
Abstract
Background
Cytoplasmic male sterility (CMS) is not only important for exploiting heterosis in crop plants, but also as a model for investigating nuclear-cytoplasmic interaction. CMS may be caused by mutations, rearrangement or recombination in the mitochondrial genome. Understanding the mitochondrial genome is often the first and key step in unraveling the molecular and genetic basis of CMS in plants. Comparative analysis of the mitochondrial genome of the hau CMS line and its maintainer line in B. juneca (Brassica juncea) may help show the origin of the CMS-associated gene orf288.
Results
Through next-generation sequencing, the B. juncea hau CMS mitochondrial genome was assembled into a single, circular-mapping molecule that is 247,903 bp in size and 45.08% in GC content. In addition to the CMS associated gene orf288, the genome contains 35 protein-encoding genes, 3 rRNAs, 25 tRNA genes and 29 ORFs of unknown function. The mitochondrial genome sizes of the maintainer line and another normal type line “J163-4” are both 219,863 bp and with GC content at 45.23%. The maintainer line has 36 genes with protein products, 3 rRNAs, 22 tRNA genes and 31 unidentified ORFs. Comparative analysis the mitochondrial genomes of the hau CMS line and its maintainer line allowed us to develop specific markers to separate the two lines at the seedling stage. We also confirmed that different mitotypes coexist substoichiometrically in hau CMS lines and its maintainer lines in B. juncea. The number of repeats larger than 100 bp in the hau CMS line (16 repeats) are nearly twice of those found in the maintainer line (9 repeats). Phylogenetic analysis of the CMS-associated gene orf288 and four other homologous sequences in Brassicaceae show that orf288 was clearly different from orf263 in Brassica tournefortii despite of strong similarity.
Conclusion
The hau CMS mitochondrial genome was highly rearranged when compared with its iso-nuclear maintainer line mitochondrial genome. This study may be useful for studying the mechanism of natural CMS in B. juncea, performing comparative analysis on sequenced mitochondrial genomes in Brassicas, and uncovering the origin of the hau CMS mitotype and structural and evolutionary differences between different mitotypes.
Similar content being viewed by others


Background
Cytoplasmic male sterility is a phenotypic trait that is widespread among plants and results in the inability of the plant to produce viable pollen [1]. Numerous studies have shown that cytoplasmic male sterility in plants is associated with aberrant recombination in the mitochondrial genome, which results in the production of chimeric ORFs that are expressed as novel polypeptides [2]. Since the first plant mitochondrial genome sequencing in Arabidopsis[3], a large number of mitochondrial genomes have been sequenced in angiosperm plants [4–10], especially in those that contain CMS cytoplasm. CMS-associated mitochondrial genome of crop species reported to date include Beta vulgaris[11], Oryza sativa[12–14], Triticumae stivum[15], Zea mays[16], Brassica napus[17, 18], Raphanus sativus[19, 20]. In this study, the mitochondrial genome of hau CMS line, its maintainer line and the normal type line “J163-4” were fully sequenced and assembled into a master circle. As in other higher plants, all three sequenced mitochondrial genomes had large sizes and distinctive features, including slow evolutionary rates, rapid rearrangement, frequent insertion, complex multipartite structures, specific modes of gene expression, cis- and trans-splicing, RNA editing and the use of universal genetic code [21]. Comparative analysis of the CMS line and its iso-nuclear maintainer line may help us verify the CMS-associated gene in hau CMS line, and contribute to a better understanding of the plant mitochondrial genome in Brassicas.
The male sterile hau CMS line (00-6-102A) emerged as a spontaneous male sterile mutant in B. juncea. The anthers in the hau CMS plants are replaced by thickened petal-like structures, and hau CMS sterility starts at the stamen primordium polarization stage, much earlier than the other four CMS systems used in Brassicas (pol, ogu, nap, and tour) [22]. A novel chimeric gene named orf288 was found to be located downstream of the atp6 gene and co-transcribed with this gene in the hau CMS sterile line. Subcellular localization analysis showed that this CMS-associated gene was translated in the mitochondria of male-sterile plants. Transgenic result also showed that ORF288 is associated with the male sterility of hau CMS in Brassica juncea[23].
In this study, we sequenced the complete mitochondrial genomes of hau CMS line, its iso-nuclear maintainer line and the normal type line “J163-4” in B. juneca using Roche/454 pyro-sequencing technology. Comparative analysis of the hau CMS mitochondrial genome further confirmed that orf288 was a cytoplasmic male sterility-associated gene in B. juneca. The sequenced mitochondrial genomes may help us identify the mechanism of natural CMS and uncover the origin and structure of the hau CMS mitotype as well as understand evolutionary differences between the different mitotypes in B. juncea. Our data give new insight into the evolution of the Brassicas mitochondrial genome.
Results
The mitochondrial genomes of the hauCMS line and its maintainer line
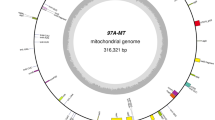
The mitochondrial genomes of the hau CMS line, its maintainer line and the normal type line “J163-4” were sequenced to an average depth of 52*, 196* and 69* multiple depths using Roche 454 FLX + pyro-sequencing technology. Sequences were assembled to 7, 3 and 4 contigs, respectively. The PCR primers used for the confirmation of contig linkage are listed in Additional file 1 and the mitochondrial DNA extracted from the etiolated seedlings in 7 days of the hau CMS line and its iso-nuclear maintainer line were used as templates. Results of the PCR amplification are presented in Additional file 2. A master circle was developed for each mitochondrial genome using a ‘parsimonious’ method [24]. Given that the mitochondrial genome of the hau CMS maintainer line and the line “J163-4” are identical to each other, we only performed comparative analysis of hau CMS line and its maintainer line in this study. Features of the hau CMS line and its maintainer line mitochondrial are shown in Table 1. The B. juncea hau CMS mitochondrial genome was assembled into a single, circular-mapping molecule with a size of 247,903 bp and GC content of 45.08%, both of which are close to the median values of other fully sequenced seed plant mitochondrial genomes. Coding sequences constitute approximately 20.52% of the genome, which contains 35 protein coding open reading frames (ORFs), 3 rRNA genes, 25 tRNA genes, and 29 unidentified ORFs (Figure 1). The size of its maintainer line is 219,863 bp, and the GC content is 45.23% with 23.43% of the sequence as coding sequence. The maintainer line mitochondrial genome contains 36 protein-coding genes, 3 rRNA genes, 22 tRNA genes, and 31 unidentified ORFs as shown in Additional file 3.

A circular diagram of the hau CMS line mitochondrial genome. From outside to inside: numbers on the outermost circle represent the physical map scaled in bp. Coding sequences transcribed in the clockwise and counterclockwise direction are drawn on the inside and outside of the second circle, respectively. Genes encoding proteins from the same complexes are similarly colored as are rRNAs and tRNAs in the inner circle. The third circle shows the locations of repeats larger than 100 bp with the most compelling evidence for recombination activity, and detailed information for repeats is shown in Additional file 5.
After the initial sequence analysis, we compared the coding regions, ORFs of unknown function and repeats of the two circular mitochondrial genomes. The electron carrier complexes I, III, IV and V genes in the hau CMS line and its maintainer line were conserved, while the rps7 gene encoding a subunit of ribosomal proteins in the hau CMS line was absent when compared with its maintainer line. Detailed information describing the tRNA gene content of the hau mitotype and its maintainer line (normal) mitotype is shown in Additional file 4. After a comparative analysis of the predicted ORFs in the two mitochondrial genomes, specific ORFs were found occurring in both mitochondrial genomes. We also plotted the syntenic regions using the bl2seq algorithm with the hau CMS line, its iso-nuclear maintainer line and the sequenced B. juncea [GenBank: JF920288] mitochondrial genome [18]. As shown in Figure 2A, the genomic arrangement of the hau CMS line mitochondrial genome was very divergent when compared with its maintainer line, with at least 14 apparent rearrangements. However, as shown in Figure 2B, when the B. juncea mitochondrial genome sequenced by Chang [18] was compared with the hau CMS maintainer line, no divergent genomic arrangement occurred except that of SNPs divergence. This result not only confirmed the accuracy of our sequence assembly but also showed that the hau CMS line mitochondrial genome was extensively rearranged when compared with its maintainer line.

Comparison syntenic region of hau CMS mitotype with its maintainer line mitotype and the normal. (A) Comparisons between the hau CMS mitochondrial genome (horizontal axis) and its maintainer line mitochondrial genome (vertical axis) indicated that the nucleotide sequences of the syntenic region are well conserved; however, the syntenic order and direction were largely rearranged. The numbers refer to the syntenic regions between the hau CMS mitochondrial genome and its iso-nuclear maintainer line mitochondrial genome. (B) Alignment of the hau CMS maintainer line mitochondrial genome and the normal mitochondrial genome [GenBank: JF920288] sequenced by Chang in Brassica juncea. Apart from SNPs, they were consistent and no rearrangement was found between them.
Repeats in the hauCMS line and its maintainer line
Repeats in the plant mitochondrial genome may be relevant to the rapid rearrangement, frequent insertion and complex multipartite structure in plant mitochondrial genomes. Based on mitochondrial genomes of the hau CMS line and its maintainer line, detailed information for repeated sequences greater than 100 bp was annotated in Additional file 5 and shown in Figure 1 and Additional file 3. The number of repeats greater than 100 bp in the hau CMS line (16 repeats) was almost two fold of that in its maintainer line (9 repeats). There were 8 direct repeats and 8 inverted repeats in the hau CMS line, as well as 5 direct and 4 inverted repeats in its maintainer line. The largest repeats in the hau CMS line were 7,396 bp and 7,404 bp with only 8 different indels between them (data not shown). The largest repeats in the maintainer line were 2293 bp, and both were direct repeats. Recombination across inverted repeats inverts the intervening sequences, whereas recombination across directly oriented repeats separates the genome into pairs of sub-genomic molecules [25]. Repeats smaller than 100 bp were also investigated in the mitochondrial genome of the hau CMS line and its maintainer line. Intriguingly, we observed many small repeats in the 2 sequenced mitochondrial genomes. In both the hau CMS line and its maintainer line, there were more than 200 short repeats smaller than 100 bp. Some large repeats also contained a few small repeats smaller than 100 bp. The largest repeat in the mitochondrial genome of the hau CMS line in Figure 3A was analyzed, which encompassed 5 small direct repeats. The border of this repeat, the initiation codon of these small repeats and the nucleotide sequences of these three small repeats were marked. The same phenomenon was also found in other large repeats in the hau CMS line and its maintainer line mitochondrial genomes. Apparently, these small repeats were part of the large repeats. However the relations between these large and small repeats remain unclear. It is known that sub-genomic molecules are produced from the MC molecule through intra-molecular recombination. Different sub-genomic molecules may constitute different mitochondrial genomes through recombination of these direct and inverted repeats. The short repeated sequences in higher plant mitochondria are usually inactive and may play key role in irreversible recombination producing a new stable mitochondrial genome structure [26]. These short repeats as part of the larger repeats in mitochondrial genome may help us to answer how the large repeats emerged, expanded and then gave rise to complex multipartite structures of plant mitochondrial genomes. Three large repeats larger than 100 bp were also found between syntenic regions of atp4 and atp6 (Figure 3B). They were located downstream of orf288, which may be relevant to the emergence of this CMS-associated gene.

The largest repetitive region and structural polymorphism surrounding orf288 in the hau CMS mitochondrial genome. (A) The largest repeats in the hau CMS mitochondrial genome are 7369 bp and 7404 bp, both of which showed 8 bp indel SNPs. There was also three small direct repeats named sR1, sR2 and sR3 in the largest repeats. A detailed nucleotide sequence of the three repeats is given above. (B) Three large repeats of more than 100 bp were located downstream of the CMS-associated orf288 gene.
The specific ORFs of different mitochondrial genomes
We further analyzed those ORFs of unknown function in the sequenced mitochondrial genomes in this study. There were 29 and 31 such ORFs in the hau CMS line and its maintainer line, respectively. By virtue of mitochondrial rearrangement and indel mutations, specific ORFs were found in the hau CMS line and its maintainer line mitochondrial genome. There were 5 mitotype specific ORFs in the hau CMS line and its maintainer line separately. Sequence analysis revealed that orf113a in the hau CMS line had only SNPs differences when compared with orf113b in its maintainer line. Although orf117b in the hau CMS line had not been detected in its maintainer line, it was detected in the sequenced mitochondrial genomes reported by Chang [18]. Thus, we chose only 3 ORFs from each of the 2 lines. Among them orf288, orf325 and orf170 were hau mitotype-specific ORFs, while orf109, orf293 and rps7 were the maintainer line specific ORFs (Table 2). PCR amplification of the mitochondrial specific SCAR markers was developed based on these specific ORFs from the two sequenced mitochondrial genomes. They were both efficient in distinguishing the hau CMS line from its maintainer line at the seedling stage. We confirmed these by PCR analysis after developing the mitochondrial specific ORF markers which are shown in Additional file 1. As demonstrated in Figure 4, the P1, P2 and P3 primers combinations were specific to the A line (hau specific mitotype) while P4, P5 and P6 primers were specific to the B line (its iso-nuclear maintainer line) after 25 cycles. However, after 30 and 35 cycles, hau CMS specific ORFs were amplified slightly in the maintainer line, and the hau CMS maintainer line specific ORFs were also amplified in the hau CMS line, especially with the P3, P5 and P6 primers. It is noteworthy that there was evidence suggesting that substoichiometrically different mitotypes coexisted in hau CMS lines and their maintainer lines as reported by Chen [9].

PCR amplification in the mitochondrial genomes of the hau CMS line and its maintainer. The hau CMS mitotype and the normal mitotype coexisted in both the hau CMS line 00-6-102A (A) and its maintainer line 00-6-102B (B), but were substoichiometrically different in these cytotypes. Different PCR cycles (25, 30, 35) were used to amplify these different open reading frames. Primer pairs P1, P2 and P3 targeted the hau CMS mitotype-specific sequences orf288, orf325 and orf170, respectively, whereas P4, P5 and P6 targeted its maintainer line (normal) mitotype-specific sequences. All PCR reactions used 50 ng mtDNA as template. The PCR assay indicated that the hau CMS mitotype and normal mitotypes coexisted in Brassica juncea, but the content was substoichiometrically different form the two cytoplasm types. M: DNA ladder.
Subsequently, total RNA was isolated from flower buds, fresh leaves, roots and hypocotyls (etiolated seedlings). We examined the transcript pattern of these specific ORFs in the hau CMS line and its maintainer line. As shown in Figure 5, orf288 was constitutively expressed in all tissues tested, while orf325 was not detectable in the roots and orf170 only appeared in leaves and the etiolated seedlings of the hau CMS line. In its maintainer line, orf293 was only expressed in the leaves, while the orf109 and rps7 were undetectable at the etiolated seedling stage. Apart from orf288, which was known to be the CMS-associated gene in the hau CMS line, understanding the function of these ORFs requires further investigation.

Expression of the unknown function ORFs in the hau CMS line and its maintainer line. (A) The expression of the hau CMS mitochondrial genome specific orf288, orf325 and orf170 in four different tissues. The CMS-associated gene orf288 is expressed in the flower, leaf, root and etiolated seedings of B. juncea and is carried by the hau CMS mitochondrial genome. However, orf325 did not appear in the root and orf170 only appeared in the leaf and etiolated seedings 7 days after germination. (B) orf109 and rps7 both appeared in the flower, leaf, and root. However, orf293, which is similar to orf288, only appeared in the leaf of hau CMS maintainer line.
The hau CMS-associated gene orf288 in Brassica juncea
CMS is often associated with specific open reading frames (ORFs) in plant mitochondrial genomes, but the origin of many CMS-associated genes and the mechanism of this phenomenon are still unclear. Utilizing next-generation sequencing, many more plant mitochondrial genomes have been released, especially for CMS-associated genomes of various crops such as rice, maize, wheat, sorghum and rapeseed (B. napus). Value of mitochondrial genome information is also well established in the study of evolutionary patterns and processes of CMS-associated genes in plants. Results from transgenics showed that orf288 was responsible for the male sterility of hau CMS in B. juncea[23]. CMS-associated genes in different mitochondrial genomes are often located downstream of the genes encoding components of the electron transport respiratory chain. For example, orf224 in the B.napus polima CMS line, orf79 in the rice BORO CMS line, and orf138 in the ogura CMS line are all located downstream of atp6. The orf522 in the sunflower PET1 CMS line is located downstream of atp1[2]. Almost all these CMS-associated genes were chimeric genes and had transmembrane domains. The transmembrane domains of these specific open reading frames in hau CMS line were predicted using TMHMM server version 2.0. This revealed that apart from orf288, orf325 also had the transmembrane domains, albeit its function is still unknown. The hau CMS-associated gene orf288 was also located downstream of atp6, but how the CMS-associated gene emerged remained unclear. The 3 large repeats located downstream of orf288 might have been relevant to the emergence of the CMS-associated gene. Like most CMS genes, orf288 has similarities to known functional mitochondrial genes. Apart from the unknown origin (1–24 bp), the chimeric orf288 gene also contains a 94 bp partial sequence of nad5, a subunit of complex I in the electron transport chain system. This is similar to the case of the 5′ region of the gene orf263 as reported in alloplasmic male sterile Brassica tournefortii[27]. The 3′ region was also similar to another predicted mitochondrial ORF, orf293 in B. juncea (Figure 6A). This result suggested that this recombinant structure evolved recently. By way of BLAST alignment analysis, it can be seen that orf263, orf286, orf293 and orf305 were homology sequenced separately from the sequenced mitochondrial genomes of B. tournefortii, B. napus, B. juncea and R. sativus in Brassicaceae. Other than orf263, the CMS-associated gene in Brassica tournefortii and orf305 was previously reported by Christopher A. Makaroff in a cytoplasmic male sterility radish [28]. The other two open reading frames only appeared in the male fertility mitochondrial genomes of B. napus and B. juncea. A maximum-likelihood inference-based dendrogram was used to generate phylogenetic trees based on conserved orf288 genes in mitochondrial genomes (Figure 6B). Molecular phylogenetic analysis indicated that orf288 bore a strong similarity to orf263 in Brassica tournefortii and the mitochondrial genomes of B. napus and B. juncea might be closer to each other than R. sativus.

The structure of orf288 and phylogenetic tree based on conserved orf288 genes in mitochondrial genomes. (A) The hau CMS-associated gene orf288 was located downstream of atp6. As for orf263 in Brassica tournefortii mitochondrial genome, the nucleotide identities (%) of the mitochondrial genome-derived fragments to the chimeric gene orf288 were shown. (B) Phylogenetic analysis of the CMS-associated gene orf288 in Brassicaceae. Orf263 was associated with cytoplasmic male sterility in Brassica tournefortii, Multiple sequence alignment used to construct the phylogenetic trees orf286, orf293 and orf305 shows they were separated in terms of sequence homology from the mitochondrial genomes of B. napus, B. juncea and R. sativus. Bootstrap values are shown at the nodes and the bar indicates the rate of nucleotide substitution per site.
We also comparatively analyzed the mitochondrial genome of hau CMS with the mitochondrial genomes of nap [AP006444], cam [JF920285], jun [JF920288], ole [JF920286], car [JF920287], polima [FR715249] and ogura [AB694744] in Brassicas[17–20]. A phylogenetic guide tree (Figure 7) and locally collinear blocks (Additional file 6) relating these eight sequenced mitochondrial genomes were calculated by using Neighbor Joining of the Mauve alignment system. The 141.8 kb segment from 173,638 bp to 315,446 bp in ole mitochondrial genome was deleted for the reason that Tandem repeats >10 kb in total length without an anchor are ignored by MAUVE. Cluster analysis of the eight mitochondrial genomes showed that the mitochondrial genomes of hau CMS and ogura CMS are both diverged from the other six mitochondrial genomes in Brassicas. And the evolutionary relationships among the six analyzed homologous mitotypes are consistent with previous research by Chang [18]. These results confirmed the hau CMS mitotype which caused male sterility in Brassicas might be heterogeneous when compared with the other related mitotypes at molecular level.

A phylogenetic tree relating the eight mitochondrial genomes sequenced in Brassicas . The hau CMS mitotype and ogura CMS mitotype were both diverged from the other six sequenced mitochondrial genomes in Brassicas. The bar indicates the rate of nucleotide substitution per site.
Discussion
Comparative analysis of the mitochondrial genome of the hauCMS line with its maintainer line
Over the past two decades, our knowledge of the organization and evolution of mitochondrial genomes has been rapidly expanding as a large number of fully sequenced mitochondrial genomes have been published along with their structure, expression and evolutionary profiles. In light of the pivotal role of the CMS line in crop breeding and its convenience for studying the cytoplasmic and nuclear interaction, we compared the mitochondrial genomes of the hau CMS line and its iso-nuclear maintainer line to investigate the origin of the hau CMS mitotype and to dissect the structural and evolutionary differences between the different mitotypes. Mitochondrial genome of the hau CMS line (247,903 bp) was larger than its maintainer line (219,863 bp) and the repeats (>100 bp) appeared noticeably more frequent than those in its maintainer line. The size of plant mitochondrial genome was relevant to the repeats it contained, and the repeats in the mitochondrial genomes also revealed the structural dynamics of the mitochondrial genome in plant development via intramolecular and intermolecular recombination. In this study, we report that small repeats (<50 bp) might contribute to larger repeats (>100 bp) in the sequenced hau CMS mitochondrial genome, but the emergence of these small repeats and their function in the hau CMS line and its maintainer line mitochondrial genomes requires further study.
As reported in rice [13], maize [29], pearl millet [30], wheat [31], B. napus[17] and cybrids in Brassicaceae[32], our results also confirmed that substoichiometrically different mitotypes coexist in mitochondrial genomes of the hau CMS line and its maintainer line in B. juncea. The ratio of the alternative genomes may be variable, but in plants, the usually prevalent main genome is accompanied by sublimons—substoichiometric mitochondrial DNA (mtDNA) molecules [33]. In plants, the relative copy number of recombination-derived sub-genomic DNA molecules within mitochondria is controlled by nuclear genes and a genomic shifting process can result in their differential copy number suppression to nearly undetectable levels [34]. Most of the mitotype-specific ORFs in one mitochondrial genome present at a substoichiometric level in the other genome. At 25 cycles of PCR amplification, only templates carrying specific ORFs were able to produce a detectable PCR product in the hau CMS line and its maintainer line, and when the cycles were up to 30 to 35, all primer pairs (P1-P6) used were able to amplify specific ORFs in hau CMS line and its maintainer line, but the pattern of amplification differed in the 2 lines. The substoichiometric amount of specific ORFs from one genome to the other is thus sufficient to be detected by PCR amplification but not enough to have been picked up by the coverage depth of the 454 sequencing that was performed [13]. Although substoichiometrically different mitotypes coexist in mitochondrial genomes of different plants, the molecular mechanism of such coexistence in different mitotypes requires further study. The coexistence of different mitotypes may play a prominent role in the coordination of nuclear and mitochondrial interaction and also make valuable contributions to the hybrid vigor in different crop plants.
Heterosis plays an irreplaceable role in China’s high-yield crop production, and male sterility, which is a prerequisite for the mass production of hybrid seeds, acts as a key factor [35]. Cytoplasmic male sterility (CMS) in plant, which is determined by the mitochondrial genome is associated with a pollen sterility phenotype and caused by mitochondrial genome mutation. Identification of a CMS-associated gene and uncovering the mechanism of this trait may facilitate plant breeding. With the sequenced mitochondrial genome, more novel mitochondrial genome types and molecular markers for cytoplasm classifications will be identified. Recently, Chang et al. [18] sequenced the mitotypes of cam (B. rape), ole (B. oleracea), jun (B. juncea) and car (B. carinata) and analyzed them together with previously sequenced mitotypes of B. napus (pol and nap) to show the evolutionary mechanism of mitochondrial genome formation in Brassica. Molecular markers such as RFLPs, AFLPs, SCARs, and SSRs were used to distinguish the CMS line from its maintainer line in the Brassicas. Based on the sequenced mitochondrial genome, specific SCAR markers (the hau CMS line specific primers combination P1, P2 and P3 and its iso-nuclear maintainer line specific primers combination P4, P5 and P6) were also developed to separate the hau CMS line from its maintainer line at the seedling stage.
The origin and emergence of the CMS-associated gene orf288 in B. juncea
A large number of CMS-associated genes have been found in crop species, but the origin and precise mechanism of CMS remains elusive. In different CMS systems, the CMS-associated genes show little or no structural relationship. Often the CMS-associated genes or loci are located close to an atp gene or contain parts of a gene encoding an ATPase subunit and are co-transcribed with flanking mt-genes [36]. Although the CMS phenotype also occurs at different stages during reproductive development, they were the root cause of male sterility. In our study, the CMS-associated gene orf288 located downstream of atp6 and is co-transcribed. The transgenic result further verified that orf288 is associated with the male sterility of hau CMS in Brassica juncea[23]. As a chimeric gene, orf288 is composed partially of nad5 and orf293 in B. juncea, and there were 3 large repeats larger than 100 bp located downstream of orf288. These repeats may be related to the formation of the CMS-associated gene. Although it has relatively high similarity with orf263 in alloplasmic male sterile Brassica tournefortii at the nucleotide level, the restorer lines for tour CMS systems were found to be ineffective for restoring fertility in the hau CMS line. This may suggest that they were different from each other [22]. A comparative analysis of the mitochondrial genome of the hau CMS line and its maintainer line further confirmed that orf288 was CMS-associated gene in hau CMS line in B. juncea.
Voluminous evidence suggests that mitochondrial gene expression can affect the function of the nuclear gene products that control floral development. In Honglian cytoplasmic male sterile rice, the CMS-associated gene orfH79 impaired mitochondrial function via interaction with P61 (a subunit of electron transport chain complex III), and resulted in an energy production dysfunction and oxidative stress in mitochondria, which may work as retrograde signals leading to abnormal pollen development [37]. In Wild Abortive CMS rice, WA352 accumulates preferentially in the tapetum of anthers, where it inhibits COX11 function in peroxide metabolism and triggers premature tapetal programmed cell death and consequent pollen abortion. These CMS models provided a mechanistic link between the gain of function of a newly identified mitochondrial CMS gene product and the loss of activity of the essential nuclear-encoded mitochondrial protein through their detrimental interaction [38]. It is thus likely that different recombinations in different plant mitochondrials gave rise to different chimeric genes that caused male sterility through interaction with genes in the anther development pathways and eventually caused male sterility in different CMS systems. This hypothesis might explain why dissimilar CMS genes in different plants all caused similar phenotypic male sterility. The nature of different CMS-associated genes that interact with the anther development pathways still needs to be studied further.
Conclusions
The hau CMS mitochondrial genome was highly rearranged as was reported for mitochondrial genomes in CMS lines of other crops. The chimeric CMS-associated gene orf288 was composed of 94 bp partial sequences of nad5 (a subunit of complex I in the electron transport respiratory chain system) and 749 bp sequences that were highly similar to orf293 in its maintainer line. Three large repeats downstream of orf288 may be related to the formation of the CMS-associated gene in the hau CMS line. These findings may help us to identify the mechanism of natural CMS in B. juncea and to uncover the origin of the hau CMS mitotype and the structural and evolutionary differences between different mitotypes.
Methods
Plant materials
The hau CMS line (00-6-102A) used in this study was originally discovered as a spontaneous male-sterile mutant in B. juncea in the experimental field at Huazhong Agricultural University in 1999. The maintainer line (00-6-102B) was iso-nuclear to the hau CMS line in B. juncea. A cultivar trilocular line, J163-4, in B. juncea with fertility anther was also used as control in our study [39]. The anthers in the hau CMS plants are replaced by thickened petal-like structures and the sterility of the hau CMS initiates at the stamen primordium polarization stage [22]. Seeds from the hau CMS line, its iso–nuclear maintainer line and the normal type line “J163-4” were harvested in an experimental field at Huazhong Agricultural University in 2011.
Isolation of mitochondrial DNA and total RNA
Discontinuous Percoll gradient centrifugation was used to separate highly purified mitochondria from 7-day-old etiolated seedlings (Additional file 7) for hau CMS line, its maintainer line and the normal type line “J163-4” from B. juncea. A 100 g sample of each seedling-stage hypocotyls from the 3 different lines were homogenized in 200 ml homogenization medium (0.4 M mannitol, 5 mM EDTA, 8 mM cysteine, 10 mM tricine, 1% BSA, 1% polyvinyl-pyrrolidone, pH 7.8). The homogenate was filtered using four pieces of Miracloth and centrifuged at 1000 g for 5 minutes. The resulting supernatants were centrifuged at 18,000 g for 15 minutes and the pellet was re-suspended in wash buffer (0.4 M mannitol, 1 mM EDTA, 10 mM MOPS-KOH and 1% BSA) to repeat the above procedure at 1000 g for 5 minutes and 18,000 g for 15 minutes. The reaction was terminated with the addition of 20 mM EDTA. The pellet was resuspended in wash buffer and layered onto a step gradient consisting of 15%, 20%, 28% and 40% Percoll in 0.4 mM mannitol, 1% BSA and 10 mM MOPS-KOH. Purified mitochondrial were removed from the 20% and 28% interphase (Additional file 7). The pellet was resuspended in wash buffer without EDTA and 25 mg/ml DNase (Roche 104159) was added at room temperature for 1-3 h. Finally, samples were centrifuged at 18,000 g for 20 minutes and resuspended in the lysis buffer (50 mM Tris–HCl, 10 mM EDTA, 1% SDS, and 200 mg/ml proteinase K (Sigma) at room temperature for 3 h [40]. The CTAB method was used to obtain the purified mitochondrial DNA [41]. Total RNA was isolated from flower buds, fresh leaves, roots, and hypocotyls using Trizol (Invitrogen) according to the manufacturer’s protocol.
The sequencing strategy
The complete mitochondrial genomes of the hau CMS line, its maintainer line and the normal type line in B. juneca were sequenced using the Roche 454 FLX + pyro-sequencing technology. The sequencing service was provided by Personal Biotechnology (Shanghai, China). Nucleotide sequences of 13,130,330 bp, 43,164,917 bp and 15,240,642 bp in total were obtained from the 00-6-102A, 00-6-102B and J163-4 lines, respectively. The average sequence depth was 52, 196 and 69. The sequence was assembled to 7, 3 and 4 contigs in the hau CMS line, its maintainer lines and the normal type line “J163-4”. The genomic PCR products sequence between contigs was obtained by Sanger sequencing.
Sequence analysis
ORF Finder, BLASTX, BLASTN, and tRNA-SE were used to identify mitochondrial genes, rRNA, and tRNA. Artemis software [42], which allowed the use of a threshold to identify ORFs, was used to identify ORFs whose function was unknown. Both of these mitochondrial genome sequences were assembled using the Seqman software (DNAStar). Bl2seq (http://www.ncbi.nlm.nih.gov/) was used to do sequence alignment to find the syntenic region in the sequenced mitochondrial genomes. ClustalW2 and MEGA 4 were used for a phylogenetic analysis of CMS associated gene orf288 in Brassicaceae. Circos was used to visualize data and information of the mitochondrial genome of the hau CMS line and its maintainer line [43]. Progressive Mauve was used for multiple alignment among the 8 sequenced mitochondrial genomes in Brassicas[44]. The mitochondrial genome sequences of the hau CMS line and its maintainer line have been deposited to the GenBank Database under accession numbers: KF736092 and KF736093.
References
Laser KD, Lersten NR: Anatomy and cytology of microsporogenesis in cytoplasmic male sterile angiosperms. Bot Rev. 1972, 38: 425-454. 10.1007/BF02860010.
Schnable PS, Wise RP: The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci. 1998, 3: 175-180. 10.1016/S1360-1385(98)01235-7.
Unseld M, Marienfeld JR, Brandt P, Brennicke A: The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat Genet. 1997, 15 (1): 57-61. 10.1038/ng0197-57.
Kubo T, Nishizawa S, Sugawara A, Itchoda N, Estiati A, Mikami T: The complete nucleotide sequence of the mitochondrial genome of sugar beet (Beta vulgaris L.) reveals a novel gene for tRNA(Cys)(GCA). Nucleic Acids Res. 2000, 28: 2571-2576. 10.1093/nar/28.13.2571.
Notsu Y, Masood S, Nishikawa T, Kubo N, Akiduki G, Nakazono M, Hirai A, Kadowaki K: The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol Genet Genomics. 2002, 268: 434-445. 10.1007/s00438-002-0767-1.
Handa H: The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 2003, 31: 5907-5916. 10.1093/nar/gkg795.
Clifton SW, Minx P, Fauron CM, Gibson M, Allen JO, Sun H, Thompson M, Barbazuk WB, Kanuganti S, Tayloe C, Louis M, Richard KW, Kathleen JN: Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiol. 2004, 136: 3486-3503. 10.1104/pp.104.044602.
Ogihara Y, Yamazaki Y, Murai K, Kanno A, Terachi T, Shiina T, Miyashita N, Nasuda S, Nakamura C, Mori N, Takumi S, Murata M, Futo S, Tsunewaki K: Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 2005, 33: 6235-6250. 10.1093/nar/gki925.
Alverson AJ, Wei XX, Rice DW, Stern DB, Barry K, Palmer JD: Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol Biol Evol. 2010, 27 (6): 1436-1448. 10.1093/molbev/msq029.
Wang J, Jiang J, Li X, Li A, Zhang Y, Guan R, Wang Y: Complete sequence of heterogenous-composition mitochondrial genome (Brassica napus) and its exogenous source. BMC Genomics. 2012, 13: 675-10.1186/1471-2164-13-675.
Satoh M, Kubo T, Nishizawa S, Estiati A, Itchoda N, Mikami T: The cytoplasmic male-sterile type and normal type mitochondrial genomes of sugar beet share the same complement of genes of known function but differ in the content of expressed ORFs. Mol Genet Genomics. 2004, 272 (3): 247-256. 10.1007/s00438-004-1058-9.
Fujii S, Kazama T, Yamada M, Toriyama K: Discovery of global genomic re-organization based on comparison of two newly sequenced rice mitochondrial genomes with cytoplasmic male sterility-related genes. BMC Genomics. 2010, 11: 209-10.1186/1471-2164-11-209.
Bentolila S, Stefanov S: A reevaluation of rice mitochondrial evolution based on the complete sequence of male-fertile and male-sterile mitochondrial genomes. Plant Physiol. 2012, 158 (2): 996-1017. 10.1104/pp.111.190231.
Igarashi K, Kazama T, Motomura K, Toriyama K: Whole genomic sequencing of RT98 mitochondria derived from Oryza rufipogon and northern blot analysis to uncover a cytoplasmic male sterility-associated gene. Plant Cell Physiol. 2013, 54 (2): 237-243. 10.1093/pcp/pcs177.
Liu H, Cui P, Zhan K, Lin Q, Zhuo G, Guo X, Ding F, Yang W, Liu D, Hu S, Yu J, Zhang A: Comparative analysis of mitochondrial genomes between a wheat K-type cytoplasmic male sterility (CMS) line and its maintainer line. BMC Genomics. 2011, 12: 163-10.1186/1471-2164-12-163.
Matera JT, Monroe J, Smelser W, Gabay-Laughnan S, Newton KJ: Unique changes in mitochondrial genomes associated with reversions of S-type cytoplasmic male sterility in maizemar. PLoS One. 2011, 6 (8): e23405-10.1371/journal.pone.0023405.
Chen J, Guan R, Chang S, Du T, Zhang H, Xing H: Substoichiometrically different mitotypes coexist in mitochondrial genomes of Brassica napus L. PLoS One. 2011, 6 (3): e17662-10.1371/journal.pone.0017662.
Chang S, Yang T, Du T, Huang Y, Chen J, Yan J, He J, Guan R: Mitochondrial genome sequencing helps show the evolutionary mechanism of mitochondrial genome formation in Brassica. BMC Genomics. 2011, 12: 497-10.1186/1471-2164-12-497.
Tanaka Y, Tsuda M, Yasumoto K, Yamagishi H, Terachi T: A complete mitochondrial genome sequence of Ogura-type male-sterile cytoplasm and its comparative analysis with that of normal cytoplasm in radish (Raphanus sativus L.). BMC Genomics. 2012, 13: 352-10.1186/1471-2164-13-352.
Park JY, Lee YP, Lee J, Choi BS, Kim S, Yang TJ: Complete mitochondrial genome sequence and identification of a candidate gene responsible for cytoplasmic male sterility in radish (Raphanus sativus L.) containing DCGMS cytoplasm. Theor Appl Genet. 2013, 126 (7): 1763-1774. 10.1007/s00122-013-2090-0.
Schuster W, Brennicke A: The plant mitochondrial genome: physical structure, information content, RNA editing and gene migration to the nucleus. Annu Rev Plant Physiol Plant Mol Biol. 1994, 45: 61-78. 10.1146/annurev.pp.45.060194.000425.
Wan Z, Jing B, Tu J, Ma C, Shen J, Yi B, Wen J, Huang T, Wang X, Fu T: Genetic characterization of a new cytoplasmic male sterility system (hau) in Brassica juncea and its transfer to B. napus. Theor Appl Genet. 2008, 116 (3): 355-362. 10.1007/s00122-007-0673-3.
Jing B, Heng S, Tong D, Wan Z, Fu T, Tu J, Ma C, Yi B, Wen J, Shen J: A male sterility-associated cytotoxic protein ORF288 in Brassica juncea causes aborted pollen development. J Exp Bot. 2012, 63 (3): 1285-1295. 10.1093/jxb/err355.
Zhang Z, Schwartz S, Wagner L, Miller W: A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000, 7 (1–2): 203-214.
Palmer JD, Shields CR: Tripartite structure of the Brassica campestris mitochondrial genome. Nature. 1984, 307: 437-440. 10.1038/307437a0.
Andre C, Levy A, Walbot V: Small repeated sequences and the structure of plant mitochondrial genomes. Trends Genet. 1992, 8 (4): 128-132. 10.1016/0168-9525(92)90370-J.
Landgren M, Zetterstrand M, Sundberg E, Glimelius K: Alloplasmic male-sterile Brassica lines containing B. tournefortii mitochondria express an ORF 3′ of the atp6 gene and a 32 kDa protein. Plant Mol Biol. 1996, 32 (5): 879-890. 10.1007/BF00020485.
Makaroff CA, Apel IJ, Palmer JD: The atp6 coding region has been disrupted and a novel reading frame generated in the mitochondrial genome of cytoplasmic male-sterile radish. J Biol Chem. 1989, 264: 11706-11713.
Small I, Suffolk R, Leaver CJ: Evolution of plant mitochondrial genomes via substoichiometric intermediates. Cell. 1989, 58 (1): 69-76. 10.1016/0092-8674(89)90403-0.
Feng X, Kaur AP, Mackenzie SA, Dweikat IM: Substoichiometric shifting in the fertility reversion of cytoplasmic male sterile pearl millet. Theor Appl Genet. 2009, 118 (7): 1361-1370. 10.1007/s00122-009-0986-5.
Hattori N, Kitagawa K, Takumi S, Nakamura C: Mitochondrial DNA heteroplasmy in wheat, Aegilops and their nucleus-cytoplasm hybrids. Genetics. 2002, 160 (4): 1619-1630.
Bellaoui M, Martin-Canadell A, Pelletier G, Budar F: Low-copy-number molecules are produced by recombination, actively maintained and can be amplified in the mitochondrial genome of Brassicaceae: relationship to reversion of the male sterile phenotype in some cybrids. Mol Genet Genomics. 1998, 257 (2): 177-185. 10.1007/s004380050637.
Woloszynska M: Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes–though this be madness, yet there’s method in’t. J Exp Bot. 2010, 61 (3): 657-671. 10.1093/jxb/erp361.
Abdelnoor RV, Yule R, Elo A, Christensen AC, Meyer-Gauen G, Mackenzie SA: Substoichiometric shifting in the plant mitochondrial genome is influenced by a gene homologous to MutS. Proc Natl Acad Sci U S A. 2003, 100 (10): 5968-5973. 10.1073/pnas.1037651100.
Sodmergen : Sterility and fertility: Keys for crop production in China. Sci China Life Sci. 2010, 53 (12): 1488-1489. 10.1007/s11427-010-4094-1.
Hanson MR, Bentolila S: Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell. 2004, 16: S154-S169. 10.1105/tpc.015966.
Wang K, Gao F, Ji YX, Liu Y, Dan ZW, Yang PF, Zhu YG, Li SQ: ORFH79 impairs mitochondrial function via interaction with a subunit of electron transport chain complex III in Honglian cytoplasmic male sterile rice. New Phytol. 2013, 198 (2): 408-418. 10.1111/nph.12180.
Luo DP, Xu H, Liu ZL, Guo JX, Li HY, Chen LT, Fang C, Zhang QY, Bai M, Yao N, Wu H, Wu H, Ji C, Zheng H, Chen Y, Ye S, Li X, Zhao X, Li R, Liu YG: A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat Genet. 2013, 45 (5): 573-577. 10.1038/ng.2570.
Xu P, Lv Z, Zhang X, Wang X, Pu Y, Wang H, Yi B, Wen J, Ma C, Tu J, Fu T, Shen J: Identification of molecular markers linked to trilocular gene (mc1) in Brassica juncea L. Mol Breeding. 2014, 33: 425-434. 10.1007/s11032-013-9960-7.
Millar AH, Liddell A, Leaver CJ: Isolation and subfractionation of mitochondria from plants. Method Cell Biol. 2007, 80: 65-90.
Fulton TM, Chunwongse J, Tanksley SD: Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol Biol Rep. 1995, 13 (3): 207-209. 10.1007/BF02670897.
Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA: Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012, 28 (4): 464-469. 10.1093/bioinformatics/btr703.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA: Circos: an information aesthetic for comparative genomics. Genome Res. 2009, 19 (9): 1639-1645. 10.1101/gr.092759.109.
Darling AE, Mau B, Perna NT: progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010, 5 (6): e11147-10.1371/journal.pone.0011147.
Acknowledgments
The authors are grateful to thank the two anonymous reviewers for their helpful comments and Dr. Jitao Zou from National Research Council Canada, Saskatoon and Mr. Wenxiang Jia for critically reading the manuscript and Xiao Lin for technical support. This study was supported by the National Natural Science Foundation of China (NSFC grant number 31271761), the Doctoral Fund of Ministry of Education of China (grant number 20120146110011) and the Fundamental Research Funds for the Central Universities (grant number 2662014PY060).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declared that they have no competing interests.
Authors’ contributions
SH, CW, BJ and ZW carried out the experiments and performed the sequence analysis. SH analyzed the data and wrote the manuscript. JW, BY, CM, JT contributed new reagents, materials and analysis tools. JS and TF designed the experiments and directed the manuscript writing. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2013_6022_MOESM2_ESM.tiff
Additional file 2: Validation of contig linkage through PCR analysis. Line number refers to the primer combinations used for validating the gap between the contigs in the hau CMS mitotype and the normal mitotype in Additional file 1. PCR confirmed the bridge sequences of the gaps in the hau CMS line and its maintainer line mitochondrial genome in A and B, respectively. (TIFF 419 KB)
12864_2013_6022_MOESM3_ESM.tiff
Additional file 3: A circular diagram of the hau CMS maintainer line mitochondrial genome in B. juneca. As for the hau CMS mitochondrial genome diagram shown in Figure 1, numbers on the outermost circle represent the physical map scaled in kb. Coding sequences transcribed in the clockwise and counterclockwise directions are drawn on the inside and outside of the second circle, respectively. Genes coding proteins from the same complexes are similarly colored as are rRNAs and tRNAs in the inner circle. The third circle shows the locations of repeats larger than 100 bp with the most compelling evidence for recombination activity, and detailed information for repeats is shown in Additional file 5. (TIFF 858 KB)
12864_2013_6022_MOESM5_ESM.xlsx
Additional file 5: Repeated sequences (>100 bp, 99% homology) in the hau CMS mitotype and its iso-nuclear maintainer mitoype.(XLSX 15 KB)
12864_2013_6022_MOESM6_ESM.tiff
Additional file 6: Locally collinear blocks identified among the eight sequenced mitochondrial genomes in Brassicas. Mauve visualization of locally collinear blocks identified among the eight sequenced mitochondrial genomes in Brassicas. Each contiguously colored region is a locally collinear block (LCB) region without rearrangement of homologous backbone sequence. LCBs below a genome’s center line are in the reverse complement orientation relative to the reference genome. Lines between genomes trace each orthologous LCB through every genome. Large gray regions within an LCB signify the presence of lineage-specific sequence at that site. (TIFF 3 MB)
12864_2013_6022_MOESM7_ESM.tiff
Additional file 7: Extraction of high-quality mitochondrial genome DNA from B. juncea. Seedlings etiolated for 7 days were used in the isolation of mitochondrial. Percoll differential centrifugation and density gradient centrifugation were used to separate the purified mitochondrial DNA. (TIFF 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Heng, S., Wei, C., Jing, B. et al. Comparative analysis of mitochondrial genomes between the hau cytoplasmic male sterility (CMS) line and its iso-nuclear maintainer line in Brassica juncea to reveal the origin of the CMS-associated gene orf288. BMC Genomics 15, 322 (2014). https://doi.org/10.1186/1471-2164-15-322
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-322