysis. The final sequencing was done for three pooled mRNA samples using the 454® Genome Sequencer FLX Titanium Series reagents according to the manufacturer’s instructions (Roche Diagnostics Corporation) by the Australian Genome Research Facility (AGRF).
The 947,623 resulting sequences and quality scores were analysed using PipeMeta, a menu based, desktop interface pipeline toolset for Windows [59, 60]. Sequences in Fasta format were screened for SMART primer (http://www.clontech.com/) using the SmartScreener script from within PipeMeta and then reads and quality scores were trimmed and quality filtered (min/max length: Avg. Length ± 2*SD; minimum avg. quality score: 20; maximum number of degenerate bases: 1). The remaining 653,814 reads and quality scores (73.6% of the original sequencing effort) were then tagged (i.e. α-numeric strings added to titles) by population to differentiate them after assembly. The sample containing the partial transcriptome of P. damicornis (type α) was not included in the final assembly, which was done using the sequences from P. damicornis (type β) only.
Assembly of the timed reads and quality scores (fasta files) was performed using commercially available assembly software (SeqMan NGen 2.1.0 build 13, DNAStar). To further assess the performance of the assembly, all resulting unigenes (Contigs and Singletons over 50 bp) were Blast aligned to a modified set of in silico predicted proteins from the starlet sea anemone Nematostella vectensis[61] and Acropora digitifera[53], the two closest related species to P. damicornis (type β) with sequenced and publically available genomes. In order to better estimate transcriptome coverage of the sequencing effort, both sets of predicted genes were modified to remove shorter protein sequences with greater than 90% similarity to longer sequences in the set.
All P. damicornis (type β) unigenes were Blast aligned to the Uniprot protein database (http://www.expasy.uniprot.org/database/download.shtml) to annotate the assembly.
Targeting SNPs in Pocillopora damicornis and Acropora millepora
The P. damicornis (type β) sequence data were searched for functional SNPs using the SNP hunter extension of the PipeMeta software (http://www.personal.psu.edu/jhm10/Vera/software.html), a tool specifically designed to find SNPs in de novo transcriptome assemblies using a variety of customizable settings and filters. About 24,000 SNP candidates were located within the partial de novo transcriptome assembly and screened to identify potential biomarkers.
The candidate SNPS were narrowed down by applying the following filters. First, all non-annotated contigs were removed, followed by any contigs that did not align to ESTs or genes of known function that were derived from other metazoans (to avoid Symbiodinium sequences). Second, all SNPs that did not have a frequency of > 10% in at least one of the two populations were removed to minimize false positives due to sequencing errors. After this, a search was made for bi-allelic SNPs for which allele frequencies differed by > 20% between the two populations (e.g., if one allele occurred at a frequency of 40% in one population, it had to have a frequency of < 20% or >60% in the other to qualify). Based on these criteria, a final list of 80 SNPs distributed among 67 contigs was created.
Primers for the 80 SNPs were designed using Primer 3 (http://frodo.wi.mit.edu/primer3/) to amplify a 75 – 150 base pair PCR product. PCR amplification was performed using DNA extracts from three colony samples of P. damicornis (type β) and the product was visualized on a 1% TAE-agarose gel. Any primer pair that produced products that were longer than predicted and hence possibly included intron sequences, yielded inconsistent amplification success or displayed any other signs of poor quality, were discarded. A final selection of eight SNPs was made to use on the larger, GBR wide sample set of P. damicornis (type β) (Table 1). Reciprocal BLAST alignment contig IDs from the Taylor-Knowles et al. [62] transcriptome were added to allow comparisons. Five of these were successfully amplified also in P. damicornis (type α).
Due to the prior knowledge and sequence data for a range of stress response genes in A. millepora, the already published transcriptome data set [51] from larvae collected at Magnetic Island, an small inshore island in the central GBR, was downloaded and mined for SNPs using PipeMeta as above. A selection of nine genes was made, based on published information relating to thermal stress responses and SNP information derived from the transcriptome sequence data [51] (Table 2). For each gene, one suitable, non-synonymous SNP site was chosen to be used on the GBR wide sample set (Table 2).
Genotyping along environmental gradients in Pocillopora damicornis and Acropora millepora
As climate change and water quality are considered among the largest threats to coral reef health on the Great Barrier Reef [5, 55], temperature and water clarity were chosen as two highly relevant parameters for stress tolerance. These two environmental variables were extrapolated across the GBR using the e-Atlas tool. Temperature data were derived from Rayner et al. [63] and can be found at http://e-atlas.org.au/content/sea-surface-temperature-hadisst-11 (Figure 2). Secchi disc depth data, which is the most widely available measure of water quality [64], were provided by scientists at the Australian Institute of Marine Science and Queensland Department of Primary Industries and Fisheries and was sourced from http://e-atlas.org.au/content/secchi-disk-depth-measure-water-clarity (Figure 3).
These datasets divide the environmental range into seven temperature and nine secchi disc depth categories, where seven is the highest temperature and nine is the deepest secchi disc depth. All temperature zones were included in the collections. Due to the photosynthetic properties of the coral-Symbiodinium symbiosis, corals are rare in highly turbid waters, hence coral collections spanned from secchi disc depth category 4 (6 – 7.8 m) to category 9 (21.7 – 28 m).
Existing samples of P. damicornis and A. millepora collected for GBR connectivity studies [65] were used for this study. Due to the current putative species delineation of P. damicornis[58] all samples of that species had to be identified using the lineage specific genetic markers mentioned above (Torda et al. submitted manuscript).
After species validations, nine populations of P. damicornis (type β) were chosen based on having a sample size of ≥ 15 colonies. The larger sample collection of P. damicornis (type α) allowed a more comprehensive selection of populations to be made for this species. Ten populations with a sample size of ≥20 colonies, spanning a broader range of thermal categories and at least two replicates of each water clarity category, were chosen for the environmental correlation analyses (Figure 1, Table 3). Eight and five SNP loci were tested in P. damicornis (type β) and (type α), respectively, against the two environmental categories, making a total of 26 tests. For A. millepora, 9 SNP loci were tested against the same environmental categories (total of 20 tests) across 17 populations, all with sample sizes of ≥ 20 (Figure 1, Table 3).
DNA was extracted from a small fragment of coral, following a slightly modified method by Wilson et al. [66]. For both P. damicornis types the SNP genotyping step was done using qPCR High Resolution Melt curve analyses (HRM) (QIAcube ©, QIAGEN) in the following manner: The PCR step was set up in 10 μL reactions incorporating Accumelt HRM supermix (Quanta Biosciences©). Each sample was set up in triplicate with a primer concentration of between 200 nM and 500 nM, and 1 μl of 1/50 diluted gDNA. No template (negative) and known genotype (positive) controls were included in all runs. PCR and HRM were carried out under the conditions recommended by the instrument manufacturer. PCR commenced with an initial 5 minute hold at 95°C followed by 40 cycles of a 10 second, 95°C denaturation and 30 second, 55°C annealing and extension. For the HRM analysis the temperature was ramped from 65°-95°C, rising by 0.1°C each step with a 90 second pre-melt conditioning cycle on the first step and a 2 second wait every cycle thereafter. Prior to HRM, the gain was optimised to set the greatest fluorescence to less than 90 fluorescence units.
Control samples of identified genotypes were included in each PCR /HRM run. CT values and amplification completeness were checked using the Rotorgene Q series software V2.02 and samples showing late or poor amplification were excluded. HRM profiles were normalised against average signatures within a pre-selected temperature range. The control samples were used as references to automatically determine sample genotypes. All genotype calls were checked manually, and confirmed by further analysis using ScreenClust (Qiagen) software.
For A. millepora, genotyping was conducted at the AGRF using a Sequenom® MassArray on an Autoflex Spectrometer using iPLEX GOLD chemistry.
Statistical analyses
Correlations between environmental variables and SNP allele frequency were tested by using a logistic regression with a logit link and a binomial error distribution to relate the probability of allele 1 in each locus to the two environmental variables, temperature and secchi depth [67]. The likelihood ratio and Wald tests were used to determine the significance of the models as in Joost et al. [68].
To test for the proportion of random correlations, 159 alleles from 11 putatively neutral microsatellite loci were also tested for these environment correlations using the already published A. millepora microsatellite data set from the same coral samples [65].
Results
Pocillopora damicornis
Transcriptome sequencing
After preliminary machine quality filtering, 947,623 sequences were obtained. Additional primer screening and quality filtering through PipeMeta removed 27.4% of all sequenced nucleotides. The remaining 653,814 reads were used in the assembly. Roughly 67% of the trimmed, filtered reads (64% of the sequenced nucleotides) assembled to form 87,520 contigs with an average length of 584 (±296) base pairs (bp), with the 244,541 remaining singletons averaging 304 (±85) bp in length (Table 4).
To further assess the quality and coverage achieved by the assembly, the unigenes were BLAST aligned to two reference gene sets from closely related species with sequenced genomes (i.e. Nematostella vectensis and Acropora digitifera modified in silico predicted proteins). Out of these, 10,021 (23.4%) and 11,148 (52.4%) were unique hits to N. vectensis and A. digitifera, respectively. Using a conservative approach to estimate transcriptome coverage length, approximately 16.0% of N. vectensis total amino acids and 25.3% of the A. digitifera total amino acids were covered (i.e. had strong alignment) by P. damicornis (type β) unigene sequence, with median coverage rates of 35.6% and 38.7% per predicted gene with a strong alignment for N. vectensis and A. digitifera proteins. Less than 6.8% (N. vectensis) or 10.2% (A. digitifera) of the above reference gene amino acid coverage was from more than one P. damicornis (type β) unigene (i.e. duplicate or overlapping amino acid coverage by multiple unigenes).
A BLAST alignment versus the Uniprot protein database produced significant hits (e-value cut off of 1×10-3; bitscore > 45) for 54,094 or 61.8% of the Contigs, out of which 44.5% had top alignments to proteins expressed in metazoans , 15.2% aligned to plant, 8.4% aligned to bacteria, and 0.3% aligned to virus (Table 4).
SNPs
From the eight SNPs that were selected from the transcriptome of P. damicornis (type β) only one was a transversion (24_CA_2390). This SNP occurred at Night and Tydeman reefs with the rare allele (A) occurring at a frequency of 18.1% and 7.6% respectively. In the original transcriptome data, this allele occurred at Wilke Islet reef, which is located in the same geographic region as Night and Tydeman (Figure 1). The remaining seven SNPs were all transitions (C/T or G/A). The resolution of the single step PCR to HRM method used in this study did not allow discrimination of A to T or C to G mutations. Hence, all the SNPs analysed by this method caused a change in the number of hydrogen bonds in the DNA chain, which produced a large enough change in the melting temperature to allow accurate scoring.
The final data set for P. damicornis (type β) was based on the allele frequencies at eight SNP loci. In P. damicornis (type α), five loci were amplified using the primers developed for P. damicornis (type B) (Table 1). The other three loci were excluded from trials in P. damicornis (type α) due to ambiguous scoring patterns of the HRM curves in this species (see Additional file 1: Figure S1 for examples of HRM curves). In P. damicornis, the SNPs were found in genes of known and unknown function and included both synonymous and non-synonymous mutations. The genes of known function included Elongation factor 1-α, β-hexosamidase, Carbonic anhydrase and the 40S ribosomal protein S3 (Table 1).
Allele frequency and environment correlations
Using the logistic regression approach, three of the eight tested SNP loci had at least one significant correlation in either P. damicornis type α or type β (Table 5; SNP allele frequencies are presented in Additional file 2: Tables S1 and Additional file 3: Table S2). Calculated as number of loci with significant frequency correlations, two out of eight (25%) were significant in P. damicornis (type β) and three out of five (60%) were significant in P. damicornis (type α). These correlations were found between temperature and SNP allele frequency in β-hexosamidase in P. damicornis (type α) and for water clarity in P. damicornis (type β). Temperature and allele frequency was also significantly correlated to SNP allele frequency in Elongation factor 1-α_2631 for type α and β as well as a gene of unknown function (Contig_24_847) in P. damicornis (type β). P. damicornis (type β) also showed a correlation between water clarity and SNP allele frequencies in Elongation factor 1-α_2631 (Table 5; Figure 4).

Scatter plots of SNP allele frequencies and environmental categories in P. damicornis (type α) and (type β).
Acropora millepora
For the A. millepora data set, the Sequinom® MassArray produced sequence data, hence allowing the inclusion of the C to G mutation that was evident in the Ligand of Numb X2 locus. One of the nine chosen loci (Coatomer) was found to be monomorphic and excluded from further analyses. The remaining eight SNPs were successfully genotyped in the majority of the colony samples. Allele frequencies in five (55%) of these loci showed a significant correlation with environmental gradients. β-gamma crystallin, Galaxin and Ubiquitin were correlated to temperature and Thioredoxin and Ligand of Numb X2 were found to correlate to water clarity (Table 6; Figures 2, 3 and 5).

Scatter plots of SNP allele frequencies and environmental categories in A. millepora.
Based on results from the microsatellite data, approximately 10% (16 out of 159 tested alleles, from 9 of the 11 tested loci) were found to have significant correlations purely by chance (Additional file 4: Table S3).
Discussion
This study presents two novel strategies to identify SNP loci that are correlated to environmental gradients, with the ultimate aim being the discovery of candidates for quantitative trait loci (QTLs) for environmental stress tolerance through subsequent correlative tests of phenotypic response. Both methods revealed likely signatures of selection in up to 55% of the loci tested. This is a considerable improvement compared to the previously reported 2-10% in non-targeted, genome-wide scans for outliers [69].
Of a small subset of SNPs screened across a large spatial scale on the Great Barrier Reef (GBR), four, two and three SNP(s) were found to be correlated to either temperature or water clarity in the reef-building coral species Acropora millepora, Pocillopora damicornis (type α) and Pocillopora damicornis (type β), respectively. These represent the first genetic markers in corals for environmental stress tolerance. After validation of these candidate loci, i.e., through laboratory or field assessment of relative stress tolerance of colonies harbouring different alleles at these loci, it is anticipated that a proportion of these loci can be used as de facto QTLs.
The risk of confounding effects (e.g. population history, neutral “surfing” allele of an expanding population, endogenous genetic barrier) causing false positives as discussed in Bierne et al. [19] is reduced by actively selecting SNPs for either function or population divergence prior to being tested for correlations. Furthermore, test on independent allelic data at 11 putatively neutral microsatellite loci in A. millepora provided an estimated the occurrence of false positives to be a maximum of ~10%.
All the SNPs that were selected in A. millepora and at least one (EF-1-α ) of the three significant SNPs in P. damicornis were non-synonymous mutations (Tables 1 and 2), hence are likely to be non-neutral makers upon which selection can act, either directly through a change in expression or function, or through linkage to other loci that are under selection. β- gamma-crystallin is mostly studied in relation to cataracts in humans and other model vertebrates [70]. The protein has also been shown to bind calcium in bacteria [71] and cephalochordates [72]. The binding of Ca2+ influences the stability of the protein and thus alters its thermal denaturation points [71], which may correlate to more successful calcification at higher temperatures. In corals, expression levels of β- gamma-crystallin has been shown to have a high heritability (h2 = 0.38), indicative of the existence of adaptive variation, and have been correlated to variations in larval responses to elevated temperatures and settlement cues [48]. Another gene that is involved in the calcification process, which exhibits allele frequency distributions that correlate to thermal gradients in the present study, is galaxin. The protein this genes encodes was originally characterised from the coral Galaxea fascicularis and is a soluble protein from the coral exoskeleton, where it is involved in calcification [73]. Three genes that are related to galaxin, amgalaxin , amgalaxin like-1 and amgalaxin like-2 have been characterised in A. millepora. The two amgalaxin like proteins are expressed exclusively during the early stages of calcification in the newly settled coral polyp, while amgalxin is continuously expressed also in the adult colony [74]. Recent studies indicate that temperature plays a pivotal role in coral calcification rates [26], supporting the correlation between temperature and proteins involved in the calcification process observed here. Ubiquitin is a protein that targets damaged proteins and marks them for destruction, a process referred to as ubiquitylation [75]. Thus, measurements of relative levels of ubiquitin provide an index of the structural integrity of cellular proteins and have been used as a biomarker for cellular stress in corals [76]. Differences in levels of ubiquitin conjugated proteins remain fixed during reciprocal transplantation of the coral Porites lobata, with corals from environmentally variable back reef sites showing consistently higher levels, regardless of transplant location [77], which adds to the current indication of a genetic difference contributing to variations to environmental stress response. SNP allele frequencies in the Ligand of numb X2 and Thioredoxin genes correlated to water clarity. Ligand of numb X2 is a close homologue to the Ligand of numb X, which is an important ligase for ubiquitin [78] while thioredoxin is involved in the sequestration of reactive oxygen species that are produced during oxidative stress [79]. Further to its putative role during oxidative stress, thioredoxin has been shown to be significantly differentially expressed along pollution gradients in the Bermudas [40], indicating that this protein is involved in the response to a range of stressors, including water quality.
In P. damicornis, two genes of known function were found to have significant correlations with temperature and/or water clarity, namely EF 1-α and β-hexosaminidase. Apart from its role in the process of cellular binding of actin [80], EF 1-α has been shown to be significantly down-regulated during cancer cell senescence [81] and during cell apoptosis associated with other cell malfunctions such as tetraploidy [82]. Hence, a possible change in function of this protein may impact the onset of apoptosis in stressed corals. β-hexosaminidase is involved in a range of functions including the glycosalation of proteins, cleaving of N-acetylgalactosamine (GalNAc) residues that form part of the cell signaling pathway, and correct protein folding mechanisms. It has also been shown to play a role in mycobacterium defense [83], which suggests it may play a role in the immune response. It is well documented that stressed corals show increased susceptibility to disease [84–86], hence a correlation between an immune response and increasing temperature or declining water quality is expected.
Although phenotypic responses are rarely determined by a single locus, but rather by the cumulative small effects of multiple loci [15], it has been observed in some studies that SNPs may be responsible for a large part of the variance in a specific phenotypic trait [11, 23, 87]. Hence, the direct utility of DNA-based QTLs is that the presence or absence of a certain allele is indicative of the relative stress tolerance of an individual or a population, as was shown to be the case of the relative proportion of one LDH-β allozyme and its correlation to increased thermal stress tolerance in populations of the killifish F. heteroclitus[22]. The use of QTLs is widespread in crop science, where extensive QTL mapping is used to direct controlled breeding for desired traits such as drought and pest resistance [88]. The link between genotype at the SNP loci presented here and the coral phenotype is currently being validated through aquarium experiments and in situ observations.
Conclusion
Developing and publishing efficient methods to screen genomes and transcriptomes for candidate genetic markers for stress tolerance remains an important scientific endeavour, and the development of temperature and water clarity-associated genetic markers in corals is both timely and relevant. Climate change does not only mean increasing sea surface temperatures, it is also predicted to come with an increasing occurrence of extreme weather events [89], including heavy rainfall, which results in increased influx of sediment, agricultural runoff and fresh water into the GBR lagoon, all of which negatively impact near shore coral reefs [90]. In addition, the GBR is currently facing a range of acute water clarity impacts such as dredging for port expansions or underwater mining operations. The value of markers such as the ones developed in this study in management practices lies in the potential to identify key reefs and populations that harbour tolerant genotypes and hence are likely to show greater resilience to increasing sea surface temperatures or declining water quality. A combination of high resilience and a role as a source reef for coral recruits would provide a solid foundation for the implementation of protection of a particular reef [91]. Other potential applications of genetic markers for stress tolerance include the potential to identify more stress tolerant colonies for seeding of degraded reefs, to initiate selective breeding programs, or to actively preserve certain genotypes for future breeding and restoration efforts.
References
IPCC: Climate change 2007: Impacts, adaptation and vulnerability. Fourth assessment report on the intergovernmental panel on climate change. 2007, Cambridge: Cambridge University Press
Baker AC, Glynn PW, Riegl B: Climate change and coral reef bleaching: an ecological assessment of long-term impacts, recovery trends and future outlook. Estuar Coast Shelf S. 2008, 80 (4): 435-471. 10.1016/j.ecss.2008.09.003.
Diaz-Pulido G, McCook LJ, Dove S, Berkelmans R, Roff G, Kline DI, Weeks S, Evans RD, Williamson DH, Hoegh-Guldberg O: Doom and Boom on a Resilient Reef: Climate Change, Algal Overgrowth and Coral Recovery. PLoS One. 2009, 4 (4): e5239-10.1371/journal.pone.0005239.
Hoegh-Guldberg O, Mumby PJ, Hooten AJ, Steneck RS, Greenfield P, Gomez E, Harvell CD, Sale PF, Edwards AJ, Caldeira K, et al: Coral reefs under rapid climate change and ocean acidification. Science. 2007, 318 (5857): 1737-1742. 10.1126/science.1152509.
Hughes TP, Baird AH, Bellwood DR, Card M, Connolly SR, Folke C, Grosberg R, Hoegh-Guldberg O, Jackson JBC, Kleypas J, et al: Climate change, human impacts, and the resilience of coral reefs. Science. 2003, 301 (5635): 929-933. 10.1126/science.1085046.
Maynard JA, Anthony KRN, Marshall PA, Masiri I: Major bleaching events can lead to increased thermal tolerance in corals. MarBiol. 2008, 155 (2): 173-182.
Holderegger R, Kamm U, Gugerli F: Adaptive vs. neutral genetic diversity: implications for landscape genetics. Landsc Ecol. 2006, 21 (6): 797-807. 10.1007/s10980-005-5245-9.
Holderegger R, Herrmann D, Poncet B, Gugerli F, Thuiller W, Taberlet P, Gielly L, Rioux D, Brodbeck S, Aubert S: Land ahead: using genome scans to identify molecular markers of adaptive relevance. Plant Ecol Divers. 2008, 1: 273-283. 10.1080/17550870802338420.
Nielsen R: Molecular signatures of natural selection. Annu Rev Genet. 2005, 39: 197-218. 10.1146/annurev.genet.39.073003.112420.
Luikart G, England PR, Tallmon D, Jordan S, Taberlet P: The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet. 2003, 4 (12): 981-994.
Gagnaire P-A, Normandeau E, Cote C, Hansen MM, Bernatchez L: The genetic consequences of spatially varying selection in the panmictic American eel (Anguilla rostrata). Genetics. 2011, 190 (2): 725-U703.
Renaut S, Nolte AW, Rogers SM, Derome N, Bernatchez L: SNP signatures of selection on standing genetic variation and their association with adaptive phenotypes along gradients of ecological speciation in lake whitefish species pairs (Coregonus spp.). Mol Ecol. 2010, 20 (3): 545-559.
Prunier J, Laroche J, Beaulieu J, Bousquet J: Scanning the genome for gene SNPs related to climate adaptation and estimating selection at the molecular level in boreal black spruce. Mol Ecol. 2011, 20 (8): 1702-1716. 10.1111/j.1365-294X.2011.05045.x.
Beaumont MA, Balding DJ: Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol. 2004, 13 (4): 969-980. 10.1111/j.1365-294X.2004.02125.x.
Flint J, Mackay TFC: Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res. 2011, 19: 723-733.
Hedrick PW, Ginevan ME, Ewing EP: Genetic polymorphism in heterogenous environments. Annu Rev Ecol Evol Syst. 1976, 7: 1-32. 10.1146/annurev.es.07.110176.000245.
Manel S, Joost S, Epperson B, Storfer A, Holderegger R, Scribner K, Rosenberg MS, Bonin A, Fortin MJ: Perspectives on the use of landscape genetics to detect genetic adaptive variation in the field. Mol Ecol. 2010, 19: 3760-3772. 10.1111/j.1365-294X.2010.04717.x.
Hedrick PW: Genetic polymorphism in heterogeneous environments: the age of genomics. Annu Rev Ecol Evol Syst. 2006, 37: 67-93. 10.1146/annurev.ecolsys.37.091305.110132.
Bierne N, Welch J, Loire E, Bonhomme F, David P: The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol. 2011, 20 (10): 2044-2072. 10.1111/j.1365-294X.2011.05080.x.
Hancock AM, Brachi B, Faure N, Horton MW, Jarymowycz LB, Sperone FG, Toomajian C, Roux F, Bergelson J: Adaptation to climate across the Arabidopsis thaliana genome. Science. 2011, 333 (6052): 83-86.
Neale DB, Savolainen O: Association genetics of complex traits in conifers. Trends Plant Sci. 2004, 9 (7): 325-330. 10.1016/j.tplants.2004.05.006.
Segal JA, Schulte PM, Powers DA, Crawford DL: Descriptive and functional characterization of variation in the Fundulus heteroclitus Ldh-B proximal promoter. J Exp Zool. 1996, 275 (5): 355-364. 10.1002/(SICI)1097-010X(19960801)275:5<355::AID-JEZ4>3.0.CO;2-O.
Korsten P, Mueller JC, Hermannstater C, Bouwman KM, Dingemanse NJ, Drent PJ, Liedvogel M, Matthysen E, van Oers K, van Overveld T, et al: Association between DRD4 gene polymorphism and personality variation in great tits: a test across four wild populations. Mol Ecol. 2010, 19 (4): 832-843. 10.1111/j.1365-294X.2009.04518.x.
Wachenfeld D, Johnson J, Skeat A, Kenshington R, Marshall P, Innes J: Introduction to the Great Barrier Reef and climate change. Climate change and the Great Barrier Reef: A vulnerability assessment. Edited by: Johnson JE, Marshall P. 2007, Townsville: Great Barrie Reef Marine Park Authority and Australian Greenhouse Office
Muscatine L: The role of symbiotic algae in carbon and energy flux in reef corals. Ecosystems of the World: Coral Reefs. 1990, 25: 75-87.
De'ath G, Lough JM, Fabricius KE: Declining coral calcification on the Great Barrier Reef. Science. 2009, 323 (5910): 116-119. 10.1126/science.1165283.
De'ath G, Fabricius K: Water Quality of the Great Barrier Reef: Distributions, Effects on Reef Biota and Trigger Values for the Protection of Ecosystem Health. 2008, Townsville: GBRMPA, 104-Research publication No 89
Furnas M: Water quality in the Great Barrier Reef Lagoon: A summary of current knowledge. Status and Trends of Water Quality and Ecosystem Health in the Great Barrier Reef World Heritage Area. 2005, Townsville: CRC Reefs, 32-53.
Berkelmans R, De'ath G, Kininmonth S, Skirving WJ: A comparison of the 1998 and 2002 coral bleaching events on the Great Barrier Reef: spatial correlation, patterns, and predictions. Coral Reefs. 2004, 23 (1): 74-83. 10.1007/s00338-003-0353-y.
Brown BE: Coral bleaching: causes and consequences. Coral Reefs. 1997, 16 (supplement): 129-138.
Glynn PW: Extensive 'bleaching' and death of reef corals on the pacific coast of Panama. Environ Conserv. 1983, 10: 149-154. 10.1017/S0376892900012248.
Goreau T, McClanahan T, Hayes R, Strong A: Conservation of coral reefs after the 1998 global bleaching event. Conserv Biol. 2000, 14 (1): 5-15. 10.1046/j.1523-1739.2000.00011.x.
Hoegh-Guldberg O: Climate change, coral bleaching and the future of the world's coral reefs. Mar Freshw Res. 1999, 50: 839-866. 10.1071/MF99078.
Oliver JK, Berkelmans R, Eakin CM: Coral bleaching in space and time. Coral Bleaching: Patterns, processes, causes and consequences, vol. 25. Edited by: Oppen MJH, Lough JM. 2009, Berlin, Heidelberg: Springer-Verlag, 178-
Berkelmans R, van Oppen MJH: The role of zooxanthellae in the thermal tolerance of corals: a 'nugget of hope' for coral reefs in an era of climate change. Proc R Soc B-Biol Sci. 2006, 273 (1599): 2305-2312. 10.1098/rspb.2006.3567.
Smith-Keune C, van Oppen M: Genetic structure of a reef-building coral from thermally distinct environments on the Great Barrier Reef. Coral Reefs. 2006, 25 (3): 493-502. 10.1007/s00338-006-0129-2.
Ulstrup KE, Berkelmans R, Ralph PJ, van Oppen MJH: Variation in bleaching sensitivity of two coral species across a latitudinal gradient on the Great Barrier Reef: the role of zooxanthellae. MarEcolProgSer. 2006, 314: 135-148.
Sharp VA, Brown BE, Miller D: Heat shock protein (hsp 70) expression in the tropical reef coral Goniopora djiboutiensis. J Therm Biol. 1997, 22 (1): 11-19. 10.1016/S0306-4565(96)00029-0.
Edge SE, Morgan MB, Gleason DF, Snell TW: Development of a coral cDNA array to examine gene expression profiles in Montastraea faveolata exposed to environmental stress. Mar Pollut Bull. 2005, 51 (5–7): 507-523.
Morgan MB, Edge SE, Snell TW: Profiling differential gene expression of corals along a transect of waters adjacent to the Bermuda municipal dump. Mar Pollut Bull. 2005, 51 (5–7): 524-533.
Kortschak R, Samuel G, Saint R, Miller D: EST analysis of the Cnidarian Acropora millepora reveals extensive gene loss and rapid sequence divergence in the model invertebrates. Curr Biol. 2003, 13: 2190-2195. 10.1016/j.cub.2003.11.030.
Technau U, Rudd S, Maxwell P, Gordon PMK, Saina M, Grasso LC, Hayward DC, Sensen CW, Saint R, Holstein TW, et al: Maintenance of ancestral complexity and non-metazoan genes in two basal cnidarians. Trends Genet. 2005, 21 (12): 633-639. 10.1016/j.tig.2005.09.007.
Császár NBM, Seneca FO, van Oppen MJH: Variation in antioxidant gene expression in the scleractinian coral Acropora millepora under laboratory thermal stress. Mar Ecol Prog Ser. 2009, 392: 93-102.
Seneca FO, Foret S, Ball EE, Smith-Keune C, Miller DJ, van Oppen MJH: Patterns of gene expression in a scleractinian coral undergoing natural bleaching. Marine Biotechnol. 2010, 12 (5): 594-604. 10.1007/s10126-009-9247-5.
van Oppen MJH, Gates RD: Conservation genetics and the resilience of reef-building corals. Mol Ecol. 2006, 15 (13): 3863-3883. 10.1111/j.1365-294X.2006.03026.x.
Bay LK, Ulstrup KE, Nielsen HB, Jarmer H, Goffard N, Willis BL, Miller DJ, Van Oppen MJH: Microarray analysis reveals transcriptional plasticity in the reef building coral Acropora millepora. MolEcol. 2009, 18 (14): 3062-3075.
Csaszar NBM, Ralph PJ, Frankham R, Berkelmans R, van Oppen MJH: Estimating the Potential for Adaptation of Corals to Climate Warming. PLoS One. 2010, 5 (3): e9751-10.1371/journal.pone.0009751.
Meyer E, Davies S, Wang S, Willis BL, Abrego D, Juenger TE, Matz MV: Genetic variation in responses to a settlement cue and elevated temperature in the reef-building coral Acropora millepora. Mar Ecol Prog Ser. 2009, 392: 81-92.
Meyer E, Wang S, Agyamova G, Matz M: Quantitative genetics and genomics of reef-building coral Acropora millepora. Integr Comp Biol. 2010, 50: E118-E118.
Souter P, Bay LK, Andreakis N, Csaszar N, Seneca FO, van Oppen MJH: A multilocus, temperature stress-related gene expression profile assay in Acropora millepora, a dominant reef-building coral. Mol Ecol Resour. 2010, 11 (2): 328-334.
Meyer E, Aglyamova GV, Wang S, Buchanan-Carter J, Abrego D, Colbourne JK, Willis BL, Matz MV: Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics. 2009, 10: article no 219-
Wang S, Zhang LL, Meyer E, Matz MV: Construction of a high-resolution genetic linkage map and comparative genome analysis for the reef-building coral Acropora millepora. Genome Biol. 2009, 10 (11): article no R126-
Shinzato C, Shoguchi E, Kawashima T, Hamada M, Hisata K, Tanaka M, Fujie M, Fujiwara M, Koyanagi R, Ikuta T, et al: Using the Acropora digitifera genome to understand coral responses to environmental change. Nature. 2011, advance online publication
Anthony KRN, Connolly SR, Hoegh-Guldberg O: Bleaching, energetics, and coral mortality risk: effects of temperature, light, and sediment regime. Limnol Oceanogr. 2007, 52 (2): 716-726. 10.4319/lo.2007.52.2.0716.
Wooldridge SA: Water quality and coral bleaching thresholds: Formalising the linkage for the inshore reefs of the Great Barrier Reef, Australia. Mar Pollut Bull. 2009, 58 (5): 745-751. 10.1016/j.marpolbul.2008.12.013.
Souter P: Hidden genetic diversity in a key model species of coral. Mar Biol. 2010, 157 (4): 875-885. 10.1007/s00227-009-1370-3.
Concepcion GT, Crepeau MW, Wagner D, Kahng SE, Toonen RJ: An alternative to ITS, a hypervariable, single-copy nuclear intron in corals, and its use in detecting cryptic species within the octocoral genus Carijoa. Coral Reefs. 2008, 27 (2): 323-336. 10.1007/s00338-007-0323-x.
Schmidt-Roach S, Lundgren P, Miller K, Muir P, Gerlach G, Noreen A, Andreakis N: Assessing hidden species diversity in the coral Pocillopora damicornis from Eastern Australia. Coral Reefs. 2012, 10.1007/s00338-00012-00959-z.
Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, Hanski I, Marden JH: Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. MolEcol. 2008, 17 (7): 1636-1647.
Vera JC: Functional transcriptomics: Ecologically important genetic variation in non-model organisms. 2012, State College: Pennsylvania State University, Ph.D. Thesis, 168p
Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV, et al: Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007, 317: 86-94. 10.1126/science.1139158.
Traylor-Knowles N, Granger BR, Lubinski TJ, Parikh JR, Garamszegi S, Xia Y, Marto A: J., Kaufman, L, Finnerty JR: Production of a reference transcriptome and transcriptomic database (PocilloporaBase) for the cauliflower coral, Pocillopora damicornis. BMC Genomics. 2011, 12 (1): 585-10.1186/1471-2164-12-585.
Rayner NA, Parker DE, Horton EB, Folland CK, Alexander LV, Rowell DP, Kent EC, Kaplan A: Global analyses of sea surface temperature, sea ice, and night marine air temperature since the late nineteenth century. Geophys Res. 2003, 108 (No. D14): 4407-4410. 1029/2002JD002670.
De'ath G, Fabricius K: Water quality as a regional driver of coral biodiversity and macroalgae on the Great Barrier Reef. Ecol Appl. 2010, 20 (3): 840-850. 10.1890/08-2023.1.
van Oppen MJH, Peplow L, Kinninmonth S, Berkelmans R: Historical and contemporary factors shape the population genetic structure of the broadcast spawning coral, Acropora millepora, on the Great Barrier Reef. Mol Ecol. 2011, 10.1111/j.1365-294X.2011.05328.x.
Wilson K, Whan V, Lehnert S, Byrne K, Moore S, Pongsomboon S, Tassanakajon A, Rosenberg G, Ballment E, Fayazi Z, et al: Genetic mapping of the black tiger shrimp Panaeus monodon with Amplified Fragment Length Polymorphisms. Aquaculture. 2002, 204: 297-309. 10.1016/S0044-8486(01)00842-0.
McCullagh P, Nelder JA: Generalized linear models. 2nd Edition. Monographs on Satistics and Applied Probability. 1989, London: Chapman & Hall
Joost S, Bonin A, Bruford M, Desperes L, Concord C, Erhart G, et al: A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Mol Ecol. 2007, 16: 3955-3969. 10.1111/j.1365-294X.2007.03442.x.
Nosil P, Funk DJ, Ortiz-Barrientos D: Divergent selection and heterogeneous genomic divergence. Mol Ecol. 2009, 18 (3): 375-402. 10.1111/j.1365-294X.2008.03946.x.
Graw J: Mouse models of cataract. J Genet. 2009, 88 (4): 469-486. 10.1007/s12041-009-0066-2.
Wenk M, Mayr EM: Myxococcus xanthus spore coat protein S, a stress-induced member of the beta gamma-crystallin superfamily, gains stability from binding of calcium ions. Eur J Biochem. 1998, 255 (3): 604-610. 10.1046/j.1432-1327.1998.2550604.x.
Kappe G, Purkiss AG, van Genesen ST, Slingsby C, Lubsen NH: Explosive expansion of beta gamma-crystallin genes in the ancestral vertebrate. J Mol Evol. 2010, 71 (3): 219-230. 10.1007/s00239-010-9379-2.
Fukuda I, Ooki S, Fujita T, Murayama E, Nagasawa H, Isa Y, Watanabe T: Molecular cloning of a cDNA encoding a soluble protein in the coral exoskeleton. Biochem Biophys Res Commun. 2003, 304 (1): 11-17. 10.1016/S0006-291X(03)00527-8.
Reyes-Bermudez A, Lin ZY, Hayward DC, Miller DJ, Ball EE: Differential expression of three galaxin-related genes during settlement and metamorphosis in the scleractinian coral Acropora millepora. BMC Evol Biol. 2009, 9: 12-10.1186/1471-2148-9-12.
Vucic D, Dixit VM, Wertz IE: Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011, 12 (7): 439-452. 10.1038/nrm3143.
Downs CA, Fauth JE, Robinson CE, Curry R, Lanzendorf B, Halas JC, Halas J, Woodley CM: Cellular diagnostics and coral health: Declining coral health in the Florida Keys. Mar Pollut Bull. 2005, 51 (5–7): 558-569.
Barshis DJ, Stillman JH, Gates RD, Toonen RJ, Smith LW, Birkeland C: Protein expression and genetic structure of the coral Porites lobata in an environmentally extreme Samoan back reef: does host genotype limit phenotypic plasticity?. Mol Ecol. 2010, 19 (8): 1705-1720. 10.1111/j.1365-294X.2010.04574.x.
Mirza M, Raschperger E, Philipson L, Pettersson RF, Sollerbrant K: The cell surface protein coxsackie- and adenovirus receptor (CAR) directly associates with the Ligand-of-Numb Protein-X2 (LNX2). Exp Cell Res. 2005, 309 (1): 110-120. 10.1016/j.yexcr.2005.05.023.
Weis VM: Cellular mechanisms of Cnidarian bleaching: stress causes the collapse of symbiosis. J Exp Biol. 2008, 211: 3059-3066. 10.1242/jeb.009597.
Furukawa R, Jinks TM, Tishgarten T, Mazzawi M, Morris DR, Fechheimer M: Elongation factor 1 beta is an actin-binding protein. Biochimica Et Biophysica Acta-General Subjects. 2001, 1527 (3): 130-140. 10.1016/S0304-4165(01)00157-X.
Byun H-O, Han N-K, Lee H-J, Kim K-B, Ko Y-G, Yoon G, Lee Y-S, Hong S-I, Lee J-S: Cathepsin D and eukaryotic translation Elongation factor 1 as promising markers of cellular senescence. Cancer Res. 2009, 69 (11): 4638-4647. 10.1158/0008-5472.CAN-08-4042.
Kobayashi Y, Yonehara S: Novel cell death by downregulation of eEF1A1 expression in tetraploids. Cell Death Differ. 2009, 16 (1): 139-150. 10.1038/cdd.2008.136.
Koo IC, Ohol YM, Wu P, Morisaki JH, Cox JS, Brown EJ: Role for lysosomal enzyme beta-hexosaminidase in the control of mycobacteria infection. Proc Natl Acad Sci USA. 2008, 105 (2): 710-715. 10.1073/pnas.0708110105.
Mydlarz LD, McGinty ES, Harvell CD: What are the physiological and immunological responses of coral to climate warming and disease?. J Exp Biol. 2010, 213 (6): 934-945. 10.1242/jeb.037580.
Littman RA, Bourne DG, Willis BL: Responses of coral-associated bacterial communities to heat stress differ with Symbiodinium type on the same coral host. Mol Ecol. 2010, 19 (9): 1978-1990. 10.1111/j.1365-294X.2010.04620.x.
Thurber RV, Willner-Hall D, Rodriguez-Mueller B, Desnues C, Edwards RA, Angly F, Dinsdale E, Kelly L, Rohwer F: Metagenomic analysis of stressed coral holobionts. Environ Microbiol. 2009, 11 (8): 2148-2163. 10.1111/j.1462-2920.2009.01935.x.
Hitchins Megan P, Rapkins Robert W, Kwok C-T, Srivastava S, Wong Justin JL, Khachigian Levon M, Polly P, Goldblatt J, Ward Robyn L: Dominantly Inherited Constitutional Epigenetic Silencing of MLH1 in a Cancer-Affected Family Is Linked to a Single Nucleotide Variant within the 52UTR. Cancer Cell. 2011, 20 (2): 200-213. 10.1016/j.ccr.2011.07.003.
Semagn K, Bjornstad A, Xu YB: The genetic dissection of quantitative traits in crops. Electron J Biotechnol. 2010, 13 (5): 45-
Huber DG, Gulledge J: Extreme weather and climate change: Understanding the link, managing the risk. 2011, Arlington, VA: Pew Centre on Global Climate Change
Haynes D, Brodie J, Waterhouse J, Bainbridge Z, Bass D, Hart B: Assessment of the water quality and ecosystem health of the Great Barrier Reef (Australia): conceptual models. Environ Manage. 2007, 40: 993-1003. 10.1007/s00267-007-9009-y.
Mumby PJ, Elliott IA, Eakin CM, Skirving W, Paris CB, Edwards HJ, Enriquez S, Iglesias-Prieto R, Cherubin LM, Stevens JR: Reserve design for uncertain responses of coral reefs to climate change. Ecol Lett. 2011, 14 (2): 132-140. 10.1111/j.1461-0248.2010.01562.x.
Acknowledgements
This work was funded by the Reef and Rainforest Research Centre (RRRC), the Great Barrier Reef Foundation (GBRF) and the Australian Institute of Marine Science (AIMS). A travel grant from the Australian Academy of Science allowed P. Lundgren to establish the collaboration and bioinformatics support from of JC Vera at Penn State University. JC Vera was funded by NSF grant IOS-0950416.
The authors would like to thank colleagues and friends who have contributed to the coral collections. Secchi depth data were provided by B. Schaffelke and M. Furnas (AIMS) and R. Coles (Queensland Department of Primary Industries and Fisheries). All interpolations were done by G. De'ath (AIMS) and original maps were vectorised by E. Lawrey (e-Atlas, AIMS). Thanks also to T. Simmonds and G. Torda for help with creating the maps and figures. S Manel was supported by the Institut Universitaire de France and was hosted by the Australian Research Council Centre of Excellence for Coral Reef Studies at James Cook University during the onset of collaborations for this project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors’ contributions
The manuscript is the outcome of the postdoctoral research of PL, who conceptualised the idea and acquired funding in collaboration with MvO, collected field material, conducted a majority of the pre-transcription laboratory work, was involved in the bioinformatics and subsequent genotyping work and did the majority of the manuscript writing. In addition to the above conceptualisation and fund acquisition, MvO provided core funds and supervision. JCV did the majority of the bioinformatics on the transcriptomes, SM provided insights and support on the statistical analyses, wrote the scripts for the logistic regression analyses and interpreted the results and LP did a large part of the HRM analyses for the P. damicornis samples. All authors read and approved the final manuscript.
Electronic supplementary material
12863_2012_1079_MOESM2_ESM.docx
Additional file 2: Table S1: SNP allele frequencies in each population of the two types of Pocillopora damicornis.(DOCX 18 KB)
12863_2012_1079_MOESM4_ESM.eps
Additional file 4: Table S3: Microsatellite alleles from the tested colonies and populations Acropora millepora that showed a significant correlation, presumably by chance. (EPS 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lundgren, P., Vera, J.C., Peplow, L. et al. Genotype – environment correlations in corals from the Great Barrier Reef. BMC Genet 14, 9 (2013). https://doi.org/10.1186/1471-2156-14-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-14-9