
Abstract
Background
TARDBP mutations have been reported in patients with amyotrophic lateral sclerosis (ALS) in different populations except Chinese. The present aim is to investigate the association between TARDBP mutations and Chinese patients with ALS.
Methods
71 SALS patients and 5 FALS families with non-SOD1 mutations were screened for TARDBP mutations via direct sequencing.
Results
A novel heterozygous variation, Ser292Asn (875G>A), was identified in the proband and 4 asymptomatic relatives including the children of the dead patient from a FALS family. Thus the dead patient, the proband's brother, was speculated to carry Ser292Asn though his sample was unavailable to the detection. This variation was not found in 200 unrelated control subjects. A homology search of the TDP-43 protein in different species demonstrated that it was highly conserved. Also, it was predicted to be deleterious to protein function with SIFT-calculated probabilities of 0.00. Therefore, Ser292Asn is predicted to be a pathogenic mutation. In addition, we have found two silent mutations (Gly40Gly and Ala366Ala) and one novel polymorphism (239-18t>c).
Conclusions
The present data have extended the spectrum of TARDBP mutations and polymorphisms, and supported the pathological role of TDP-43 in Chinese ALS patients.
Similar content being viewed by others


Background
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease and is characterized by progressive loss of upper and lower motor neurons from the spinal cord, brain stem and cerebral cortex, leading to paralysis and eventually death which is due to respiratory failure within 3-5 years after symptom onset [1]. Approximately 5-10% of ALS cases are familial (FALS) whereas the remaining patients are sporadic (SALS) [2]. About 15-20% of patients with autosomal dominant FALS have mutations in the copper-zinc superoxide dismutase 1 gene (SOD1), while mutations in other genes including alsin (ALS2), senataxin (SETX, ALS4), dynactin (DCTIN1), angiogenin (ANG), synaptobrevin-associated membrane protein B (VAPB, ALS8) and Fused in Sarcoma (FUS, ALS6) are described as rare causes of FALS [2–5]. Recently, a novel senataxin mutation has been reported in a SALS patient [6]. Ubiquitin-positive tau-negative neuronal cytoplasmic inclusion is the common pathological feature in frontotemporal lobar degeneration (FTLD) and ALS. TAR DNA-binding protein 43 (TDP-43), which is a 414-amino-amino-acid nuclear protein encoded by TARDBP on chromosome 1p36.22, was recognized as a major constituent of these neuronal cytoplasmic inclusions [7, 8]. To date, a total of 30 TARDBP mutations have been reported in ALS patients [9–20]. These mutations affect the C-terminal region of TDP-43 and may influence protein-protein interaction, exon skipping and splicing inhibitory activity, thus, may influence the proper function or transport of TDP-43. Here, we first reported the screen for TARDBP mutations in Chinese patients with ALS and we identified one novel missense mutation, two silent mutations and one novel polymorphism.
Methods
Subjects
Seventy-one unrelated SALS patients and probands from 5 FALS families with non-SOD1 mutations from the Han ethnic group were enrolled in our study between 12 December 2007 and 3 March 2009 from department of Neurology at Huashan Hospital. Of them, 56 were males and 20 were females, and the average age of symptom onset was 52 years (range, 32-76 years). All patients had been examined by at least two neurologists. Neurological examinations including electromyography (EMG) and magnetic resonance imaging (MRI) of the cervical cord were performed. Medical history and demographic information were collected by a specially-assigned person, and records were reviewed by two senior neurologists. All patients were diagnosed as definite ALS according to the Airlie House criteria [21]. 16 patients presented with bulbar-onset disease and 60 patients presented with spinal-onset disease. Two hundred unrelated aged individuals (≥60 years) consisted of 100 men and 100 women with no known history of ALS were selected as a control group. All of them are Han people from Southern China. This study was approved by the local ethics committee, and informed consent was obtained from the participants or their legal surrogates prior to inclusion in the study. Genomic DNA was extracted from peripheral EDTA blood with a TIANamp Blood DNA kit (TIANGEN Biotech, Beijing).
Mutation scanning
The coding region of TARDBP, exons 2-6, including the intron-exon boundaries, were analyzed using primer combinations designed based on the intronic sequences of TARDBP. PCR amplification was performed using a GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA, USA) with standard conditions. The sequence of the primers and the annealing temperatures are shown in the Table 1. Amplified products were purified and subjected to direct sequencing, and the procedure is as previously reported [22]. Obtained sequences were compared with the genomic DNA sequence of TARDBP (NCBI Sequence Viewer NT_021937.18), and nucleotide changes were numbered corresponding to their position in TARDBP mRNA (NCBI Sequence Viewer NM_007375.3).
Results
Mutations and polymorphisms of TARDBP gene identified in the present study
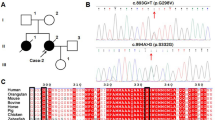
After screening mutations of TARDBP in 5 FALS patients and 71 SALS patients, a heterozygous variation Ser292Asn (875G>A) which has not been reported previously was identified in a FALS patient. The chromatogram is shown in figure 1A. This variation was not found in 200 unrelated control subjects, reducing the likelihood that it represents a polymorphism. Moreover, a homology search of the TDP-43 protein in different species demonstrated that this variation was highly conserved (figure 1B). At the same time, the program SIFT (Sorting Intolerant From Tolerant) [23] has been applied to predict whether this sequence change could affect protein function, and it was predicted to be deleterious to protein function with SIFT-calculated probabilities of 0.00, thus Ser292Asn is a novel mutation and most probably a pathogenic one.

Chromatogram of Ser292Asn (A) and conservation of Ser292Asn in different species (B).
In addition, two silent mutations, Gly40Gly (120G>A) and Ala366Ala (1098C>G) were detected. The Gly40Gly which has not been reported previously was found in one SALS patient and none of 200 unrelated control individuals, indicating that it is a rare silent mutation. The Ala366Ala which has been previously reported in Japanese SALS patients [20] was found in one SALS patient and one control individual, thus it was thought to be a benign polymorphism. We also have detected a novel polymorphism 239-18t>c in one SALS patient and one control individual. The chromatograms of them are shown in additional file 1.
Clinical features of the FALS patient carrying Ser292Asn mutation
The Ser292Asn mutation was identified in a 67-year-old male FALS patient (II2). He had some difficulty in speech at the age of 64 and gradually presented dysarthria, dysphagia and atrophy of lingual muscle. One year later, he began to have some difficulty in using chopsticks and lifting his right arm, and gradually developed weakness of both upper limbs and atrophy of muscles. However, the fasciculation was slight. After another 4 months, he began to feel weakness in both lower extremities and started having difficulties in walking up and down stairs. Neurological examination showed muscle weakness involving all the extremities with motor power graded as follows: upper limbs, 3/4; lower limbs, 4/5. However, the sensation was intact. Atrophy of lingual muscle and distal muscles of upper limbs were evident. There was positive Babinski sign, and the deep-tendon reflexes were increased in both lower limbs whereas decreased in both upper limbs. EMG testing showed reduced amplitudes of compound muscle-action potentials in right median nerve, positive sharp waves, fibrillations, and normal motor and sensory NCV. He was treated with coenzyme Q10 60 mg daily and followed with follow-up examinations every three months. At the last follow-up 2 months ago, neurological examination showed progression of disease. The powers of upper limbs were both 2/3 grades and powers of lower limbs were both 3/4 grades. Dysarthria and dysphagia were more and more evident, whereas the patient refused Bi-level positive airway pressure (BiPAP) and percutaneous endoscopic gastrostomy (PEG).
Pedigree analysis of the ALS family with Ser292Asn mutation
The pedigree of this family is shown in figure 2. The proband's younger brother (II5) developed right lower limb weakness at the age of 58, and gradually extended to muscles of both lower limbs and upper limbs. Two years later, he developed bulbar symptoms and died of respiratory failure at the age of 61.

The pedigree with Ser292Asn. "*" indicates the asymptomatic relatives carrying Ser292Asn.
Direct sequencing was performed to screen for Ser292Asn in the proband's mother (I2), three sisters (II7, II9 and II13), one brother (II11), and their children (III3, III5, III6, III7, III8, III11). Four of them (II7, III6, III7, III8) were detected to have Ser292Asn. They haven't any clinical symptoms presently. II7 is 60 years old while III6, III7 and III8 are less than 40 years old. Because the 85-year-old mother (I2) doesn't have Ser292Asn, we speculated that Ser292Asn might be come from the dead father who was asymptomatic. III6 and III7 are the children of the dead patient (II5), thus we speculate that II5 carried the Ser292Asn mutation, although his sample is unavailable for confirmation.
Discussion
The TDP-43, which is a 414-amino-amino-acid nuclear protein encoded by TARDBP on chromosome 1p36.22, has been identified as the major disease protein in ALS. It is evolutionary conserved and its structure consists of a glycine-rich domain and two RNA recognition motifs [24]. It is known to bind DNA and RNA, such as human immunodeficiency virus type 1 TAR DNA sequence motifs [25], and to be involved in the regulation of messenger RNA splicing and exon skipping [26]. The importance of the glycine-rich C-terminal domain of TDP-43 in mediating its exon skipping and splicing inhibitory ability has been demonstrated and has been observed to correlate with its ability to interact with other members of the heterogeneous nuclear ribonuclear A and B protein families with well-known splicing inhibitory properties [27]. However, the exact function remains unclear.
Recently different pathogenic TARDBP mutations have been described in different cohorts of patients with FALS and SALS, supporting a direct role of TDP-43 in neurodegeneration [9–20]. However, two additional studies by Gijselinck I et al and Guerreiro RJ et al failed to find mutations in Belgian and North American patients with ALS [28, 29]. Among the 30 previously reported mutations [9–20], 29 are missense mutations, and one is frameshift mutation (Tyr374Term) which creates a premature stop codon, consequently leading to the expression of a truncated protein [17]. None of them is homozygous. Met337Val, Gly348Cys and Ala382Thr are the most common mutations. Additionally, except for the Asp169Gly mutation, all other TARDBP mutations are located in exon 6 encoding for the C-terminus of TDP-43. Considering all the previous studies, the frequency of TARDBP mutations is 3.6% in FALS and 1.0% in SALS [16]. Most ALS patients carrying TARDBP mutations have an Italian or French origin, suggesting a higher frequency of TARDBP mutations in Southern Europe (average 3.4%; 8% in France and 2.7% in Italy) than in other Caucasian populations (average 0.7%) [16].
In the present study, after screening TARDBP mutations in 71 unrelated SALS patients and probands from 5 FALS families in Chinese population, we have identified one missense mutation in a FALS family and 2 silent mutations in SALS patients. This frequency is lower than those of previous studies, which may be due to the cohort of ALS patients analyzed here is small compared to previous studies. Also, different races may refer to different genetic backgrounds. The Ser292Asn mutation was located in the highly conserved region, exon6, encoding for the C-terminus of TDP-43. In the FALS family with Ser292Asn, we speculated that I1 and II5 might both carry Ser292Asn though their samples are unavailable to the detection. The proband presented clinical symptoms at the age of 64. However, the other four family members (II7, III6, III7, III8) carried the same mutation as the proband did were asymptomatic presently. III6, III7 and III8 are in their thirties and obviously haven't reached the onset age of ALS. II7 is 60 years old though she is clinically asymptomatic. We suggest detecting EMG examination to her but she refused.
Conclusion
In conclusion, our data have extended the spectrum of TARDBP mutations and polymorphisms, and supported the pathological role of TDP-43 in ALS. Further studies are needed to shed light on the pathophysiological link between Ser292Asn and ALS. Competing interestsThe authors declare that they have no competing interests.
References
Cleveland DW, Rothstein JD: From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001, 2: 806-819. 10.1038/35097565.
Pasinelli P, Brown RH: Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006, 7: 710-723. 10.1038/nrn1971.
Gros-Louis F, Gaspar C, Rouleau GA: Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochimica et Biophysica Acta. 2006, 1762: 956-972.
Valdmanis PN, Rouleau GA: Genetics of familial amyotrophic lateral sclerosis. Neurology. 2008, 70: 144-152. 10.1212/01.wnl.0000296811.19811.db.
Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE: Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009, 323: 1208-1211. 10.1126/science.1165942.
Zhao ZH, Chen WZ, Wu ZY, Wang N, Zhao GX, Chen WJ, Murong SX: A novel mutation in the senataxin gene identified in a Chinese patient with sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Sc. 2009, 10: 118-122. 10.1080/17482960802572673.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM: Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006, 314: 130-133. 10.1126/science.1134108.
Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM, Trojanowski JQ: Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007, 61: 427-434. 10.1002/ana.21147.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE: TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008, 319: 1668-1672. 10.1126/science.1154584.
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Velde Vande C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA: TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008, 40: 572-574. 10.1038/ng.132.
Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ, Lee VM, Schellenberg GD, Yu CE: TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008, 7: 409-416. 10.1016/S1474-4422(08)70071-1.
Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ: TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008, 63: 535-538. 10.1002/ana.21344.
Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, Eguchi H, Tsujino A, Ikeuchi T, Kakita A, Okamoto K, Nishizawa M, Takahashi H, Onodera O: TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008, 63: 538-542. 10.1002/ana.21392.
Kühnlein P, Sperfeld AD, Vanmassenhove B, Van Deerlin V, Lee VM, Trojanowski JQ, Kretzschmar HA, Ludolph AC, Neumann M: Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch Neurol. 2008, 65: 1185-1189. 10.1001/archneur.65.9.1185.
Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH, Geschwind DH, Knopman DS, Mitsumoto H, Petersen RC, Cashman NR, Hutton M, Shaw CE, Boylan KB, Boeve B, Graff-Radford NR, Wszolek ZK, Caselli RJ, Dickson DW, Mackenzie IR, Petrucelli L, Rademakers R: Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genetics. 2008, 4: e1000193-10.1371/journal.pgen.1000193.
Corrado L, Ratti A, Gellera C, Buratti E, Castellotti B, Carlomagno Y, Ticozzi N, Mazzini L, Testa L, Taroni F, Baralle FE, Silani V, D'Alfonso S: High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat. 2009, 30: 688-694. 10.1002/humu.20950.
Daoud H, Valdmanis PN, Kabashi E, Dion P, Dupré N, Camu W, Meininger V, Rouleau GA: Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J Med Genet. 2009, 46: 112-114. 10.1136/jmg.2008.062463.
Lemmens R, Race V, Hersmus N, Matthijs G, Bosch Van Den L, Van Damme P, Dubois B, Boonen S, Goris A, Robberecht W: TDP-43 M311V mutation in familial amyotrophic lateral sclerosis. J Neurot Neurosurq Psychiatry. 2009, 80: 354-355. 10.1136/jnnp.2008.157677.
Del Bo R, Ghezzi S, Corti S, Pandolfo M, Ranieri M, Santoro D, Ghione I, Prelle A, Orsetti V, Mancuso M, Sorarù G, Briani C, Angelini C, Siciliano G, Bresolin N, Comi GP: TARDBP (TDP-43) sequence analysis in patients with familial and sporadic ALS: identification of two novel mutations. Eur J Neurol. 2009, 16: 727-732. 10.1111/j.1468-1331.2009.02574.x.
Kamada M, Maruyama H, Tanaka E, Morino H, Wate R, Ito H, Kusaka H, Kawano Y, Miki T, Nodera H, Izumi Y, Kaji R, Kawakami H: Screening for TARDBP mutations in Japanese familial amyotrophic lateral sclerosis. J Neurol Sci. 2009, 284 (1-2): 69-71. 10.1016/j.jns.2009.04.017.
Brooks BR, Miller RG, Swash M, Munsat TL: World Federation of Neurology Research Group on Motor Neuron Diseases. EI Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000, 1: 593-599.
Wu ZY, Zhao GX, Chen WJ, Wang N, Wan B, Lin MT, Murong SX, Yu L: Mutation analysis of 218 Chinese patients with Wilson disease revealed no correlation between the canine copper toxicosis gene MURR1 and Wilson disease. J Mol Med. 2006, 84: 438-442. 10.1007/s00109-005-0036-y.
Ng PC, Henikoff S: SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31: 3812-3814. 10.1093/nar/gkg509.
Wang HY, Wang IF, Bose J, Shen CK: Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004, 83: 130-139. 10.1016/S0888-7543(03)00214-3.
Ou SH, Wu F, Harrich D, Garcıá-Martıńez LF, Gaynor RB: Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995, 69: 3584-3596.
Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE: TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005, 280: 37572-37584. 10.1074/jbc.M505557200.
Buratti E, Baralle FE: Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008, 13: 867-878. 10.2741/2727.
Gijselinck I, Sleegers K, Engelborghs S, Robberecht W, Martin JJ, Vandenberghe R, Sciot R, Dermaut B, Goossens D, Zee van der J, De Pooter T, Del-Favero J, Santens P, De Jonghe P, De Deyn PP, Van Broeckhoven C, Cruts M: Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol Aging. 2009, 30: 1329-1331. 10.1016/j.neurobiolaging.2007.11.002.
Guerreiro RJ, Schymick JC, Crews C, Singleton A, Hardy J, Traynor BJ: TDP-43 is not a common cause of sporadic amyotrophic lateral sclerosis. PLoS ONE. 2008, 3: e2450-10.1371/journal.pone.0002450.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/11/8/prepub
Acknowledgements
We sincerely thank the ALS patients and their family members. The authors also thank the reviewers for improving the manuscript. This work was supported by grant from Huashan Hospital for special professorship of Fudan University, Shanghai, and grant FMU-RT002 of program for Innovative Research Team in Science and Technology in the Fujian Province University and grant JS6037 from Fujian Medical College for professorship, Fuzhou, China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
Hui-Ling Xiong carried out the molecular genetic studies, participated in the analysis of the data and drafted the manuscript. Jin-Yang Wang and Yi-Min Sun collected demographic data and risk factor profiles of subjects and participated in analysis and interpretation of data. Jian-Jun Wu, Yan Chen and Kai Qiao analyzed the clinical data of all subjects. Gui-xian Zhao and Qiao-Juan Zheng participated in the acquisition of data. Zhi-Ying Wu conceived of the study, and participated in its design and coordination, and revising the manuscript critically for important intellectual content. All authors have read and approved the final manuscript.
Hui-Ling Xiong, Jin-Yang Wang, Yi-Min Sun contributed equally to this work.
Electronic supplementary material
12881_2009_569_MOESM1_ESM.JPEG
{kind=link}
Additional file 1: Chromatograms of silent mutations (Gly40Gly and Ala366Ala) and the novel polymorphism (239-18t>c). The normal sequence is shown in the upper half and the corresponding mutation is shown below. (JPEG 262 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Xiong, HL., Wang, JY., Sun, YM. et al. Association between novel TARDBP mutations and Chinese patients with amyotrophic lateral sclerosis. BMC Med Genet 11, 8 (2010). https://doi.org/10.1186/1471-2350-11-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-11-8