
Abstract
Background
Phellinus sulphurascens is a fungal pathogen that causes laminar root rot in conifers, one of the most damaging root diseases in western North America. Despite its importance as a forest pathogen, this fungus is still poorly studied at the genomic level. An understanding of the molecular events involved in establishment of the disease should help to develop new methods for control of this disease.
Results
We generated over 4600 expressed sequence tags from two cDNA libraries constructed using either mycelia grown on cellophane sheets and exposed to Douglas-fir roots or tissues from P. sulphurascens-infected Douglas-fir roots. A total of 890 unique genes were identified from the two libraries, and functional classification of 636 of these genes was possible using the Functional Catalogue (FunCat) annotation scheme. cDNAs were identified that encoded 79 potential virulence factors, including numerous genes implicated in virulence in a variety of phytopathogenic fungi. Many of these putative virulence factors were also among 82 genes identified as encoding putatively secreted proteins. The expression patterns of 86 selected fungal genes over 7 days of infection of Douglas-fir were examined using real-time PCR, and those significantly up-regulated included rhamnogalacturonan acetylesterase, 1,4-benzoquinone reductase, a cyclophilin, a glucoamylase, 3 hydrophobins, a lipase, a serine carboxypeptidase, a putative Ran-binding protein, and two unknown putatively secreted proteins called 1 J04 and 2 J12. Significantly down-regulated genes included a manganese-superoxide dismutase, two metalloproteases, and an unknown putatively secreted protein called Ps0058.
Conclusions
This first collection of Phellinus sulphurascens EST sequences and its annotation provide an important resource for future research aimed at understanding key virulence factors of this forest pathogen. We examined the expression patterns of numerous fungal genes with potential roles in virulence, and found a collection of functionally diverse genes that are significantly up- or down-regulated during infection of Douglas-fir seedling roots by P. sulphurascens.
Similar content being viewed by others


Background
The basidiomycete Phellinus sulphurascens (Ps) is the causal agent of laminated root rot (LRR), one of the most damaging root diseases affecting conifers in northwestern America [1]. Douglas-fir (DF; Pseudotsugata menziezii) and grand fir (Abies grandis) are particularly susceptible to this disease [2]. Like most root pathogens, Ps can live as both a parasite and saprotroph. As a parasite, it initiates infection by colonizing the live bark and cambium of its host, then progressively moves inward to kill phloem and cambial tissues, spreading both proximally and distally from the point of infection. It then enters and initiates decay in the xylem, and finally advances into the heartwood and upward in the stem [3]. Wounding the roots facilitates but is not essential for parasitic colonization by the fungus [3]. As a saprotroph, Ps can remain viable in stumps, fallen stems, and roots for as long as 50 years [4]. New infections most commonly begin when new roots of susceptible hosts contact infested stumps or roots left in the soil from the previous stand [3]. It has been estimated that LRR decreases stand volume by 40 – 70% on infected sites [5].
As recommended in a recent report [6], a better understanding of the DF – Ps pathosystem should help to develop new methods for control. While significant progress has been made on genomic and proteomic aspects of the response by Douglas-fir to Ps infection [7–9], the fungus itself is poorly characterized at the molecular level. Aside from taxonomic and population studies comparing ITS regions [10], no gene sequences from this fungus have yet been reported. A proteomics study of DF response to Ps infection revealed one protein of fungal origin, 1,4-benzoquinone reductase, that was up-regulated during infection [9]. In the white rot fungus Phanerochaete chrysosporium this protein plays a critical role in vanillin metabolism, which is a key intermediate step in lignin biodegradation [11]. This finding suggests that lignin degradation may play an important role in the infection strategy of Ps[9].
A small but growing number of studies have examined genes expressed in conifer pathogens, including Armillaria solidipes, a white-rot root disease pathogen [12], Grosmannia clavigera, a mountain pine beetle-associated pathogen of lodgepole pine [13, 14], and Heterobasidion annosum sensu lato, a complex of closely related white-rot basidiomycetes that causes wood-decay in pine, spruce and fir trees [15–17], reviewed in [18]. In this study, we utilized expressed sequence tag (EST) sequencing to discover some of the fungal genes expressed during the Ps-DF interaction. To gain insight into the processes underlying P. sulphurascens pathogenesis and development, we constructed and analyzed two cDNA libraries: a pure fungal library of mycelia grown on cellophane sheets and exposed to Douglas-fir roots (Ps library), and a mixed library of DF roots infected with Ps (Ps-DF library). Our data represent the first available sequence information on genes expressed by this basidiomycetous fungus, and expand the public database of genes expressed in forest pathogens. The host plant genes expressed from the Ps-DF library have been reported separately [7, 8]. We identified several Ps genes putatively involved in pathogenicity based on homology to genes from other phytopathogens that have been shown to play a role in the infection process. Also, because many virulence factors are secreted, we identified which genes are likely to be part of the secretory pathway in Ps. Including two Ps genes sequenced separately from the cDNA libraries, 74 putative virulence factors and 46 other putatively secreted proteins were identified. We designed primers for 86 of these 120 genes to examine their expression patterns during the first 7 days of Ps infection of DF seedlings. Genes that showed significant up- or down-regulation in the initial screen were further assessed using 3 biological replicates. This work has identified 16 genes potentially important during Ps infection of DF. Future functional studies on the proteins encoded by these 16 genes will more definitively establish their role during the infection process.
Results
Titre of cDNA libraries
The primary Ps-DF library had a titre of 106 cfu ml−1, and after amplification the titre was 3 × 109 cfu ml−1. The titre of the Ps library was low, with only 118,000 clones in total. However, we checked a small sample of the clones, and estimated that 90% were recombinant, with insert sizes ranging from approximately 200 bp to 3 kb. Thirty randomly-selected clones were sequenced and we obtained 26 different high quality reads representing a variety of genes from several different functional categories.
Contig assembly and analysis of P. sulphurascensESTs
After assembly, the 1007 high quality sequences from the Ps library yielded 713 unisequences of which 579 were singletons and 134 were contigs containing 2 to 21 sequences. For the Ps-DF library, BLASTx analysis identified 443 sequences with significant hits to fungal genes. Assembly of these 443 sequences yielded 270 unisequences of which 205 were singletons and 65 were contigs containing 2 to 11 sequences. There were no non-overlapping sequences among the 5′ and 3′ fungal transcripts from the Ps-DF library. To assess the overlap between the Ps and Ps-DF libraries, we assembled all of the high quality fungal sequences from both libraries using Sequencher 4.8, and confirmed manually that 93 of the contigs contained sequences from both libraries. Thus 93 of 270 (34.4%) of the unisequences from the Ps-DF library were also present in the Ps library, and 93 of 713 (13%) of the unisequences from the Ps library were also present in the Ps-DF library. The 890 individual high quality fungal EST sequences from the two libraries were submitted to the dbEST database at NCBI: JK341455, JK341456, and JK316298 through JK317123 (unisequences from single-pass reads), and JO317757 through JO317818 (computationally assembled unisequences). A categorized list of all of the genes from the Ps and Ps-DF libraries are found in Additional files 1 and 2, respectively.
After trimming vector and poor quality sequence ends, the mean length of all the Ps reads was 652 bp, and the mean length of all the fungal Ps-DF Forward reads was 516 bp. Based on comparisons with homologous proteins in the public databases, 31% of the Ps library unisequences included complete open reading frames (ORFs), and 44% of the fungal unisequences from the Ps-DF library contained complete ORFs.
Functional classification of P. sulphurascensgenes expressed during induction by Douglas-fir roots
Functional annotation was possible for 72% of the 1007 ESTs (70% of the 713 unisequences) in the Ps library. In classifying the predicted proteins, we used the MIPS FunCat scheme with a few minor modifications. ‘Cell wall degradation’ , which in FunCat falls under the subcategory ‘Extracellular metabolism’ , was here designated as its own separate category; likewise ‘Ribosomal proteins’ , normally a subcategory of ‘Protein Synthesis’ , were also given their own category. The 1007 ESTs were divided among 18 broad functional categories, including an ‘Unclassified’ and a ‘No significant match’ category. Not including these last two groups, approximately 16.7% of the total annotated proteins were involved in metabolism, 13.1% in protein fate, 11.2% in transport, 8.6% in energy, 5.0% in protein synthesis, 3.6% in cell wall degradation, 3.5% in cell rescue, disease, and virulence, and 14.9% were identified as ribosomal proteins (Figure 1). Of the 25.8% of ESTs that could not be assigned to functional categories, 15.7% were similar to predicted proteins or to proteins for which a function has yet to be determined, and were designated as “unclassified”, and 10.1% did not exhibit significant similarity to any other sequences in the databases.

Functional classification of 730 ESTs from the P. sulphurascens library with significant similarities to genes available in GenBank. The putative genes were classified into functional categories following the Munich Information Center for Protein Sequences Functional Categories (MIPS FunCat) annotation scheme [56].
The most prevalent transcript in the Ps library, with 21 ESTs, encoded a predicted small (98 aa), secreted protein with no matches in the databases. The second and third most prevalent transcripts, with 15 and 14 ESTs, respectively, encoded two nearly identical hydrophobins. Four other hydrophobins (1 to 4 transcripts each) were also identified for a total of 38 hydrophobins. Other prevalent (≥5) transcripts included those with similarities to a putative galactose mutarotase-like protein (11 transcripts), translation elongation factor 1a (9), two unclassified proteins (7 transcripts of one, 6 of another that is putatively secreted), guanine nucleotide binding protein beta subunit (6 transcripts), manganese superoxide dismutase (5 transcripts), peptidyl-prolyl cis-trans isomerase (5 transcripts), an unclassified, putatively secreted protein (5 transcripts), and several ribosomal proteins (up to 7 transcripts each) (Table 1). A number of sequences matching endoglucanases and ubiquitin-conjugating enzyme E2 were identified, but most of these corresponded to families of related enzymes rather than identical copies of the same transcript. Eleven transcripts comprising five groups of metalloproteases were also identified.
Functional classification of P. sulphurascensgenes expressed during infection of Douglas-fir roots
Of the 446 ESTs from the Ps-DF library that shared significant similarity with fungal sequences in the public databases, functional annotation was possible for 85% (83% of the unisequences). We excluded all sequences with no significant similarities to known sequences (E-value > 10−5) in our analyses of this library, because it was uncertain whether they originated from Ps or DF. Of the 380 annotated ESTs, approximately 9.7% were involved in metabolism, 10.5% in protein fate, 6.6% in transport, 11.8% in energy, 5.8% in protein synthesis, 1.8% in cell wall degradation, 4.2% in cell rescue, disease, and virulence, and 37.9% were ribosomal proteins (Figure 2). Unclassified proteins accounted for 15% of the total fungal ESTs from this library.

Functional classification of 378 ESTs from the P. sulphurascens- Douglas-fir library with significant similarities to fungal genes available in GenBank. The putative genes were classified into functional categories following the Munich Information Center for Protein Sequences Functional Categories (MIPS FunCat) annotation scheme [56].
The most prevalent fungal transcript in the Ps-DF library, with 11 copies, was a predicted protein that shared putative homology (E-value of 10−14) with DNA-binding helix-turn-helix proteins, which are transcriptional regulators. Other transcripts in relatively high abundance included a cytochrome-c oxidase subunit (5 transcripts), ubiquitin-conjugating enzyme E2 (5 transcripts), an unclassified protein (5 transcripts), a ubiquinol-cytochrome c reductase complex protein (4 transcripts), an unclassified protein (4 transcripts), and numerous ribosomal proteins (up to 9 transcripts each) (Table 2).
ESTs predicted to be involved in virulence
We identified genes for which there is evidence for a role in the infection process in other pathogens, based on a search of the relevant literature. Seventy-nine Ps genes identified as candidate virulence factors are listed in Additional file 3. We considered all proteins classified in the functional categories “Cell wall degradation” and “Cell rescue, disease and virulence” to be potential virulence factors. Because proteins directly involved in virulence tend to be small and secreted, we identified all open reading frames with a N-terminal signal peptide that make them eligible for secretion from the fungus. Eighty-two (9.2%) of the 890 unisequences from the two libraries were predicted to encode secreted proteins (Additional file 4).
Two additional Ps sequences encoding for putatively secreted proteins identified apart from the current study (H.L.Williams, unpublished data) were added to the list of potential virulence factors: a 1,4 benzoquinone reductase, originally identified as a protein up-regulated during infection of DF [9] (partial gene sequence in Additional file 5), and a cyclophilin, which was identified as a prevalent protein in Ps mycelia grown on malt extract [GenBank: JX848541].
In most cases, the putative function given in Tables 1 and 2 and Additional files 1 through 4 is that of the protein with the lowest E-value (highest similarity) in the BLASTx search. Where the highest similarity was to a hypothetical protein of unknown function, the putative function of the top match having an assigned name (if available) and its E-value are given. For redundant ESTs, the clone with the lowest E-value is reported.
Expression of putative virulence genes
The outcome of our initial screen for infection-responsive expression patterns in 86 candidate virulence genes resulted in the identification of 16 candidate fungal genes that may be important in the virulence of Ps. These 16 genes were significantly up- or down- regulated during infection of DF seedling roots (Table 3 and Figures 3, 4 and 5). The earliest up-regulated genes at 12 hpi were cyclophilin 1 (14.4×, p = 0.0005), lipase (4.3×, p = 0.0430), 1,4 benzoquinone reductase (4.4×, p < 0.0001), a putative Ran-binding protein (6.7×, p < 0.0001) and an unknown gene called 2 J12 (13.8×, p = 0.0056). While the increase in lipase levels was short-lived, cyclophilin 1, the putative Ran-binding protein, and 2 J12 sustained high levels of up-regulation during the initial 36 hpi, and 1,4 benzoquinone reductase maintained fairly steady levels throughout the 7 day infection period. The greatest increase in transcription levels was for the glucoamylase gene, which gradually increased from 4.9× at 2 dpi (p = 0.0842) to 26.8× at 7 dpi (p = 0.0003). Similarly, serine carboxypeptidase (SC) showed a gradual up-regulation beginning around 3 dpi and reaching 9.6× at 7 dpi (p = 0.0021). Rhamnogalacturonan acetylesterase (RGA) was also highly up-regulated (15.6×, p <0.0001) at the final 7 dpi time point. Three hydrophobins showed varying patterns of up-regulation; Hyd0345 expression levels were too low for detection at 12 hpi and 24 hpi and then sustained at approximately 6-fold up-regulation levels for the rest of the time course (p < 0.017), while Hyd0002 and Hyd0173 were briefly up-regulated to 6.9× (p = 0.0002) and 6.7× (p < 0.0001), respectively, during the first 24 hpi. A gene called 1 JO4 with low homology to a MAPK-interacting protein maintained somewhat stable albeit low levels of up-regulation throughout the infection time course (Table 3; Figures 3 and 5).

Gene expression profiles of 12 P. sulphurascens genes up-regulated in infected Douglas-fir seedling roots. By qRT-PCR with ubiquitin conjugating enzyme E2 (Ps uceE2) as an internal control gene, relative quantities of the 12 genes were measured in vegetative mycelia and infected DF seedling roots collected at eight time points from 12 hours to 7 days post-inoculation. This experiment was performed with three biological replicates with triplicate technical replicates. Error bars represent standard deviations. RGA = rhamnogalacturonan acetylesterase, 1,4BQ = 1,4 benzoquinone reductase, SC = serine carboxylesterase, Cyp = cyclophilin, Hyd = hydrophobin, RanBP10 = Ran-binding protein 10.

Gene expression profiles of four P. sulphurascens genes down-regulated in infected Douglas-fir seedling roots. By qRT-PCR with ubiquitin conjugating enzyme E2 (Ps uceE2) as an internal control gene, relative quantities of the four genes were measured in vegetative mycelia and infected DF seedling roots collected at eight time points from 12 hours to 7 days post-inoculation. This experiment was performed with three biological replicates with triplicate technical replicates. Error bars represent standard deviations. Met = metalloprotease, Mn-SOD = manganese superoxide dismutase.

Heat map showing expression levels of 16 P. sulphurascens genes during infection of Douglas-fir seedling roots. Expression levels were determined by qRT-PCR and are relative to those in vegetative mycelia. Full gene names and numerical values for the fold changes and P-values are shown in Table 3.
A metalloprotease called Met0030 was immediately and intensely repressed approximately 25-fold throughout the time course, while 3 highly homologous metalloproteases (Met0115 a-c), were slightly repressed during the second half of the infection trial. A manganese-superoxide dismutase (Mn-SOD) was down-regulated throughout the first 2 dpi, and an unknown gene called Ps0058 was increasingly down-regulated from 24 hpi to 7 dpi (Table 3; Figures 4 and 5).
Discussion
A number of studies have assessed defence-related gene expression in coniferous trees in response to root and butt rot fungi eg. [7, 8, 19]. Less is known about the fungal component of these pathosystems, although recent studies have begun to shed light on molecular strategies utilized by the pathogens [12–19]. The current study represents the first analysis of genes expressed by the causal agent of laminated root rot, Phellinus sulphurascens. The aim of this study was to enhance our understanding of LRR and improve management of the disease in affected forests.
Genes up-regulated during the initial phase of plant infection
In hemibiotrophic and biotrophic pathogens, genes that regulate the development of infection structures and prevent elicitation of host defenses are among the first to be induced, whereas in nectrotrophic fungi, toxins and cell wall degrading enzymes are often among the first to be up-regulated [20]. In this study we were most interested in the genes induced during initial penetration of Ps into the roots of living trees and the early parasitic phase, with the goal of identifying candidate virulence factors that may be manipulated to halt or attenuate the symptoms of laminated root rot.
Our expression analyses indicated significant up-regulation for three hydrophobins in the initial stages of parasitic infection [GenBank: JK317027 & JK317028, JK316976] with one having sustained up-regulation throughout the seven day infection time course [GenBank: JK317085]. Hydrophobins are small, secreted fungal proteins found on the outer surfaces of cell walls of hyphae and conidia. These proteins assemble at hydrophobic/hydrophilic interfaces and thus may be involved in adhesion of fungi to host structures [21]. It has also been suggested that hydrophobins might act as ‘stealth’ factors, protecting an invading fungus from detection and rejection by its host plant [22]. In the rice-blast fungus M. grisea, two hydrophobin genes play essential roles in surface hydrophobicity and infection-related fungal development, and are required for virulence of M. grisea[23, 24]. In H. annosum s.s, a conifer pathogen with a similar life history to Ps, 16 hydrophobin-encoding genes with different regulatory patterns were observed. Several were highly up-regulated during saprotrophic growth on bark, sapwood and/or heartwood of Scots pine, suggesting that this pathogen employs different hydrophobins during growth on diverse wood components [25]. A separate study found that two H. annosum hydrophobin genes are up-regulated in aerial hyphae but are down-regulated during early infection of pine seedling roots [26]. It is clear that some hydrophobins play a crucial role in fungal pathogenesis, while others are needed for other biological processes.
Cyclophilins are peptidyl-prolyl cis–trans isomerases that are highly conserved throughout eukaryotes and have been implicated in a wide variety of cellular processes, including the response to environmental stresses and the control of transcriptional repression [27]. In M. grisea, disruption of cyclophilin highly expressed during plant infection resulted in a mutant strain impaired in appressorium turgor generation, penetration peg formation, and ability to infect rice seedlings [28]. In the chestnut blight fungus Cryphonectria parasitica, disruption of a cyclophilin gene revealed that it is required for virulence [29]. We identified a cyclophilin homolog (cyp2) in our Ps library, and previous protein work in our laboratory identified a paralogous cyclophilin sharing 71% coding sequence identity with the one from the Ps library (H.L.Williams, unpublished data). This cyclophilin (cyp1) [GenBank: JX848541] showed significant up-regulation during the earliest stages of the infection process, indicating that it may contribute to the virulence of Ps.
One of two Ps lipase genes [GenBank: JK316329] was significantly up-regulated at the beginning of the infection process, and may be involved in using DF triglycerides as a carbon source. In Fusarium graminearum, targeted gene disruption demonstrated that a secreted lipase encoded by FLP1 is a major virulence factor of this fungus on wheat and maize [30], and in two obligate biotrophic rust fungi, secreted lipase transcripts were strikingly enriched (≥10-fold) in planta[31].
A gene sharing significant homology with Ran-binding protein 10 (RanBP10) [GenBank: JK316879] from Rhizoctonia solani was immediately up-regulated and sustained at high levels for the first 36 hpi. RanBP10 acts as a guanine nucleotide exchange factor for RAN GTPase, a master regulatory switch that controls shuttling of proteins between nuclear and cytoplasm compartments. Although the function of RanBP10 in fungi is not well studied, up-regulation of this gene in Ps during the initial stages of infection of DF suggests that it may play a key role in mobilizing intracellular signalling pathways required for the fungus to invade host tissues.
Given that secreted effectors tend to be small and poorly conserved between pathogens [31, 32], the characterization of small unknown secreted proteins may be fruitful in discovering novel virulence factors. We identified 20 functionally unclassified proteins with predicted signal peptides, three of which have no known homolog in the sequence databases. An unknown predicted 224 aa protein called 2 J12 [GenBank: JK316741] was highly up-regulated early in the infection of DF seedlings, and a predicted 220 aa protein called 1 JO4 with slight homology to a MAPK-interacting protein [GenBank: JK316546] maintained a low level of up-regulation throughout the infection time course. We aim to further characterize these unknown proteins in order to determine what role they play in the virulence of Ps. In particular, 2 J12 was cumulatively the most highly up-regulated of all the candidate genes over the seven day infection period, and was immediately up-regulated at high levels that were sustained for the first 36 hours of the infection time course. The 2 J12 protein is highly homologous to several unclassified proteins from Fomitiporia mediterranea, a wood decaying fungus belonging to the same order as Phellinus.
Genes induced and expressed during the parasitic growth phase
White-rot fungi are unique among most microorganisms in their capacity to depolymerize and metabolize lignin. Phellinus pini (current name: Porodaedalea pini) and H. annosum are examples of fungi that remove the lignin at a faster rate than cellulose or the hemicelluloses in early decay stages [33]. This may also be the case for P. sulphurascens, given its close relationship and similar life history to both of these species. Our previous work on proteins regulated in DF in response to Ps[9] identified an up-regulated fungal protein that shared homology with a fungal 1,4-benzoquinone reductase, an enzyme involved in degradation of lignin in its host [11]. Expression analysis in our pathosystem confirmed that this gene was significantly up-regulated throughout the 7 day infection time course. We did not identify any lignin-degrading enzymes in the cDNA libraries, even though lignin is an important component of DF cell walls, possibly because the DF seedling roots were too young for substantial lignification to have occurred. In nature, Ps causes severe damage on mature, suberized roots, and some differences are expected between infection events in sterile three-week old seedlings and in larger roots in the forest. However, the fact that our study verified immediate up-regulation of a fungal enzyme involved in lignin degradation in young, scantly lignified seedlings suggests that lignin degradation could be important during the parasitic phase of Ps growth in DF. In H. annosum P strains, which are more aggressive pathogens and wood decayers than S strains, the oxidizing ligninolytic enzyme laccase is produced 5 to 6 times more than in S strains [34].
In addition to 1,4BQ, several other genes that were immediately induced remained up-regulated well after initial penetration of the host (i.e. after 2 dpi), including one hydrophobin (Hyd0345), the putative Ran-binding protein 10, cyclophilin 1, and the unknown secreted proteins 2 J12 and 1 J04. In addition to possible roles in recognition, adhesion or penetration of the host, the proteins encoded by these genes may be important in sustaining infection during the parasitic phase of Ps on DF roots.
Genes induced during the later stage of parasitic growth
Our EST libraries revealed an array of glycoside hydrolase enzymes involved in extracellular plant cell wall degradation (CWDE), including the pectin-degrading enzymes exopolygalacturonase, pectinesterase, and rhamnogalacturonan acetylesterase, the hemicellulose-degrading enzyme endoxylanase, and the cellulose-degrading enzymes β-glucosidase and endoglucanase. We also identified four members of the recently characterized AA9 copper dependent LPMOs (formerly GH61), which support the activity of glycoside hydrolase enzymes through an oxidative degradation of cellulose [35]. However, our initial gene expression screen indicated induction of only one of enzyme involved in cell wall degradation: rhamnogalacturonan acetylesterase (RGA) [GenBank: JO317758], which was highly up-regulated at the end of the seven-day time course. Rhamnogalacturonan is a pectin polymer that constitutes a major portion of conifer cell walls [36], and RGAs are enzymes which in Aspergillus aculeatus have been shown to act in synergy with rhamnogalacturonases to degrade plant cell wall rhamnogalacturonan [37]. CWDE have been demonstrated to be virulence factors in several plant pathogenic fungi reviewed in [38]; however partially degraded polymers released by CWDE can act as elicitors of plant defense [39]. Biotrophic and hemibiotrophic fungi may need to carefully time the release of CWDE, possibly delaying their up-regulation until a sufficient mass of hyphae have penetrated the host tissues using other routes. This may be the case in Ps, given that the one induced enzyme was only up-regulated at 7 dpi. In the young seedlings used in our study, 7 dpi was considered to be part of the later parasitic stage, because at this point some of the DF roots were becoming weak and easily torn. The large increase in RGA expression suggests that degradation of plant cell wall pectin may be an important aspect of host colonization, and it is likely that a number of other CWDEs, including those not identified in this study, are also involved in degrading DF cell walls during the later parasitic and saprotrophic phases.
Much research on parasitism has focused on understanding pathogenicity and virulence factors, while the strategies used by parasites to acquire nutrients and adapt to changing nutritional environments are less well studied. A Ps glucoamylase [GenBank: JK317004] showed increasing up-regulation levels throughout the seven day infection period, and was the most highly up-regulated Ps gene at 7 dpi, indicating that this enzyme may be associated with the virulence of this fungus in DF. A glucoamylase secreted by Sclerotinia sclerotiorum was proposed to be important in providing energy to this phytopathogen via plant starch degradation during infection [40].
Extracellular proteases can play an active role in pathogenesis, and we observed a putatively secreted serine carboxypeptidase (SC) [GenBank: JK316612] was increasingly up-regulated from 3 dpi to the end of the infection time course. SCs have diverse biological functions, including supplying amino acids and energy to an entomopathogenic fungus during starvation and pathogenesis [41]. In the rice blast fungus Magnaporthe grisea, a serine carboxypeptidase was up-regulated during inductive appressorium formation [42]. A recent genome-wide association study of the necrotrophic forest pathogen Heterobasidion annosum sensu stricto (s.s.) yielded an SNP marker associated with virulence that was positioned beside a serine carboxypeptidase gene, suggesting a possible role for this enzyme in virulence [43].
Psgenes down-regulated during infection of DF
One of the earliest plant responses to pathogen attack is a rapid, transient production of a large amount of reactive oxygen species (ROS), termed an ‘oxidative burst’ [44]. This represents a potentially toxic insult that may serve not only to protect the plant from the invading pathogen, but also as a signal to activate further plant defense reactions [45]. We identified several Ps ESTs corresponding to putative proteins that respond to oxidative stress, including a manganese-superoxide dismutase (Mn-SOD) [GenBank: JK317052]. Superoxide dismutases are believed to be responsible for directly inactivating ROS, although gene inactivation of an SOD gene in Botrytis cinerea indicated that this H2O2-generating enzyme is a virulence factor [46], contributing to the accumulation of phytotoxic H2O2 levels. However, our expression analysis of the Ps Mn-SOD gene showed that it was significantly down-regulated immediately upon infection of DF roots, followed by a gradual increase back to baseline (negative control) levels from two days to the end of the seven day infection period. This contrasts with findings in a similar pathosystem, where after 72 hours in contact with pine seedlings, Mn-SOD expression in H. annosum was almost double compared to that in controls [47]. However, when H. parviporum was inoculated on Norway spruce tissue cultures, the SOD expression profile was almost identical to ours [19]. The authors hypothesized that the SOD functions in protecting the fungal cell against superoxide produced as a by-product of oxidative metabolism, and that SOD activity should correlate with the nutritional status of the fungus, which is expected to be high during growth on the medium used for the controls and during growth on dead host cells in the later stages of host colonization. This would also explain the immediate repression and then gradual increase in Mn-SOD transcript levels over our infection time course. Mn-SOD therefore may not be a virulence factor in Ps, but may be important during nutrient acquisition in the later parasitic phase of its growth in DF.
We identified several other proteins involved in stress response and detoxification of xenobiotic substances, including thioredoxins, glutathione transferases, heat shock proteins, and cytochrome P450s, but due to sample limitations they have not yet been assessed for responsiveness during infection of DF using qRT-PCR. Our future work will examine whether antioxidant and detoxification gene expression is modulated when Ps is introduced to DF seedlings.
A number of transcripts comprising five groups of zinc-dependent metalloproteases were identified in the Ps library. Metalloproteases from two diverse plant pathogens are associated with virulence [48, 49], and given their prevalence in the Ps library and putative secretory signals, we considered these enzymes to be prime candidates as virulence factors. It was surprising that a Ps metalloprotease was greatly down-regulated throughout the entire seven day time course [GenBank: JO317766], and a second set of three nearly identical metalloproteases [GenBank: JK317066, JK317067, JO317767] were significantly down-regulated in the latter half of the infection trial. Although their down-regulation during the infection time course indicates these enzymes lack a key role in the pathogenic process, the answer to the question of why they are repressed during pathogenesis could be illuminating to our understanding of the pathogenic process. Metalloproteases are a broad and highly diverse group of enzymes, and it is difficult to hypothesize why they might be down-regulated in Ps during infection of DF. We found no evidence of similar repression of these enzymes in the literature.
The most prevalent transcript identified from the Ps library, a putatively secreted protein called Ps0058 [GenBank: JK317046], was increasingly and substantially down-regulated throughout the infection time course. This transcript shared no homology with any sequences in the NCBI database.
Future work
Future studies aimed at the functional characterization of genes reported here via expression vector and/or targeted deletion experiments will help to elucidate whether the corresponding proteins are important in the virulence of Ps. We aim to identify genes that could prove to be effective targets for control of this pathogen. Additionally, these candidate genes may contribute to our understanding of the defense mechanisms and gene response of DF to Ps. This information may be useful for the development of molecular markers for marker-assisted breeding and screening of DF that are tolerant to Ps.
Conclusions
In this study we report the first set of annotated P. sulphurascens EST sequences, a total of 890 unique sequences from two cDNA libraries. The putative identity of the majority these transcripts revealed many potential candidate genes that may be essential in the Ps/DF pathogenicity process. Analysis of the expression patterns of 86 of these genes during infection using real-time PCR revealed 16 fungal genes that are significantly up- or down-regulated during infection of DF. In conclusion, this work provides an important inventory of P. sulphurascens genes for exploring cellular events related to host-pathogen interactions.
Methods
Plant and fungal isolates and inoculation technique
Seed from coastal DF family 60686 were obtained from the Tree Seed Centre of the British Columbia Ministry of Forests and Range, located in Surrey, BC, Canada and stored in plastic bags at -20°C. To germinate the seeds, they were first soaked in ddH2O for 24 h, stratified at 4°C for 21 days, surface sterilized in 35% aqueous H2O2 (vol/vol) for 15 min with vigorous agitation, and rinsed four times with sterile H2O. Sterilized seeds were placed in petri plates (~20 seeds per plate) containing 10 g/l water agar (Sigma-Aldrich Canada Ltd), the plates were sealed with paraffin film, and the seeds were allowed to germinate in darkness at 24 ± 1C for 14 to 20 days, until most of the roots were at least ~ 4 cm long and the needles were no longer encased in the seed coat.
P. sulphurascens isolate PFC-581 was originally collected from the infected stem of a mature DF tree growing near Cowichan Lake on Vancouver Island, British Columbia (BC) in 1993. The fungus was isolated from the DF stem onto petri plates containing 15% malt extract agar (Sigma-Aldrich Canada Ltd, Oakville, ON, Canada) and then stored as mycelial plugs in cryovials filled with sterile double-distilled water at 3°C, at the Pacific Forestry Centre in Victoria, BC. Isolate PFC-583 was originally collected from a DF tree at Shawnigan Lake BC in 1993, and was cultured and stored in the same manner as PFC-581.
In preparation for creating a purely fungal cDNA library (Ps library), PFC-581 mycelial plugs were transferred to 15 g/l malt extract agar petri plates overlaid with a sterile cellophane sheet (Bio-Rad Laboratories, Mississauga, ON) and allowed to grow in darkness at room temperature for 3 weeks. The cellophane sheets (covered in mycelia) were then transferred to 15 g/l water agar plates and incubated at room temperature for 2 days. A portion of cellophane and the water agar beneath were cut and discarded (approx. 1/3 of each plate), and 12 DF seedlings were placed with their roots resting on the mycelia and their foliage resting in the empty portion of the plate. A second semi-circle of water agar with cellophane and mycelia was placed upside down over the roots of all the seedlings to form a sandwich. Petri plates were sealed with paraffin film, the portion containing the roots was covered with aluminum foil, and the plates were incubated in a growth chamber at 20°C with a photoperiod of 16 h (50 to 60 μmol m−2 s−1). We used cellophane sheets because they allowed sufficient quantities of fungal mycelia to be easily collected without inadvertent agar contamination. Additionally, cellophane sheets are composed of regenerated cellulose, and therefore may mimic some of the characteristics of plant cell walls.
We also created a P. sulphurascens-infected Douglas-fir cDNA library (Ps-DF library), the details of which have been described [8]. The inoculation method was essentially the same as for the Ps library, except the mycelial plugs were laid directly onto petri plates containing 1% water agar with no cellophane. The plates were incubated in darkness at room temperature for 20 days before seedlings were added.
RNA isolation and cDNA library construction
Previous studies in our laboratory with scanning electron microscopy showed that using our infection method, Ps hyphae had penetrated DF seedling epidermal tissues by 2 dpi [50]. Necrosis of DF tissues was visible at 5 dpi, and light microscopy of prepared slides of Ps-infected roots stained with toluidine blue showed extensive mycelial colonization and hyphal penetration in the root cortex at 7 dpi [51]. These results suggested that optimal detection of pathogenicity-related gene expression would be between 2 and 7 dpi.
For the induced, purely fungal library (Ps library), infection sandwiches were dismantled, seedlings were discarded, and mycelia was scraped from both the upper and lower cellophane sheets using an RNase-free spatula. The mycelia was immediately frozen in liquid nitrogen, ground to a fine powder using a mortar and pestle with liquid nitrogen, and total RNA was extracted using the RNeasy Plant mini kit (Qiagen, Valencia, CA). Separate extractions were done for 2, 4 and 6 dpi induced mycelia, and the RNA was checked for quality and quantity using a Nanodrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE) and 1.2% agarose gel. To eliminate most of the ribosomal RNA, mRNA was isolated from total RNA by two successive mRNA extraction cycles with Dynabeads (Invitrogen Canada Inc, Burlington, ON). Again, the concentration and purity of the mRNA was checked using the Nanodrop spectrophotometer, and its integrity was verified on a 1.2% agarose gel.
For the Ps -infected DF library, RNA was extracted from infected DF roots at 3, 5, and 7 dpi using an RNA extraction protocol designed for use with recalcitrant plant (eg. conifer) tissues [8].
Both the purely fungal library and Ps-infected DF library were constructed using the Creator SMART cDNA library construction kit (Clontech Laboratories Inc, Mountain View, CA). For the fungal library, equal amounts of the 2, 4, and 6 dpi mRNA were pooled for a total of 1 μg mRNA. For the infected DF library, the 3, 5, and 7 dpi total RNA were pooled for a total of ~ 100 ng total RNA. These RNA samples were used for cDNA synthesis following the instructions of the manufacturer. Recombinant pDNR-LIB plasmids containing cloned cDNAs were transformed into Escherichia coli (ElectroMAX DH10B electrocompetent cells; Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. To evaluate the quality of the cDNA libraries, 50 individual cDNA clones were randomly picked from each library and screened using M13 primers to determine the percentage of recombinant clones. We also initially sequenced 30 clones from the Ps library and 100 clones from the Ps-DF library, to ensure that the libraries contained a wide variety of gene sequences. The library titres were calculated following Clontech’s instruction manual. The mixed Ps-DF library was amplified following Clontech’s instruction manual.
DNA sequencing, editing, and bioinformatics
We randomly selected 1400 E. coli clones from our induced Ps library and grew overnight cultures in 3 ml LB + 30 μg/ml chloramphenicol media at 37°C at 200 rpm. One μl of each overnight culture was used as PCR template with the M13 primer set. Clones with no insert or very short inserts (< ~100 bp) were discarded, and the remaining 1117 clones were made into glycerol stocks by adding filter-sterilized glycerol to a final concentration of 15% to each overnight culture. A portion of each glycerol stock was sent to the BC Cancer Agency in Vancouver, BC for unidirectional sequencing (M13 Forward primer), and the remainder was stored at −80°C for future potential functional analysis studies. As previously described [8], the Ps-infected DF library was sent to the BC Cancer Agency where 3517 clones were sequenced. The first 1213 sequences were unidirectional (M13 Forward primer) and the rest were bi-directional (M13 Forward and Reverse primers).
cDNA sequences were edited using Sequencher version 4.8 (Gene Codes Corporation, Ann Arbor, MI) by trimming the vector sequence and ambiguous base pairs at the ends of the sequences under the default settings of the program. The few remaining sequences that were less than 100 bp were deleted. For each of the two libraries, the sequence sets were assembled into contigs using the automatic assembly function in Sequencher. For the Ps-DF library, overlapping sequences (from both 5′ and 3′ ends of the same transcript) were evaluated and aligned into full consensus sequence contigs, but were reported as a singleton when no other cDNAs were part of the contig. All the unisequences were compared to the protein database at NCBI using the BLASTx algorithm [52]. Similarities were classified as hits indicating significant homology when the expected E-value was equal to or less than 10−5.
To identify open reading frames, all the Ps library sequences and the fungal matches from the Ps-DF library were analyzed using the ORF Finder [53]. Protein sequences corresponding to the putative open reading frames were searched using the NCBI BLASTp algorithms [52] to corroborate the identity of each ORF by confirming that matches were the same as for the BLASTx search and the predicted start codon aligned with that of homologous proteins. The translated sequences of cDNAs with positively identified start codons were analyzed for signal peptidase sites using SignalP 3.0 [54]. When both algorithms employed by this program (SignalP-NN and SignalP-HMM) predicted the same signal peptide and the SignalP-HMM signal peptide probability was ≥ 0.98, the gene was considered to be a candidate member of the secretome of Ps. We corroborated the hypothesized extracellular destination of these proteins using WoLF PSORT [55] for fungal proteins. When no ORF was detected because the 5′ portion of the gene was missing, the top three BLASTx matches were analyzed using SignalP, and if all three matches predicted a signal peptide (average SignalP-HMM signal peptide probability ≥ 0.98), the Ps sequence in question was also hypothesized to be a candidate member of the secretome of Ps.
Each unisequence was assigned to a functional category following the Munich Information Center for Protein Sequences Functional Categories (MIPS FunCat) annotation scheme [56]. The assignment of ESTs to these categories was aided in large part by COGEME [57]: a tBLASTx search (translated nucleotide database search using translated nucleotide query) of the COGEME database with the Ps sequences produced ranked matches as well as their functional classification group(s), which used a scheme adapted from that of MIPS [56]. The percentage of genes in each functional category was calculated as the number of individual sequences in that group divided by the total number of annotated fungal ESTs from the library. Relatively short ESTs (<350 bp) with no significant matches, poly-A tails, and negligible ORFs (none or <50 aa) were assumed to comprise 3′ untranslated regions, and were deleted from the data set. Although the start codon of longer ORFs with no significant matches could not be verified by comparison with homologues, those containing predicted signal peptidase sites were assumed to begin with the correct start codon, and were considered potential genes of interest.
RNA extraction and cDNA synthesis for qRT-PCR
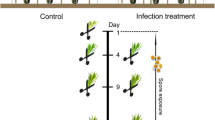
In total, 48 RNA samples were extracted from control mycelia and Ps-infected DF seedling roots over 8 time points: 12, 18, 24, and 36 h, and 2, 3, 5, and 7 days post-infection. Three independent biological replicates were prepared in triplicate for each time point. We used PFC-583 for qRT-PCR experiments. Each individual extraction included 10 to 14 seedlings for ~ 100 mg of plant tissue from infected roots, or approximately 5 petri plates of mycelia for ~ 80 mg fungal tissue. The control mycelia was grown on agar without cellophane, and mycelia were scraped from the plates using a liquid-nitrogen cooled spatula which was gently pulled across the surface of each plate to collect mycelia while avoiding any contamination from the agar. The RNeasy Plant mini kit (Qiagen) supplemented with Plant Isolation Aid (Ambion, Austin, TX) was used to extract total RNA. The extracted RNA samples were precipitated with 50% (v/v) 2.5 M LiCl at -20°C overnight, cleaned with 70% EtOH, resuspended in RNAse-free water, and treated with TURBO DNase (Ambion), and the SUPERase RNase Inhibitor (Ambion) was added to the recovered RNA. Total RNA was quantified using the NanoDrop spectrophotometer.
First-strand cDNA was synthesized from 500 ng of total RNA using the Transcriptor First-Strand cDNA Synthesis Kit (Roche Applied Science, Mannheim, Germany) following the manufacturer’s instructions.
qRT-PCR
To analyze the expression of each candidate virulence gene, primer pairs were designed for each gene using Roche’s LightCycler Probe Design Software 2.0 (version 1.0; Roche). qRT-PCR was conducted with the synthesized cDNA using the LightCycler 2.0 System (Roche) according to the manufacturer’s instructions. Two microliters of 10× diluted cDNA sample was used as the template for each reaction, and 500 nM of each primer was added to the reaction mix. PCR cycling parameters were 95°C for 10 min; followed by 45 cycles of 95°C for 10s, 60°C for 10s, and 72°C for 10s; one cycle of 95°C to generate a melting curve; and finally, 40°C for cooling. Fluorescent PCR products were generated from cDNA using the LightCycler FastStart DNA MasterPLUS SYBR Green I kit (Roche) and relative quantification was performed using the LightCycler 4.1 software (Roche). Water was used as a no template control, and pure DF cDNA was used to ensure products were amplified from Ps rather than DF cDNA.
We considered actin and ubiquitin-conjugating enzyme E2 (uceE2) as candidate internal control genes. The variance of expression of each candidate control over each time-point during the 7-day infection were tested using GeNorm [58] and Normfinder [59]. GeNorm analysis calculated uceE2 to be more stable than actin with a lower stabilization factor (M) of 1.18 compared to 1.47 respectively. GeNorm suggests genes with M values below 1.5 can be considered to have sufficiently stable expression [58]. NormFinder further validates the use of uceE2, with a lower stability value (0.444) than actin (0.494). Numerous qPCR studies have found ubiquitin conjugating enzyme to be a suitable internal control gene in plants [60] as well as in the oomycete, Phytopthora parasitica, during different life cycles including plant pathogenesis [61].
To determine the amplification efficiencies for the genes of interest and the uceE2 gene, a concentrated cDNA sample was serially diluted over a 4 to 5 log range to cover the threshold cycles (Ct) observed in the experimental samples. qRT-PCR was performed for these dilutions and the Ct values were recorded at threshold and baseline parameters standardized for each of the transcripts. The data were plotted against the log of starting template concentration. The slope curve of the Ct cycles is dependent upon amplification efficiency (E) calculated by the equation E = 10[−1/slope] [62]. The expression level (R) of each gene of interest in relation to uceE2 in control and infected samples was calculated from the Ct values and PCR amplification efficiencies using the following equation [62]:
To analyze and compare expression profiles for our two treatments (control versus infected) at different time points, transcript levels for control samples at 12 h were considered to be the basal levels during expression analysis and the corresponding transcript levels were given the value of 1.00. Ct values for all control and infected transcripts were calculated based on this basal value. As for each gene of interest, three separate biological replicates were each amplified in triplicate for the internal reference gene.
We performed an initial screen of qRT-PCR gene expression analysis for 86 putative virulence genes from Ps (see Additional files 3 and 4), testing single PCR reactions for each time point with one or two biological replicates. Based on this screen we focussed on genes that showed significant up- or-down regulation during infection, and for these genes we repeated qRT-PCR experiments for each of three biological replicates in triplicate. The primer sequences for genes that showed significant up- or down-regulation are shown in Additional file 6.
Availabililty of supporting data
The data sets supporting the results of this article are included within the article and its additional files.
Authors’ information
Abul K M Ekramoddoullah worked at the Pacific Forestry Centre with the other authors, but he is now retired.
Abbreviations
- Ps :
-
Phellinus sulphurascens
- DF:
-
Douglas-fir
- EST:
-
Expressed sequence tag
- BLAST:
-
Basic local alignment search tool
- aa:
-
Amino acids
- dpi:
-
Days post inoculation
- hpi:
-
Hours post inoculation
- qRT-PCR:
-
Quantitative reverse transcriptase-polymerase chain reaction.
References
Thies WG, Sturrock RN: Laminated root rot in Western North America. 1995, Portland, OR: USDA Forest Service, Pacific Northwest Research Station General Technical Report, PNW-GTR-349:32 p
Nelson EE, Sturrock RN: Susceptibility of western conifers to laminated root rot (Phellinus weirii) in Oregon and British Columbia field tests. West J Appl For. 1993, 8: 67-70.
Wallis GW, Reynolds G: The initiation and spread of Poria weirii root rot of Douglas-fir. Can J Bot. 1965, 43: 1-9. 10.1139/b65-001.
Buckland DC, Molnar AC, Wallis GW: Yellow laminated root rot of Douglas-fir. Can J Bot. 1954, 32: 69-81. 10.1139/b54-009.
Sturrock RN, Zeglen S, Turner J: Laminated root rot forest health stand establishment decision aid. BC J Ecosyst Manage. 2006, 7: 41-43.
Cook RJ, Edmonds RL, Klopfenstein NB, Littke W, McDonald G, Omdal D, Ripley K, Shaw CG, Sturrock RN, Zambino P: A report from the Washington State Academy of Sciences, In cooperation with the Washington State Department of Natural Resources. Opportunities for addressing laminated root rot caused by Phellinus sulphurascens in Washington’s forests. 2013, Olympia, WA: Washington State Academy of Sciences
Islam MA, Sturrock RN, Williams HL, Ekramoddoullah AKM: Identification, characterization, and expression analyses of class II and IV chitinase genes from Douglas-fir seedlings infected by Phellinus sulphurascens. Phytopathology. 2010, 100: 356-366. 10.1094/PHYTO-100-4-0356.
Islam MA, Sturrock RN, Ekramoddoullah AKM: Gene expression profiling of a compatible interaction between Douglas-fir and the root rot fungal pathogen Phellinus sulphurascens. Phytopathology. 2013, 103: 585-593.
Islam MA, Sturrock RN, Ekramoddoullah AKM: A proteomics approach to identify proteins differentially expressed in Douglas-fir seedlings infected by Phellinus sulphurascens. J Proteomics. 2008, 71: 425-438. 10.1016/j.jprot.2008.06.004.
Lim YW, Sturrock RN, Leal I, Pellow KW, Yamaguchi T, Breuil C: Distinguishing homokaryons and heterokaryons in Phellinus sulphurascens using pairing tests and ITS polymorphisms. Antonie Van Leeuwenhoek. 2008, 93: 99-110. 10.1007/s10482-007-9184-0.
Shimizu M, Yuda N, Nakamura T, Tanaka H, Wariishi H: Metabolic regulation at the tricarboxylic acid and glyoxylate cycles of the lignin-degrading basidiomycete Phanerochaete chrysosporium against exogenous addition of vanillin. Proteomics. 2005, 5: 3919-3931. 10.1002/pmic.200401251.
Ross-Davis AL, Steward JE, Hanna JW, Kim M-S, Knaus BJ, Cronn R, Rai H, Richardson BA, McDonald GI, Klopfenstein NB: Transcriptome of an Armillaria root disease pathogen reveals candidate genes involved in host substrate utilization at the host-pathogen interface. For Path. 2013, 43: 468-477. 10.1111/efp.12056.
DiGuistini S, Ralph SG, Lim YW, Holt R, Jones S, Bohlmann J, Breuil C: Generation and annotation of lodgepole pine and oleoresin-induced expressed sequences from the blue-stain fungus Ophiostoma clavigerum, a Mountain Pine Beetle-associated pathogen. FEMS Microbiol Lett. 2007, 267: 151-158. 10.1111/j.1574-6968.2006.00565.x.
Hesse-Orce U, DiGuistini S, Keeling CI, Wang Y, Li M, Henderson H, Docking TR, Liao NY, Robertson G, Holt RA, Jones SJM, Bohlmann J, Breuil C: Gene discovery for the bark beetle-vectored fungal tree pathogen Grosmannia clavigera. BMC Genomics. 2010, 11: 536-10.1186/1471-2164-11-536.
Karlsson M, Olson Å, Stenlid J: Expressed sequences from the basidiomycetous tree pathogen Heterobasidion annosum during early infection of Scots pine. Fungal Genet Biol. 2003, 39: 51-59. 10.1016/S1087-1845(02)00586-8.
Olson Å, Aerts A, Asiegbu F, Belbahri L, Bouzid O, Broberg A, Canbäck B, Coutinho PM, Cullen D, Dalman K, Deflorio G, van Diepen LTA, Dunand C, Duplessis S, Durling M, Gonthier P, Grimwood J, Fossdal CG, Hansson D, Henrissat B, Hietala A, Himmelstrand K, Hoffmeister D, Högberg N, James TY, Karlsson M, Kohler A, Kües U, Lee Y-H, Lin YC, et al: Insight into trade-off between wood decay and parasitism from the genome of a fungal forest pathogen. New Phytol. 2012, 194: 1001-1013. 10.1111/j.1469-8137.2012.04128.x.
Yakovlev IA, Hietala AM, Steffenrem H, Solheim H, Fossdal CG: Identification and analysis of differentially expressed Heterobasidion parviporum genes during natural colonization of Norway spruce stems. Fungal Genet Biol. 2008, 45: 498-513. 10.1016/j.fgb.2007.10.011.
Asiegbu FO, Adomas A, Stenlid J: Conifer root and butt rot caused by Heterobasidion annosum (Fr.) Bref. s.l. Mol Plant Pathol. 2005, 6: 395-409. 10.1111/j.1364-3703.2005.00295.x.
Karlsson M, Hietala AM, Kvaalen H, Solheim H, Olson Å, Stenlid J, Fossdal CG: Quantification of host and pathogen DNA and RNA transcripts in the interaction of Norway spruce with Heterobasidion parviporum. Physiol Mol Plant Pathol. 2007, 70: 99-109. 10.1016/j.pmpp.2007.07.006.
Tudzynski P, Sharon A: Fungal pathogenicity genes. Appl Mycol Biotechnol. 2003, 3: 187-212.
Wessels JGH: Fungal hydrophobins: proteins that function at an interface. Trends Plant Sci. 1996, 1: 9-15.
Whiteford JR, Spanu PD: Hydrophobins and the interactions between fungi and plants. Mol Plant Pathol. 2002, 3: 391-400. 10.1046/j.1364-3703.2002.00129.x.
Talbot NJ, Ebbole DJ, Hamer JE: Identification and characterization of MPG1, a gene involved in pathogenicity from the rice blast fungus Magnaporthe grisea. Plant Cell. 1993, 5: 1575-1590. 10.1105/tpc.5.11.1575.
Kim S, Ahn I-P, Rho H-S, Lee Y-H: MHP1, a Magnaporthe grisea hydrophobin gene, is required for fungal development and plant colonization. Mol Microbiol. 2005, 57: 1224-1237. 10.1111/j.1365-2958.2005.04750.x.
Mgbeahuruike AC, Kovalchuk A, Chen H, Ubhayasekera W, Asiegbu FO: Evolutionary analysis of hydrophobin gene family in two wood-degrading basidiomycetes, Phlebia brevispora and Heterobasidion annosum s.l. BMC Evol Biol. 2013, 13: 240-10.1186/1471-2148-13-240.
Karlsson M, Stenlid J, Olson A: Two hydrophobin genes from the conifer pathogen Heterobasidion annosum are expressed in aerial hyphae. Mycologia. 2007, 99: 227-231. 10.3852/mycologia.99.2.227.
Andreeva L, Heads R, Green CJ: Cyclophilins and their possible role in the stress response. Int J Exp Pathol. 1999, 80: 305-315.
Viaud MC, Balhadère PV, Talbot NJ: A Magnaporthe grisea cyclophilin acts as a virulence determinant during plant infection. Plant Cell. 2002, 14: 917-930. 10.1105/tpc.010389.
Chen MM, Jiang M, Shang J, Lan X, Yang F, Huang J, Nuss D, Chen B: CYP1, a hypovirus-regulated cyclophilin, is required for virulence in the chestnut blight fungus. Mol Plant Pathol. 2011, 12: 239-246. 10.1111/j.1364-3703.2010.00665.x.
Voigt CA, Schäfer W, Salomon S: A secreted lipase of Fusarium graminearum is a virulence factor required for infection of cereals. Plant J. 2005, 42: 364-375. 10.1111/j.1365-313X.2005.02377.x.
Duplessis S, Cuomo CA, Lin YC, Aerts A, Tisserant E, Veneault-Fourrey C, David L, Joly DL, Stéphane Hacquard S, Amselem J, Cantarel BL, Chiu R, Coutinho PM, Feau N, Field M, Frey P, Gelhaye E, Goldberg J, Grabherr MG, Kodira CD, Kohler A, Kües U, Lindquist EA, Lucas SM, Mago R, Mauceli E, Morin E, Murat C, Pangilinan JL, Park R, et al: Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc Natl Acad Sci U S A. 2011, 108: 9166-9171. 10.1073/pnas.1019315108.
Tyler BM, Tripathy S, Zhang X, Dehal P, Jiang RHY, Aerts A, Arredondo FD, Baxter L, Bensasson D, Beynon JL, Chapman J, Damasceno CMB, Dorrance AE, Dou D, Dickerman AW, Dubchak IL, Garbelotto M, Gijzen M, Gordon SG, Govers F, Grunwald NJ, Huang W, Ivors KL, Jones RW, Kamoun S, Krampis K, Lamour KH, Lee M-K, McDonald WH, Medina M, et al: Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science. 2006, 313: 1261-1266. 10.1126/science.1128796.
Zabel RA, Morrell JJ: Wood microbiology: decay and its prevention. 1992, San Diego: Academic Press
Daniel GF, Asiegbu F, Johansson M: The saprotrophic wood-degrading abilities of Heterobasidium annosum intersterility groups P and S. Mycol Res. 1998, 1028: 991-997.
Hemsworth GR, Henrissat B, Davies GJ, Walton PH: Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat Chem Biol. 2014, 10: 122-126.
Sarkar P, Bosneaga E, Auer M: Plant cell walls throughout evolution: towards a molecular understanding of their design principles. J Exp Bot. 2009, 60: 3615-3635. 10.1093/jxb/erp245.
Kauppinen S, Christgau S, Kofod LV, Halkier T, Dörreich K, Dalbøge H: Molecular cloning and characterization of a rhamnogalacturonan acetylesterase from Aspergillus aculeatus. J Biol Chem. 1995, 270: 27172-27178. 10.1074/jbc.270.45.27172.
ten Have A, Tenberge KB, Benen JAE, Tudzynski P, Visser J, van Jan AL, Kan JAL: The contribution of cell wall degrading enzymes to pathogenesis of fungal plant pathogens. Agricultural Applications. 2002, Berlin Heidelberg: Springer, 341-358.
Cervone F, Hahn MG, De Lorenzo G, Darvill A: Albersheim P. Host-pathogen interactions XXXIII. a plant protein converts a fungal pathogenesis factor into an elicitor of plant defense responses. Plant Physiol. 1989, 90: 542-548. 10.1104/pp.90.2.542.
Khare KB, Bompeix G: Activités proteolytiques des Sclerotinia sclerotiorum et S minor: rôle possible lors de la pathogénèse. Rev Mycol. 1976, 40: 65-84.
St. Leger RJ, Bidochka MJ, Roberts DW: Characterization of a novel carboxypeptidase produced by the entomopathogenic fungus Metarhizium anisopliae. Arch Biochem Biophys. 1994, 314: 392-398. 10.1006/abbi.1994.1458.
Kim ST, Yu S, Kim SG, Kim HJ, Kang SY, Hwang DH, Jang YS, Kang KY: Proteome analysis of rice blast fungus (Magnaporthe grisea) proteome during appressorium formation. Proteomics. 2004, 4: 3579-3587. 10.1002/pmic.200400969.
Dalman K, Himmelstrand K, Olson A, Lind M, Brandström-Durling M, Stenlid J: A genome-wide association study identifies genomic regions for virulence in the non-model organism Heterobasidion annosum s.s. PLoS One. 2013, 8 (1): e53525-10.1371/journal.pone.0053525.
Sutherland MW: The generation of oxygen radicals during host plant responses to infection. Physiol Mol Plant Pathol. 1991, 39: 79-93. 10.1016/0885-5765(91)90020-I.
Tenhaken R, Levin A, Brisson LF, Dixon RA, Lamb C: Function of the oxidative burst in hypersensitive disease resistance. Proc Natl Acad Sci U S A. 1995, 92: 4158-4163. 10.1073/pnas.92.10.4158.
Rolke Y, Liu S, Quidde T, Williamson B, Schouten A, Weltring KM, Siewers V, Tenberge KB, Tudzynski B, Tudzynski P: Functional analysis of H2O2-generating systems in Botrytis cinerea: the major Cu-Zn-superoxide dismutase (BCSOD1) contributes to virulence on French bean, whereas a glucose oxidase (BCGOD1) is dispensable. Mol Plant Pathol. 2004, 5: 17-27. 10.1111/j.1364-3703.2004.00201.x.
Karlsson M, Stenlid J, Olsen Å: Identification of a superoxide dismutase gene from the conifer pathogen Heterobasidion annosum. Physiol Mol Plant P. 2005, 66: 99-107. 10.1016/j.pmpp.2005.05.004.
Dow JM, Davies HA, Daniels MJ: A metalloprotease from Xanthomonas campestris that specifically degrades proline/hydroxyproline-rich glycoproteins of the plant extracellular matrix. MPMI. 1998, 11: 1085-1093. 10.1094/MPMI.1998.11.11.1085.
Orbach MJ, Farrall L, Sweigard JA, Chumley FG, Valent B: A telomeric avirulence gene determines efficacy for the rice blast resistance gene Pi-ta. Plant Cell. 2000, 12: 2019-2032. 10.1105/tpc.12.11.2019.
Islam MA, Sturrock RN, Holmes TA, Ekramoddoullah AKM: Ultrastructural studies of Phellinus sulphurascens infection of Douglas-fir roots and immunolocalization of host pathogenesis-related proteins. Mycol Res. 2009, 113: 700-712. 10.1016/j.mycres.2009.02.006.
Sturrock RN, Islam MA, Ekramoddoullah AKM: Host-pathogen interactions in Douglas-fir seedlings infected by Phellinus sulphurascens. Phytopathology. 2007, 97: 1406-1414. 10.1094/PHYTO-97-11-1406.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Open reading frame finder. http://www.ncbi.nlm.nih.gov/projects/gorf/,
SignalP 3.0 Server. http://www.cbs.dtu.dk/services/SignalP/,
WoLF PSORT. http://wolfpsort.org/,
Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Güldener U, Mannhaupt G, Münsterkötter M, Mewes HW: The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 2004, 32: 5539-5545. 10.1093/nar/gkh894.
Consortium for the Functional Genomics of Microbial Eukaryotes (COGEME). http://cogeme.ex.ac.uk/index.html,
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F: Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3: 1-11.
Anderson CL, Jensen JL, Orntoft TF: Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64: 5245-5250. 10.1158/0008-5472.CAN-04-0496.
Khanlou KM, Van Bockstaele E: A critique of widely used normalization software tools and an alternative method to identify reliable reference genes in red clover (Trifolium pratense L.). Planta. 2012, 236: 1381-1393. 10.1007/s00425-012-1682-2.
Yan HZ, Liou RF: Selection of internal control genes for real-time quantitative RT-PCR assays in the oomycete plant pathogen Phytophthora parasitica. Fungal Genet Biol. 2006, 43: 430-438. 10.1016/j.fgb.2006.01.010.
Pfaffl MW: A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29: 2002-2007.
Acknowledgements
Funding for this study was provided by Natural Resources Canada (NRCan), Canadian Forest Service (CFS), Genomics Research & Development Initiative (GRDI) funds to R. Sturrock and by NRCAN, CFS funds. The funding sources were not involved in the study design, collection, analysis, or interpretation of data, nor in the writing of the manuscript or decision to submit the manuscript for publication. We thank B. Foord and G. Ross for their help preparing samples and generating qRT-PCR data, K. Pellow for his help germinating seedlings and subculturing P. sulphurascens, K. Quazi, Associate Director, Quintiles Canada Inc., Quebec, Canada for his assistance with statistical analysis of gene expression data, and the British Columbia Ministry of Forests and Range for providing Douglas-fir seeds.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HLW created the Ps cDNA library, annotated the Ps and Ps-DF library sequences, prepared samples for qRT-PCR and analyzed the data, and drafted the manuscript. CH performed qRT-PCR, statistically analyzed the data, and assisted with completion of the manuscript. RNS and AKME conceived of the study and provided a critical review of the manuscript. MAI created the Ps-DF cDNA library, assisted with statistical analysis, and provided comments on the manuscript. IL helped create the Ps cDNA library and assisted with writing and critically reviewing the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2014_6292_MOESM1_ESM.xlsx
Additional file 1: Genes from a cDNA library constructed using P. sulphurascens induced by exposure to Douglas-fir seedling roots. 713 unique sequences from the Ps library, grouped into functional categories. (XLSX 93 KB)
12864_2014_6292_MOESM2_ESM.xlsx
Additional file 2: Genes from a cDNA library constructed using P. sulphurascens -infected Douglas-fir seedling roots. 270 unique sequences from the Ps-DF library, grouped into functional categories. (XLSX 43 KB)
12864_2014_6292_MOESM3_ESM.xlsx
Additional file 3: P. sulphurascens genes potentially involved in pathogenicity. Seventy-nine Ps genes considered to be candidate virulence factors. (XLSX 35 KB)
12864_2014_6292_MOESM4_ESM.xlsx
Additional file 4: P. sulphurascens genes encoding predicted secreted proteins. Eighty-two Ps genes with putative secretion signals. (XLSX 29 KB)
12864_2014_6292_MOESM5_ESM.docx
Additional file 5: Partial gene sequence of P. sulphurascens putative 1,4 benzoquinone reductase. Putative 1,4 benzoquinone reductase gene sequence obtained separately from the cDNA libraries. (DOCX 11 KB)
12864_2014_6292_MOESM6_ESM.xlsx
Additional file 6: qRT-PCR primers for P. sulphurascens genes regulated during infection of DF seedlings and for two internal reference genes. Primers corresponding to 16 P. sulphurascens genes up- or down-regulated during infection of DF and two internal reference genes. (XLSX 14 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Williams, H.L., Sturrock, R.N., Islam, M.A. et al. Gene expression profiling of candidate virulence factors in the laminated root rot pathogen Phellinus sulphurascens. BMC Genomics 15, 603 (2014). https://doi.org/10.1186/1471-2164-15-603
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-603