
Abstract
Background
Bacterial genomes possess varying GC content (total guanines (Gs) and cytosines (Cs) per total of the four bases within the genome) but within a given genome, GC content can vary locally along the chromosome, with some regions significantly more or less GC rich than on average. We have examined how the GC content varies within microbial genomes to assess whether this property can be associated with certain biological functions related to the organism's environment and phylogeny. We utilize a new quantity GCVAR, the intra-genomic GC content variability with respect to the average GC content of the total genome. A low GCVAR indicates intra-genomic GC homogeneity and high GCVAR heterogeneity.
Results
The regression analyses indicated that GCVAR was significantly associated with domain (i.e. archaea or bacteria), phylum, and oxygen requirement. GCVAR was significantly higher among anaerobes than both aerobic and facultative microbes. Although an association has previously been found between mean genomic GC content and oxygen requirement, our analysis suggests that no such association exits when phylogenetic bias is accounted for. A significant association between GCVAR and mean GC content was also found but appears to be non-linear and varies greatly among phyla.
Conclusions
Our findings show that GCVAR is linked with oxygen requirement, while mean genomic GC content is not. We therefore suggest that GCVAR should be used as a complement to mean GC content.
Similar content being viewed by others


Background
The knowledge of the chemical basis for nucleic acids goes back more than a hundred years, to the work of Miescher [1]. By the early 1950's, it was known that the relative frequency of the four DNA bases ("base composition") was different for different organisms [2], and in general the number of A's was equal to the number of T's, and the number of G's was the same as the number of C's; this is known as 'Chargaff's first parity rule' [3]. Further, for nearly all genomes studied, the parity rule appears to extend to each strand of the chromosome, when averaged over long distances [4], although in bacterial chromosomes, there is a clear bias of G's towards the replication leading strand, and for some genomes (many Firmicutes, for example) the A's are also biased towards the leading strand [5]. For a circular chromosome with the replication origin and terminus on exactly opposite sides, this bias of G's towards the replication leading strand will average out to near zero, when one only looks at the DNA sequence in the GenBank file, and the sequence will appear to conform to Chargaff's second rule.
From the Genbank database at NCBI http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi it can be seen that GC content in prokaryotes ranges from 16.6% in Carsonella ruddii strain Pv to 74.9% in Anaeromyxobacter dehalogenans Strain 2CP-C. Within a given genome, the GC content along the chromosome can vary, although since most bacterial genomes have a high coding density, usually the variation is less than that found in eukaryotes [6]. The average genomic GC content is an important property in microbial genomes and has been associated with properties such as genome size [7], oxygen, and nitrogen exposure [8, 9] and specific habitats [10–13]. For instance, intracellular bacteria have, on average, smaller genomes and are mostly AT rich, while soil bacteria tend to have larger genomes and higher %GC [14]. Higher AT content in intracellular bacteria may be attributed to a loss of repair genes; this loss will eventually lead to an increase in mutation rates from cytosine to thymine [15, 16]. Genes not expressed will eventually lead to reduced genome sizes [15, 16]. Higher GC content in soil bacteria may be due to the increased availability of nitrogen [9]. However, increased nitrogen in the soil does not explain why GC rich bacteria often have larger genomes. The base composition in GC rich genomes might reflect stronger selective forces than AT rich genomes [17–19]. This may indicate that GC rich microbes live in more complex environments than intracellular bacteria [20]. The reasons for stronger selective forces and GC richness is not known, but may be connected to the fact that considerably more energy is required to de-stack GC rich DNA sequences than AT rich DNA sequences [21].
Although GC content has been found to vary only slightly within prokaryotic genomes some regions differ more than others. A large region flanking the replication origin, for instance, is more GC rich than the average genomic GC content [22] whereas the region around replication terminus is more AT rich [5]. Surface proteins and RNA genes often have GC content that differs from the average genomic GC content [22], and protein coding regions have been found to be, on average, approximately 5% more GC rich than non-coding regions [18]. In addition to being more GC rich, coding regions have been found to be more homogeneous in terms of base composition than non-coding regions [18]. The GC heterogeneity in coding regions has, however, been found to be associated with mean genomic AT content in non-coding regions [18, 23]. In other words, GC content variability tends to increase with higher mean genomic AT content in non-coding regions.
Horizontally transferred DNA may have a different fraction of GC than the host genome as a result of different evolutionary pressures [6, 24–26]. Since horizontally transferred DNA is often linked to pathogenesis in microbes [27], detection of such regions is of great importance. The GC content of foreign DNA will, however, become progressively more similar to the host genome in a process known as amelioration [24] making such regions more difficult to detect as time progress [25]. The conformation of base compositional patterns from foreign DNA to host DNA may be related to the finding that a particular subunit of the DNA polymerase III, the Pol III α subunit, appears to be driving genomic GC content in prokaryotes [28].
There is a considerable amount of research and documentation related to mean genomic GC content in prokaryotes demonstrating that this property is the result of many factors interacting in a highly complex manner [29]. On the other hand, analysis of genomic GC content variability within microbial chromosomes, has received much less attention. A more recent overview of methodology used to analyze GC content variation within genomes can be found in Bernaola-Galvan et. al., [30], and a study of how intra-genomic GC content variation affects codon usage is described by Daubin et. al. [31]. In the present work, we introduce the GCVAR measure to examine GC content variability within prokaryotic genomes. The GCVAR metric gives a measure of how GC content varies within a given genome with respect to the mean genomic GC content. A low GCVAR thus points to little GC content variation, or GC content homogeneity, within the genome, while a high GCVAR designates varying GC content, or GC content heterogeneity.
To the best of our knowledge, no study has examined the interplay between environmental factors and GC content homogeneity in prokaryotes. In the present study the aim was therefore to examine whether GC content homogeneity in prokaryotes, measured here using the GCVAR measure, could be related to specific factors in the environment such as temperature and oxygen, as well as the broader properties implicated in phylogeny and GC content. To do this, regression analyses were performed using GCVAR as the response variable. The response variable was fitted to the following variables: oxygen requirement (a categorical variable defined as either aerobic, anaerobic or facultative), phylum, genomic GC content, genome size, growth temperature (a categorical variable used to define psychrophiles, mesophiles and thermophiles), pathogenicity (a dichotomous variable describing whether the microbe is pathogenic or not) and habitat (a categorical variable describing the environment where the microbe is found, i.e. aquatic, host-associated, multiple, specialized and terrestrial). The dataset consisted of 488 genomes (526 chromosomes) with similar strains and species removed from the analysis to reduce phylogenetic bias.
Results and Discussion
GC distribution within genomes
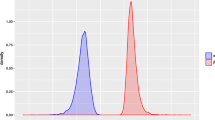
The histograms in Figure 1 shows the statistical distributions of GC content differences, D i = GC i - GC (Equation (1), Methods section), within four AT-rich and four GC-rich genomes. The statistical distributions shown in Figure 1 are based on the differences, or residuals, between the GC content of a 100 bp non-overlapping sliding window and mean genomic GC content for each of the 8 genomes. Figure 1 therefore shows the statistical distributions of how GC contents differences are distributed within each of the described genomes. With the exception of Carsonella rudii, one of the smallest bacterial genomes currently sequenced (~160 kbp), all empirical distributions follow the bell shaped Gaussian curve. This indicates that GC difference within prokaryotic genomes appears to be a sum of many independent processes, giving a Gaussian like distribution according to the central limit theorem (see, for instance, [32]). Thus, it seems likely that for most prokaryotic genomes intra-genomic GC content variation appears to follow a random, white-noise like pattern, devoid of any complex and long-range interacting factors.

The distributions of GC difference within genomes. The histograms show the distribution of GC difference, D i , (Equation (1) in the Methods section) for eight different microbial genomes. The blue curves are empirical density estimates, while the red curves are Gaussian densities using based on the same means and standard deviations as the empirical estimates. The upper panels show the statistical distributions for four AT rich genomes, while the lower panels show the distributions for four GC rich genomes.
The GCVAR regression model
We define GCVAR as a measure of the intra-genomic GC variation in a genome. A linear regression model was fitted to data for 526 prokaryote chromosomes with GCVAR as the response and with GC content, size, phylum, oxygen requirement, growth temperature, pathogenicity and habitat as covariates (Equation (3) in the Methods section). The results of the GCVAR regression model can be observed in Table 1, and in Figure 2 we show the 95% confidence intervals for the significant effects. The variables: size, growth temperature, pathogenicity and habitat had no significant influence on GCVAR, and were therefore discarded from further analyses.

Significant effects on GC variation. The bars indicate 95% confidence intervals for the effects of various phyla (top panel) and oxygen requirements (lower panel) based on the regression model described by Equation (3) in the Methods section. Note that the values on the horizontal axis are scaled differently in the two panels. Categories with non-overlapping intervals can be said to differ significantly at a 5% level. Only significant effects are included.
GCVAR in phyla
Table 1 shows that GCVAR is significantly influenced by phylum. We find that 10 phyla have GCVAR significantly above the average phylum, and 4 phyla have GCVAR significantly below average. The two phyla, Crenarchaeota and Euryarcheota, (both archaea) are among the groups with an above average GCVAR. The archaea domain, consisting predominantly of organisms living in extreme environments, had a significantly higher GCVAR than bacteria (p < 0.001). The highest GCVAR are found in the aquatic group Cyanobacteria, which is largely populated with species capable of photosynthesis [33]. The lowest GCVAR are found in the phylum of the aquatic Planctomycetes, but this group is only based on one single genome, therefore no conclusions can be assumed at the phylum level.
Environmental factors and phylogenetic bias
To examine how GCVAR was affected by phylogenetic bias a regression model similar to the one described above was fitted, i.e. GCVAR was the response variable, with mean genomic GC content, oxygen requirement, habitat, optimal growth temperature, and genome size as predictors. In addition, an interaction term between GC content and phylum was added to account for more similar GC content within phyla (Equation (5) in the Methods section). Using this regression model we found that oxygen requirement was the only significant factor (p < 0.001). GCVAR was significantly higher in the genomes of anaerobic microbes (103 chromosomes) as compared to the genomes of aerobic microbes, meaning that the genomes of anaerobic microbes tend to have a more heterogeneous distribution of GC content than genomes of aerobic microbes (246 chromosomes). Facultative microbes were found to have GCVAR values in the region between aerobic and anaerobic microbes (see Figure 2).
The associations between mean genomic GC content, GCVAR and oxygen requirement
The regression models described above indicates that aerobic microbes have genomes with more homogeneous GC content than those of organisms with facultative and anaerobic oxygen requirement (see Figure 2). It has been shown that GC rich genomes tend to be more homogeneous in terms of base composition than AT rich genomes [18, 19, 34]. Aerobic microbes have been associated with GC rich genomes [8]. This result is supported by our linear regression model only when we ignore phylogenetic bias is (p < 0.001). However, adding phyla as a predictor (Equation (5) in the Methods section) fails to demonstrate such an association (p ~0.9).
We found that mean genomic GC content was associated with GCVAR, but there was no linear relationship between mean genomic GC content and GCVAR (Figure 3), although this does not exclude a non-linear relationship.

Overall relation between GC content and GCVAR. The Figure shows GCVAR on the vertical axis versus %GC on the horizontal axis. The trend line is made using standard loess smoother.
GCVAR and DNA uptake
There are many indications that mean genomic GC content is as much affected by the environment as by phyla [10–13]. It is also well known that chromosomally integrated foreign DNA may differ in base composition as compared to host DNA. The difference in base composition between foreign and host DNA is assumed to be the result of exposure to different selective pressures. It is thought that such genetic regions may be acquired from horizontal transfer or other means of DNA uptake [6, 24–26]. Since pathogenesis is often associated with horizontally transferred DNA, i.e. pathogenicity islands, [27], establishing a link between any genomic property and horizontal transfer is of considerably interest. However, no significant association (p ~ 0.25) was found between the dichotomous pathogenicity factor and GCVAR using the regression model that included all covariates discussed above (Equation (3) in the Methods section).
Base composition and oxygen requirement
The introduction of atmospheric oxygen is presumed to have had profound effects on environment and life [35]. Increase in atmospheric oxygen is believed to have influenced cellular compartmentalization and thus to have been instrumental in the evolution of eukaryotes [36]. Prokaryotes were also affected by the introduction of oxygen [35] in that while some remained anaerobic others adapted to an aerobic metabolism [37].
The precise effect of increase in atmospheric oxygen on prokaryotic genomes is debated [37, 38]. A negative correlation has been found between proteomic oxygen content and genomic GC content [37]. Although it has been suggested that genomic GC content is also affected by an aerobic lifestyle [8], the effects on prokaryote genome composition has remained unclear [37, 38]. Indeed, our own results presented above do not support any connection between genomic GC content and aerobiosis. Our results did, however, find a significant association between GCVAR and oxygen requirement. This greater GC content homogeneity found in aerobes implies that the genomes of these organisms have been subjected to stronger selective pressures than the genomes of anaerobes. This is supported by the recent report that metabolic networks of aerobic bacteria are more complex than those of anaerobic bacteria [35]. From Figure 2 it can be seen that GCVAR appears to be progressively decreasing in facultative and aerobic prokaryotes, respectively.
Conclusion
In summary, we found that GCVAR was associated with oxygen requirement. It is possible that GCVAR is associated with GC content, but from Figure 3 it appears to be a highly non-linear relationship. Other factors such as genome size, habitat and growth temperature were not found significant in the GCVAR model. GCVAR was however found to be higher in archaea than bacteria. By adding an interaction term to model the closer similarity between the genomes in the same phylogenetic group, we found that oxygen requirement was not significantly associated with mean genomic GC content in microbes.
The different results obtained for the models describing GCVAR and mean genomic GC content imply that these properties are governed by different influences, or are interrelated in a non-linear manner. Thus, our findings suggest that GCVAR is linked with oxygen requirement, while mean genomic GC content is not.
Methods
All genomes and related information were gathered from the NCBI web site http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi. The statistical package R[39] was used for statistical analyses and graphical representations.
The GCVAR measure
To calculate GC variation within a prokaryotic genome, the number of guanine and cytosine nucleotides in a chromosome were counted and divided by chromosome size, giving the mean chromosomal GC content GC. A similar counting was performed for all 100 bp non-overlapping windows along the chromosome, giving the mean GC content GC i for window i. The difference, D i , between the mean GC content of window i, and the mean chromosomal GC content, can therefore be written as:

The quantity GCVAR is then defined as the log-transformed average of the absolute value of the difference between the mean GC content of each non-overlapping sliding window i and mean chromosomal GC content:

N is the maximum number of non-overlapping 100 bp sliding windows that can fit into the chromosome that is being analyzed. The log-transformation makes GCVAR s empirical distribution more Gaussian-like, for convenience in subsequent linear regression model fitting and statistical inference. Since the optimal sized sliding window varies from genome to genome [18], different window lengths were tested. The sliding window width of 100 bp was chosen to make the test as sensitive as possible. The other sliding window lengths tested contained 500, 1000, and 2000 bp. The 100 bp sliding window was found to be large enough to carry genome specific information without discarding weak genomic signals as noise. Since the aim of this study was to examine GC content difference within genomes, non-overlapping sliding windows were used to avoid bias and interactions from neighboring genetic regions.
Linear models
Linear regression analysis was used to examine influences affecting GCVAR. In our first analysis we made a regression of GCVAR onto GC and Size (genome size in Mb), also including the categorical variables phylum (22 phylogenetic groups), required oxygen (aerobic, facultative, anaerobic), growth temperature (psychrophilic, mesophilic and thermophilic), pathogenicity (pathogenic, non-pathogenic) and habitat (aquatic, host-associated, multiple, specialized and terrestrial) as predictors or explanatory variables. The model can be written as:

where μ is the overall intercept, α y , y = 1,...,22, are the effects of phylum, δ o , o = 1,2,3, are the effects of oxygen requirement, κ t , t = 1,2,3, are the effects of growth temperature, λ a , a = 1,2, are the effects of pathogenicity and ηh,h = 1,...4, are the effects of habitat. β and γ are the regression coefficients for the continuous variables GC and Size, respectively.
Based on the inference using the regression model described by (3) we eliminated the non-significant variables and obtained a reduced set of predictors: GC, phylum and oxygen requirement. In this reduced model, we included phyla only as an interaction with GC. The reason for this is that genomes within the same phylum tend to have similar GC content. Hence, a main effect of phylum may actually be a phylum-dependent GC effect. The model formulated as follows:

The α y in this model are defined as regression coefficients for each of the 22 phylum categories.
To test for possible associations between aerobiosis and mean genomic GC content, a regression model was fitted with GC as the response variable and aerobiosis as a group variable:

μ, α y , δ o are the same effects as those described for Equation (3).
References
Levine PA, Bass LW: Chapter VIII. Nucleic Acids. 1931, J.J. Little and Ives Company
Chargaff E: Structure and function of nucleic acids as cell constituents. Fed Proc. 1951, 10 (3): 654-659.
Elson D, Chargaff E: Regularities in the composition of pentose nucleic acids. Nature. 1954, 173 (4413): 1037-1038. 10.1038/1731037a0.
Karkas JD, Rudner R, Chargaff E: Seapration of B. subtilis DNA into complementary strands. II. Template functions and composition as determined by transcription with RNA polymerase. Proc Natl Acad Sci USA. 1968, 60 (3): 915-920. 10.1073/pnas.60.3.915.
Worning P, Jensen LJ, Hallin PF, Staerfeldt HH, Ussery DW: Origin of replication in circular prokaryotic chromosomes. Environ Microbiol. 2006, 8 (2): 353-361. 10.1111/j.1462-2920.2005.00917.x.
Sueoka N: On the genetic basis of variation and heterogeneity of DNA base composition. Proc Natl Acad Sci USA. 1962, 48: 582-592. 10.1073/pnas.48.4.582.
Mitchell D: GC content and genome length in Chargaff compliant genomes. Biochem Biophys Res Commun. 2007, 353 (1): 207-210. 10.1016/j.bbrc.2006.12.008.
Naya H, Romero H, Zavala A, Alvarez B, Musto H: Aerobiosis increases the genomic guanine plus cytosine content (GC%) in prokaryotes. J Mol Evol. 2002, 55 (3): 260-264. 10.1007/s00239-002-2323-3.
McEwan CE, Gatherer D, McEwan NR: Nitrogen-fixing aerobic bacteria have higher genomic GC content than non-fixing species within the same genus. Hereditas. 1998, 128 (2): 173-178. 10.1111/j.1601-5223.1998.00173.x.
Chen LL, Zhang CT: Seven GC-rich microbial genomes adopt similar codon usage patterns regardless of their phylogenetic lineages. Biochem Biophys Res Commun. 2003, 306 (1): 310-317. 10.1016/S0006-291X(03)00973-2.
Foerstner KU, von MC, Hooper SD, Bork P: Environments shape the nucleotide composition of genomes. EMBO Rep. 2005, 6 (12): 1208-1213. 10.1038/sj.embor.7400538.
Willenbrock H, Hallin PF, Wassenaar TM, Ussery DW: Characterization of probiotic Escherichia coli isolates with a novel pan-genome microarray. Genome Biol. 2007, 8 (12): R267-10.1186/gb-2007-8-12-r267.
Schloss PD, Handelsman J: A statistical toolbox for metagenomics: assessing functional diversity in microbial communities. BMC Bioinformatics. 2008, 9: 34-10.1186/1471-2105-9-34.
Wassenaar TM, Bohlin J, Binnewies TT, Ussery DW: Genome Comparison of Bacterial Pathogens. Genome Dyn. 2009, 6: 1-20. full_text.
Moran NA: Microbial minimalism: genome reduction in bacterial pathogens. Cell. 2002, 108 (5): 583-586. 10.1016/S0092-8674(02)00665-7.
Rocha EP, Danchin A: Base composition bias might result from competition for metabolic resources. Trends Genet. 2002, 18 (6): 291-294. 10.1016/S0168-9525(02)02690-2.
Reva ON, Tummler B: Global features of sequences of bacterial chromosomes, plasmids and phages revealed by analysis of oligonucleotide usage patterns. BMC Bioinformatics. 2004, 5: 90-10.1186/1471-2105-5-90.
Bohlin J, Skjerve E, Ussery DW: Investigations of oligonucleotide usage variance within and between prokaryotes. PLoS Comput Biol. 2008, 4 (4): e1000057-10.1371/journal.pcbi.1000057.
Barkovskii EV, Khrustalev VV: Inverse correlation between GC-content of bacterial genomes and the level of preterminal codons usage in them. Mol Gen Mikrobiol Virusol. 2009, 1 (1): 16-21.
Cases I, de Lorenzo V, Ouzounis CA: Transcription regulation and environmental adaptation in bacteria. Trends Microbiol. 2003, 11 (6): 248-253. 10.1016/S0966-842X(03)00103-3.
Sinden RR: DNA Structure and Function. 1994, Academic Press, New York
Ussery D, Wassenaar TM, Borini S: Computing for Comparative Microbial Genomics: Bioinformatics for Microbiologists. 2009, Springer, London
Chen SL, Lee W, Hottes AK, Shapiro L, McAdams HH: Codon usage between genomes is constrained by genome-wide mutational processes. Proc Natl Acad Sci USA. 2004, 101 (10): 3480-3485. 10.1073/pnas.0307827100.
Lawrence JG, Ochman H: Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol. 1997, 44 (4): 383-397. 10.1007/PL00006158.
Karlin S: Global dinucleotide signatures and analysis of genomic heterogeneity. Curr Opin Microbiol. 1998, 1 (5): 598-610. 10.1016/S1369-5274(98)80095-7.
Baran RH, Ko H: Detecting horizontally transferred and essential genes based on dinucleotide relative abundance. DNA Res. 2008, 15 (5): 267-276. 10.1093/dnares/dsn021.
Fournier PE, Drancourt M, Raoult D: Bacterial genome sequencing and its use in infectious diseases. 2007, 7 (11): 711-723.
Zhao X, Zhang Z, Yan J, Yu J: GC content variability of eubacteria is governed by the pol III alpha subunit. Biochem Biophys Res Commun. 2007, 356 (1): 20-25. 10.1016/j.bbrc.2007.02.109.
Vetsigian K, Goldenfeld N: Genome rhetoric and the emergence of compositional bias. Proc Natl Acad Sci USA. 2009, 106 (1): 215-220. 10.1073/pnas.0810122106.
Bernaola-Galvan P, Oliver JL, Carpena P, Clay O, Bernardi G: Quantifying intrachromosomal GC heterogeneity in prokaryotic genomes. Gene. 2004, 333: 121-133. 10.1016/j.gene.2004.02.042.
Daubin V, Perriere G: G+C3 structuring along the genome: a common feature in prokaryotes. Mol Biol Evol. 2003, 20 (4): 471-483. 10.1093/molbev/msg022.
Ewens WJ, Grant GR: Statistical Methods in Bioinformatics. 2001, Springer, New York
Willenbrock H, Friis C, Juncker AS, Ussery DW: An environmental signature for 323 microbial genomes based on codon adaptation indices. Genome Biol. 2006, 7 (12): R114-10.1186/gb-2006-7-12-r114.
Reva ON, Tummler B: Differentiation of regions with atypical oligonucleotide composition in bacterial genomes. BMC Bioinformatics. 2005, 6: 251-10.1186/1471-2105-6-251.
Raymond J, Segre D: The Effect of Oxygen on Biochemical Networks and the Evolution of Complex Life. Science. 2006, 311 (5768): 1764-1767. 10.1126/science.1118439.
Acquisti C, Kleffe J, Collins S: Oxygen content of transmembrane proteins over macroevolutionary time scales. Nature. 2007, 445 (7123): 47-52. 10.1038/nature05450.
Vieira-Silva S, Rocha EP: An assessment of the impacts of molecular oxygen on the evolution of proteomes. Mol Biol Evol. 2008, 25 (9): 1931-1942. 10.1093/molbev/msn142.
Sasidharan R, Smith A, Gerstein M: Transmembrane protein oxygen content and compartmentalization of cells. PLoS One. 2008, 3 (7): e2726-10.1371/journal.pone.0002726.
Acknowledgements
The authors thank Colleen Ussery for critically reading the manuscript and the reviewers for their insightful suggestions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
JB wrote the paper and carried out analyses. LS, ABK, JB, ES carried out statistical analyses. TD suggested the study. DWU, SPH, ES, JB, KL performed biological analyses. TD, KL, ES, SPH and DWU critically drafted and revised the manuscript. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bohlin, J., Snipen, L., Hardy, S.P. et al. Analysis of intra-genomic GC content homogeneity within prokaryotes. BMC Genomics 11, 464 (2010). https://doi.org/10.1186/1471-2164-11-464
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-11-464