
Abstract
Background
Pierre-Robin sequence (PRS) is defined by micro- and/or retrognathia, glossoptosis and cleft soft palate, either caused by deformational defect or part of a malformation syndrome. Neurofibromatosis type 2 (NF2) is an autosomal dominant syndrome caused by mutations in the NF2 gene on chromosome 22q12.2. NF2 is characterized by bilateral vestibular schwannomas, spinal cord schwannomas, meningiomas and ependymomas, and juvenile cataracts. To date, NF2 and PRS have not been described together in the same patient.
Case presentation
We report a female with PRS (micrognathia, cleft palate), microcephaly, ocular hypertelorism, mental retardation and bilateral hearing loss, who at age 15 was also diagnosed with severe NF2 (bilateral cerebellopontine schwannomas and multiple extramedullary/intradural spine tumors). This is the first published report of an individual with both diagnosed PRS and NF2. High resolution karyotype revealed 46, XX, del(22)(q12.1q12.3), FISH confirmed a deletion encompassing NF2, and chromosomal microarray identified a 3,693 kb deletion encompassing multiple genes including NF2 and MN1 (meningioma 1).
Five additional patients with craniofacial dysmorphism and deletion in chromosome 22-adjacent-to or containing NF2 were identified in PubMed and the DECIPHER clinical chromosomal database. Their shared chromosomal deletion encompassed MN1, PITPNB and TTC28. MN1, initially cloned from a patient with meningioma, is an oncogene in murine hematopoiesis and participates as a fusion gene (TEL/MN1) in human myeloid leukemias. Interestingly, Mn1-haploinsufficient mice have abnormal skull development and secondary cleft palate. Additionally, Mn1 regulates maturation and function of calvarial osteoblasts and is an upstream regulator of Tbx22, a gene associated with murine and human cleft palate. This suggests that deletion of MN1 in the six patients we describe may be causally linked to their cleft palates and/or craniofacial abnormalities.
Conclusions
Thus, our report describes a NF2-adjacent chromosome 22q12.2 deletion syndrome and is the first to report association of MN1 deletion with abnormal craniofacial development and/or cleft palate in humans.
Similar content being viewed by others

Background
Pierre-Robin sequence (PRS) refers to a combination of micrognathia or retrognathia, glossoptosis and respiratory distress, with or without cleft palate, named after the French stomatologist, Pierre Robin [1–5]. The incidence of PRS is estimated at 1 in 8,500-14,000 births and continues to be associated with high morbidity secondary to a compromised airway, feeding difficulties, and speech problems [6–9]. While the clinical presentation is well defined, pathogenesis is heterogeneous and not completely understood. In half or more of patients PRS occurs in isolation [2, 4, 5, 7]. Isolated PRS is often thought to be due to intrauterine fetal constraint where extrinsic physical forces (e.g., oligohydramnios, breech position or abnormal uterine anatomy) inhibit normal mandibular growth. Micrognathia in early fetal development may then cause the tongue to remain between the palatal shelves, thus interfering with palate closure [2, 10]. However, this mechanism has been challenged by some [11]. Isolated PRS can also be associated with chromosomal deletions such as 2q24.1-33.3, 4q32-qter, 11q21-23.1, and 17q21-24.3 [6]. Recent reports of isolated PRS have additionally found mutations in highly conserved non-coding elements surrounding SOX9 at 17q24 [6, 12, 13]. Frequently, PRS is associated with additional clinical findings that may or may not constitute part of a recognized syndrome. The most common PRS-associated syndromes include Stickler syndrome, velo-cardio-facial syndrome (VCFS, associated with chromosome 22q11.2 mutations), fetal alcohol syndrome and trisomy 18 [3–5].
Neurofibromatosis type 2 (NF2) is an autosomal dominant disorder that predisposes to multiple neoplastic lesions. Birth incidence is estimated to be between 1 in 25,000 to 1 in 40,000 births [14–18]. The distinctive features of NF2 are eighth cranial nerve schwannomas, which occur bilaterally in over 90% of NF2 patients [14, 16, 19]. Patients may also develop schwannomas in other locations, particularly the spine, as well as meningiomas, ependymomas, and juvenile cataracts. NF2 is typically caused by truncating mutations (nonsense or frameshift) of the NF2 gene on chromosome 22q12.2, which encodes the intracellular membrane-associated tumor suppressor protein, merlin, also called schwannomin [20–23]. While most NF2 patients are diagnosed in their second or third decades of life, cataract, hearing loss, facial weakness, headache or ataxia may occur during childhood.
To date, there have been no published reports of patients diagnosed with both PRS and NF2. Here we provide the first description of clinical association between PRS (including cleft palate) and NF2 in a patient in whom we found a 22q12.2 deletion. Interestingly in addition to NF2, this deletion also encompasses Meningioma1 (MN1), a gene that is important in murine craniofacial development, and whose haploinsufficiency in mice causes skull abnormalities and secondary cleft palate, but which to date, has not been associated with malformations in humans.
Case presentation
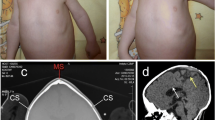
Our patient (PRS-NF2) was born to healthy non-consanguineous parents (mother 39 years old) at week 36 by emergency cesarean section due to premature contractions and early decelerations. Pregnancy was otherwise uncomplicated, without report of oligohydramnios or breech presentation. Cleft palate, micrognathia, microcephaly and ocular hypertelorism were noted at birth and the patient suffered multiple apneic episodes. For the first year she was fed by nasogastric tube due to poor weight gain. Motor development was delayed (rolled at 8 months, sat at 10 months, walked at 18 months), and she was later diagnosed with mental retardation. From preschool age she had bilateral severe conductive hearing loss requiring hearing aids that was attributed to bony changes associated with her congenital dysmorphic features. Sensorineural hearing was only mildly affected at that time. At age 14 she had severe to profound mixed hearing loss to which the bony conductance only contributed mild to moderate impairment. Her dysmorphic features, including micrognathia and hypertelorism, persisted into early adulthood and are shown in Figure 1A-B.

Patient PRS-NF2, clinical presentation. A-B) PRS-NF2 at age 17 years with severe micrognathia, ocular hypertelorism and microcephaly. Measurements: outer canthal distance 8.3 cm, 25th %ile; inner canthal distance 4 cm, 3 S.D. above the mean; interpupillary distance 5.5 cm, 25th %ile; head circumference 52 cm, 2nd %ile, weight 41.8 kg, 2-1.4 S.D. below the mean; height 156 cm, 10th-25th %ile. C-D) MRI of brain shows bilateral cerebellopontine angle tumors (left: 3.1 × 4.6 × 6.1 cm, right: 1 × 1.2 × 1.1 cm; arrows) with mass effect by the left tumor on the brain stem, pons and cerebellum, with associated effacement of the 4th ventricle, enlargement of the 3rd and lateral ventricles, enlarged cervical cord and tonsillar herniation. E-F) MRI of spine shows holocord syrinx with large neurofibroma at C7-T1 (E), causing cord compression and canal stenosis. Some of the other extramedullary intradural neurofibromas described in the text are also shown. G-H) Histological analysis of the resected left cerebellopontine angle tumor shows spindled nuclei with dense to finely granular chromatin and cells with long tapering cytoplasmic processes with inconspicuous outer membranes. Panel G demonstrates both short fascicular bundles with dense Antoni type A area (right upper corner) and loose hypocellular Antoni type B area (middle to left lower corner) characteristic of schwannomas (H&E, G-100×). Panel H is 400 × magnification of the inset from panel G).
At age 15 years PRS-NF2 presented for the first time with a 2 week history of headache and ataxia. Brain MRI showed bilateral large acoustic masses and a left trigeminal mass (Figure 1C-D). Corpus callosum was normal. Spine MRI showed multiple extramedullary intradural lesions ranging from 1 to 7 mm in diameter (approximately 12 lesions identified from the C4-C5 level to the cauda equina; Figure 1E-F). A left-sided C7-T1 extradural lesion and a separate left-sided C7-T1 neurofibroma were also seen, and increased in size in subsequent years. No definite intramedullary lesions were identified. A holocord syrinx, likely related to a secondary Chiari malformation (inferior cerebellar tonsil herniation), was also evident. The left cerebellopontine tumor was partially-resected and identified as a schwannoma, consistent with diagnosis of NF2 (Figure 1G-H). Hence, we designated her as patient PRS-NF2. The high tumor burden, tumor progression and the patient's young age at onset of tumors fit a classification of severe NF2 [14, 21–25].
Due to the unique combination of severe NF2, facial dysmorphism and mental retardation in patient PRS-NF2, genetic evaluation was initiated. G-banding high resolution karyotype on her blood revealed 46, XX, del(22)(q12.2), indicative of an interstitial deletion of band q12.2 in the long arm of chromosome 22 (Figure 2A, arrow). FISH demonstrated a deletion of at least 1,607 kb in the 22q12.2 region (Figure 2B). Parental testing was declined. Chromosomal microarray analysis (CMA; Affymetrix SNP 6.0 array) to better delineate the mutation detected a chromosome 22q12.2 deletion of approximately 3.7 Mb (3,693 kb) spanning the NF2 gene, with break points at base pairs 25937541 and 29635842 (UCSC Genome Browser version hg18, release name NCBI build 36.1) (Figure 2C).

3,693 kb deletion of 22q12.2 in patient PRS-NF2. A) G-banding high resolution karyotype reveals interstitial deletion of the light band q12.2 in the long arm of chromosome 22 (arrowhead - intact normal region; arrow - deleted region). B) Fluorescence in situ hybridization (FISH) studies with EWSR1 (22q12.2) dual color break apart probe revealed one entire fusion signal missing indicating heterozygous deletion in 22q12.2. FISH studies with flanking probes: TUPLE1 (22q11.2) and the BCR(22q11.2)/ABL(9q34) probes showed no deletions of TUPLE or BCR. C) Affymetrix Genotyping Console™ (GTC) Software display of the 22q12.2 deletion identified using chromosomal microarray (CMA; Affymetrix Genome-wide Human SNP Array 6.0).
Discussion
This is the first published report of a patient diagnosed with both PRS and NF2. Our patient's diagnosis of PRS, based on micrognathia and cleft palate, represents only part of her phenotype, which also includes microcephaly, ocular hypertelorism and mental retardation. Molecular analysis revealed a 22q12.2 deletion spanning approximately 3.7 Mb and encompassing the NF2 gene locus. The chromosomal microdeletion associated with velo-cardio-facial syndrome (22q11.2 locus) was intact in our patient. Therefore, we propose that we have identified a novel NF2-adjacent 22q12.2 microdeletion syndrome.
Review of the literature revealed only five other patients who presented with clinical features distinct from the standard diagnostic criteria of NF2 [24, 26] and who had large 22q12 deletions encompassing the NF2 gene (Table 1, Figure 3). Of these five patients three are described by Bruder et al [19, 27]. The first (JP) had a 7.4 Mb deletion of chromosome 22q12, bilateral vestibular schwannomas before the age of 25 years, mental retardation, and grand mal seizure at age 9 years, but no report of facial dysmorphism [19]. A second patient (p41) had NF2, facial dysmorphic features, mental retardation, developmental delay and a large 22q12 deletion with unknown span due to an unclear proximal deletion breakpoint [27]. The third patient (p12) had moderate NF2, mild mental retardation, and a 530 kb deletion encompassing NF2 [27]. Two other described patients, pX and TC, with large deletions at chromosome 22q12 that included NF2 also had severe craniofacial dysmorphism, but were too young to show stigmata of NF2 [28, 29]. The first was a 10.5 month old female (pX) with facial dysmorphism including hypertelorism, mandibular hypoplasia, cleft palate, severe neurodevelopmental delay and 8 Mb deletion of chromosome 22q12.2-q12.3 [28]. Her mandibular hypoplasia and cleft palate suggest likely diagnosis of PRS. The second young patient (TC) had Toriello-Carey syndrome, including PRS with large cleft palate, micrognathia, hypertelorism, agenesis of the corpus callosum, and 6 Mb deletion overlapping much of the deletion of pX and PRS-NF2 [29]. Patient TC only walked at 22 months [29].

Chromosome 22q12.2 deletions reveal loss of MN1 in six patients with facial dysmorphism and/or cleft palate and/or abnormal cranium. Schema of the deletions on chromosome 22q12.2 in the eight patients described in the text and Table 1. Black bars depict the six patients with abnormal cranium and/or facial dysmorphism, and/or cleft palate. pX and TC were too young to determine whether they manifest clinical NF2. Gray bars depict the two patients without these abnormalities but with NF2. Dashed black line indicates the approximated proximal deletion in patient p41. Genes: NF2-purple, TTC28-green, PITPNB-orange, MN1-blue. Translucent vertical bars align the location of the deleted genes in relation to the chromosomal deletion found in each patient.
The young age at which PRS-NF2 was diagnosed with bilateral vestibular schwannomas (15 years), the high tumor burden that included multiple spinal tumors, and the progressive nature of some of these tumors qualify her as having a severe form of NF2 [14, 23, 24]. Several reports have demonstrated that nonsense/frameshift mutations are associated with a more severe phenotype than are deletions of the NF2 gene. This difference is thought to be due to nonsense/frameshift mutations causing a truncated protein that may have dominant negative effect, versus deletion of one copy of the NF2 gene, that results in decreased merlin protein expression [14, 22, 23, 31]. Watson et al provided the first report on a large deletion (600-700 kb) encompassing NF2 that was associated with mild disease without mental retardation or facial dysmorphism [32]. Bruder et al described patient p12, who had a 530 kb deletion encompassing NF2 (partially overlapping with the patient reported by Watson et al) and who had moderate NF2 [27] (Figure 3, Table 1). Others support that complete deletion of the NF2 gene is usually associated with mild disease [33–35]. It is therefore interesting that PRS-NF2 (our patient) and patient JP [19], both of whom had large deletions encompassing the NF2 gene, had severe NF2 phenotype (Figure 3). Bruder et al hypothesized that the high severity of NF2 in JP may be due to loss of a putative second tumor suppressor gene within the 7.4 Mb deletion, that usually remained intact in patients with smaller chromosomal deletions that encompass all of NF2 [19]. Deletion of this putative tumor suppressor may have similarly changed a generally mild phenotype associated with complete deletion of NF2 to the severe phenotype seen in PRS-NF2.
The craniofacial dysmorphism in PRS-NF2 and the three other patients whose deletion overlaps her deletion (pX, p41, TC; Table 1, Figure 3) suggested a causal link between loss of one or more genes within the overlapping region of the NF2-adjacent 22q12.2 deletion and these clinical findings. To further investigate this association we searched the public database DECIPHER, which contains clinical information of individuals with chromosomal anomalies with the aim of increasing medical and scientific knowledge about these chromosomal defects [30]. DECIPHER reported two patients with chromosomal deletions in the region deleted in PRS-NF2 (Figure 3, patients 4110 and 999). Although the information is brief, there are similarities to PRS-NF2, pX, p41 and TC (Table 1): Both 4110 and 999 had mental retardation; patient 4110 had a submucous cleft palate and patient 999 had speech delay and microcephaly.
To determine genes potentially associated with the craniofacial dysmorphism and/or palatal defects in these six patients we aligned their deletions (Figure 3). The deleted area common to all six patients includes three recognized genes: MN1, PITPNB and TTC28, none of which has been previously linked in humans to palatal defects, calvarial abnormalities or craniofacial dysmorphism. Of these, MN1, initially reported as a candidate gene for sporadic meningioma and later found to be involved in acute myeloid leukemia (fusion TEL/MN1) and as an oncogene in murine hematopoiesis, was recently implicated in murine calvarial osseous development, facial membranous bone ossification, and palatal development [36–38]. Homozygous Mn1 -/- mice had calvarial bone defects with secondary cleft palate and died at birth or shortly thereafter [38, 39]. Heterozygous Mn1 +/- mice showed hypoplastic membranous calvarium with incomplete penetrance of cleft palate [38], consistent with the demonstrated role of Mn1 in maintaining maturation and function of calvarial osteoblasts [40]. Furthermore, Mn1 is a transcriptional activator that regulates expression of Tbx22 during palatal development in mice [39]. Loss of murine Tbx22, which regulates intramembranous bone formation in the posterior hard palate, causes submucosal cleft palate with ankyloglossia in mice [41]. Importantly, TBX22 was recently also implicated in cleft palate formation in humans in that up to 5% of children with non-syndromic cleft palate have mutations in TBX22 [42], and TBX22 loss in humans is associated with X-linked cleft palate with ankyloglossia (CPX) [43]. Interestingly, the six patients we identified with MN1 deletion (Figure 3, Table 1) all had either craniofacial dysmorphism and/or palatal defects. Taken together, this suggests that loss of MN1 in these six patients may be the underlying cause for, or contributing factor to, their craniofacial dysmorphism and/or cleft palate, and supports investigation of a role for MN1 in human craniofacial dysmorphism and/or cleft palate.
Conclusions
This is the first report of association between haploinsufficiency of the human MN1 gene (a gene associated with murine craniofacial development) and human craniofacial malformations, supporting a more in-depth investigation of a role for MN1 in human craniofacial development. Additionally, the overlapping phenotype of the patients described here suggests that a 22q12.2-contiguous gene deletion syndrome should be considered in individuals with hearing loss, developmental deficits and craniofacial dysmorphism that includes Pierre-Robin Sequence or isolated micrognathia with or without cleft palate. As microarray testing replaces chromosome and FISH testing, large deletions such as the one found in our patient are more likely to be found when interrogating cases of PRS. This may allow earlier diagnosis of similar contiguous gene deletions and provide for earlier surveillance of serious clinical disorders such as NF2.
Consent
Written informed permission for use and disclosure of this patient's protected health information for research purposes and permission to obtain and release the accompanying photographs were obtained from the patient's parents. Copies of the consents are available for review by the Editor-in-Chief of this journal.
Abbreviations
- MN1:
-
Meningioma1
- NF2:
-
Neurofibromatosis type 2
- PRS:
-
Pierre-Robin Sequence.
References
Robin P: La chute de la base de la langue consideree comme une nouvelle cause de gene dans la respiration naso-pharyngienne. Bull Acad Med (Paris). 1923, 89: 37-
Evans AK, Rahbar R, Rogers GF, Mulliken JB, Volk MS: Robin sequence: a retrospective review of 115 patients. Int J Pediatr Otorhinolaryngol. 2006, 70 (6): 973-980. 10.1016/j.ijporl.2005.10.016.
van den Elzen AP, Semmekrot BA, Bongers EM, Huygen PL, Marres HA: Diagnosis and treatment of the Pierre Robin sequence: results of a retrospective clinical study and review of the literature. Eur J Pediatr. 2001, 160 (1): 47-53. 10.1007/s004310000646.
Holder-Espinasse M, Abadie V, Cormier-Daire V, Beyler C, Manach Y, Munnich A, Lyonnet S, Couly G, Amiel J: Pierre Robin sequence: a series of 117 consecutive cases. J Pediatr. 2001, 139 (4): 588-590. 10.1067/mpd.2001.117784.
Butow KW, Hoogendijk CF, Zwahlen RA: Pierre Robin sequence: appearances and 25 years of experience with an innovative treatment protocol. J Pediatr Surg. 2009, 44 (11): 2112-2118. 10.1016/j.jpedsurg.2009.04.018.
Jakobsen LP, Knudsen MA, Lespinasse J, Garcia Ayuso C, Ramos C, Fryns JP, Bugge M, Tommerup N: The genetic basis of the Pierre Robin Sequence. Cleft Palate Craniofac J. 2006, 43 (2): 155-159. 10.1597/05-008.1.
Printzlau A, Andersen M: Pierre Robin sequence in Denmark: a retrospective population-based epidemiological study. Cleft Palate Craniofac J. 2004, 41 (1): 47-52. 10.1597/02-055.
Bush PG, Williams AJ: Incidence of the Robin Anomalad (Pierre Robin syndrome). Br J Plast Surg. 1983, 36 (4): 434-437. 10.1016/0007-1226(83)90123-6.
Glynn F, Fitzgerald D, Earley MJ, Rowley H: Pierre Robin sequence: an institutional experience in the multidisciplinary management of airway, feeding and serous otitis media challenges. Int J Pediatr Otorhinolaryngol. 2011, 75 (9): 1152-1155. 10.1016/j.ijporl.2011.06.009.
Schubert J, Jahn H, Berginski M: Experimental aspects of the pathogenesis of Robin sequence. Cleft Palate Craniofac J. 2005, 42 (4): 372-376. 10.1597/03-166.1.
Rintala A, Ranta R, Stegars T: On the pathogenesis of cleft palate in the Pierre Robin syndrome. Scand J Plast Reconstr Surg. 1984, 18 (2): 237-240. 10.3109/02844318409052844.
Benko S, Fantes JA, Amiel J, Kleinjan DJ, Thomas S, Ramsay J, Jamshidi N, Essafi A, Heaney S, Gordon CT, et al: Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet. 2009, 41 (3): 359-364. 10.1038/ng.329.
Jakobsen LP, Ullmann R, Christensen SB, Jensen KE, Molsted K, Henriksen KF, Hansen C, Knudsen MA, Larsen LA, Tommerup N, et al: Pierre Robin sequence may be caused by dysregulation of SOX9 and KCNJ2. J Med Genet. 2007, 44 (6): 381-386. 10.1136/jmg.2006.046177.
Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, Strachan T, Harris R: A genetic study of type 2 neurofibromatosis in the United Kingdom. II. Guidelines for genetic counselling. J Med Genet. 1992, 29 (12): 847-852. 10.1136/jmg.29.12.847.
Evans DG, Huson SM, Donnai D, Neary W, Blair V, Teare D, Newton V, Strachan T, Ramsden R, Harris R: A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992, 29 (12): 841-846. 10.1136/jmg.29.12.841.
Evans DG: Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis. 2009, 4: 16-10.1186/1750-1172-4-16.
Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, Lalloo F: Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010, 152A (2): 327-332. 10.1002/ajmg.a.33139.
Evans DG, Moran A, King A, Saeed S, Gurusinghe N, Ramsden R: Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005, 26 (1): 93-97. 10.1097/00129492-200501000-00016.
Bruder CE, Ichimura K, Blennow E, Ikeuchi T, Yamaguchi T, Yuasa Y, Collins VP, Dumanski JP: Severe phenotype of neurofibromatosis type 2 in a patient with a 7.4-MB constitutional deletion on chromosome 22: possible localization of a neurofibromatosis type 2 modifier gene?. Genes Chromosomes Cancer. 1999, 25 (2): 184-190. 10.1002/(SICI)1098-2264(199906)25:2<184::AID-GCC15>3.0.CO;2-B.
Evans DG, Sainio M, Baser ME: Neurofibromatosis type 2. J Med Genet. 2000, 37 (12): 897-904. 10.1136/jmg.37.12.897.
Parry DM, MacCollin MM, Kaiser-Kupfer MI, Pulaski K, Nicholson HS, Bolesta M, Eldridge R, Gusella JF: Germ-line mutations in the neurofibromatosis 2 gene: correlations with disease severity and retinal abnormalities. Am J Hum Genet. 1996, 59 (3): 529-539.
Evans DG, Trueman L, Wallace A, Collins S, Strachan T: Genotype/phenotype correlations in type 2 neurofibromatosis (NF2): evidence for more severe disease associated with truncating mutations. J Med Genet. 1998, 35 (6): 450-455. 10.1136/jmg.35.6.450.
Ruttledge MH, Andermann AA, Phelan CM, Claudio JO, Han FY, Chretien N, Rangaratnam S, MacCollin M, Short P, Parry D, et al: Type of mutation in the neurofibromatosis type 2 gene (NF2) frequently determines severity of disease. Am J Hum Genet. 1996, 59 (2): 331-342.
Baser ME, Friedman JM, Wallace AJ, Ramsden RT, Joe H, Evans DG: Evaluation of clinical diagnostic criteria for neurofibromatosis 2. Neurology. 2002, 59 (11): 1759-1765.
Baser ME, Kuramoto L, Joe H, Friedman JM, Wallace AJ, Gillespie JE, Ramsden RT, Evans DG: Genotype-phenotype correlations for nervous system tumors in neurofibromatosis 2: a population-based study. Am J Hum Genet. 2004, 75 (2): 231-239. 10.1086/422700.
Baser ME, Friedman JM, Evans DG: Increasing the specificity of diagnostic criteria for schwannomatosis. Neurology. 2006, 66 (5): 730-732. 10.1212/01.wnl.0000201190.89751.41.
Bruder CE, Hirvela C, Tapia-Paez I, Fransson I, Segraves R, Hamilton G, Zhang XX, Evans DG, Wallace AJ, Baser ME, et al: High resolution deletion analysis of constitutional DNA from neurofibromatosis type 2 (NF2) patients using microarray-CGH. Hum Mol Genet. 2001, 10 (3): 271-282. 10.1093/hmg/10.3.271.
Barbi G, Rossier E, Vossbeck S, Hummler H, Lang D, Flock F, Terinde R, Wirth J, Vogel W, Kehrer-Sawatzki H: Constitutional de novo interstitial deletion of 8 Mb on chromosome 22q12.1-12.3 encompassing the neurofibromatosis type 2 (NF2) locus in a dysmorphic girl with severe malformations. J Med Genet. 2002, 39 (2): E6-10.1136/jmg.39.2.e6.
Said E, Cuschieri A, Vermeesch J, Fryns JP: Toriello-Carey syndrome with a 6 Mb interstitial deletion at 22q12 detected by array CGH. Am J Med Genet A. 2011, 155A (6): 1390-1392.
DECIPHER. accessed March 1; 2011, [http://decipher.sanger.ac.uk/]
Selvanathan SK, Shenton A, Ferner R, Wallace AJ, Huson SM, Ramsden RT, Evans DG: Further genotype-phenotype correlations in neurofibromatosis 2. Clin Genet. 2010, 77 (2): 163-170. 10.1111/j.1399-0004.2009.01315.x.
Watson CJ, Gaunt L, Evans G, Patel K, Harris R, Strachan T: A disease-associated germline deletion maps the type 2 neurofibromatosis (NF2) gene between the Ewing sarcoma region and the leukaemia inhibitory factor locus. Hum Mol Genet. 1993, 2 (6): 701-704. 10.1093/hmg/2.6.701.
Zucman-Rossi J, Legoix P, Der Sarkissian H, Cheret G, Sor F, Bernardi A, Cazes L, Giraud S, Ollagnon E, Lenoir G, et al: NF2 gene in neurofibromatosis type 2 patients. Hum Mol Genet. 1998, 7 (13): 2095-2101. 10.1093/hmg/7.13.2095.
Lopez-Correa C, Zucman-Rossi J, Brems H, Thomas G, Legius E: NF2 gene deletion in a family with a mild phenotype. J Med Genet. 2000, 37 (1): 75-77. 10.1136/jmg.37.1.75.
Sanson M, Marineau C, Desmaze C, Lutchman M, Ruttledge M, Baron C, Narod S, Delattre O, Lenoir G, Thomas G, et al: Germline deletion in a neurofibromatosis type 2 kindred inactivates the NF2 gene and a candidate meningioma locus. Hum Mol Genet. 1993, 2 (8): 1215-1220. 10.1093/hmg/2.8.1215.
Buijs A, van Rompaey L, Molijn AC, Davis JN, Vertegaal AC, Potter MD, Adams C, van Baal S, Zwarthoff EC, Roussel MF, et al: The MN1-TEL fusion protein, encoded by the translocation (12;22)(p13;q11) in myeloid leukemia, is a transcription factor with transforming activity. Mol Cell Biol. 2000, 20 (24): 9281-9293. 10.1128/MCB.20.24.9281-9293.2000.
Lekanne Deprez RH, Riegman PH, Groen NA, Warringa UL, van Biezen NA, Molijn AC, Bootsma D, de Jong PJ, Menon AG, Kley NA, et al: Cloning and characterization of MN1, a gene from chromosome 22q11, which is disrupted by a balanced translocation in a meningioma. Oncogene. 1995, 10 (8): 1521-1528.
Meester-Smoor MA, Vermeij M, van Helmond MJ, Molijn AC, van Wely KH, Hekman AC, Vermey-Keers C, Riegman PH, Zwarthoff EC: Targeted disruption of the Mn1 oncogene results in severe defects in development of membranous bones of the cranial skeleton. Mol Cell Biol. 2005, 25 (10): 4229-4236. 10.1128/MCB.25.10.4229-4236.2005.
Liu W, Lan Y, Pauws E, Meester-Smoor MA, Stanier P, Zwarthoff EC, Jiang R: The Mn1 transcription factor acts upstream of Tbx22 and preferentially regulates posterior palate growth in mice. Development. 2008, 135 (23): 3959-3968. 10.1242/dev.025304.
Zhang X, Dowd DR, Moore MC, Kranenburg TA, Meester-Smoor MA, Zwarthoff EC, MacDonald PN: Meningioma 1 is required for appropriate osteoblast proliferation, motility, differentiation, and function. J Biol Chem. 2009, 284 (27): 18174-18183. 10.1074/jbc.M109.001354.
Pauws E, Hoshino A, Bentley L, Prajapati S, Keller C, Hammond P, Martinez-Barbera JP, Moore GE, Stanier P: Tbx22null mice have a submucous cleft palate due to reduced palatal bone formation and also display ankyloglossia and choanal atresia phenotypes. Hum Mol Genet. 2009, 18 (21): 4171-4179. 10.1093/hmg/ddp368.
Marcano AC, Doudney K, Braybrook C, Squires R, Patton MA, Lees MM, Richieri-Costa A, Lidral AC, Murray JC, Moore GE, et al: TBX22 mutations are a frequent cause of cleft palate. J Med Genet. 2004, 41 (1): 68-74. 10.1136/jmg.2003.010868.
Braybrook C, Doudney K, Marcano AC, Arnason A, Bjornsson A, Patton MA, Goodfellow PJ, Moore GE, Stanier P: The T-box transcription factor gene TBX22 is mutated in X-linked cleft palate and ankyloglossia. Nat Genet. 2001, 29 (2): 179-183. 10.1038/ng730.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/13/19/prepub
Acknowledgements
The authors are grateful to the patient and her parents who made this report possible. We thank Dr. Jonathan Finlay for critically reviewing the manuscript and providing input. PA S-L is supported by the Harold Amos Faculty Development Program through the Robert Wood Johnson Foundation and the CHLA-USC Child Health Research Career Development Program (NIH K12-HD05954). AE-E is supported in part by Grayson's Gift supporting pediatric brain tumor research and by the Rachel Ann Hage Neuro-Oncology Fund.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors reviewed the manuscript critically for its content, revised and edited it, and approved the final version. Additionally, specific author contributions are as follows: TBD coordinated data acquisition, analyzed and interpreted the data and drafted the manuscript. PA S-L participated in analysis and interpretation of the molecular data, search of the DECIPHER database and drafting of the manuscript. LMR and MDK acquired and interpreted the clinical data and drafted the manuscript. SW performed and interpreted the karyotype and FISH analysis. AP analyzed and interpreted the MRI imaging data. HS analyzed and interpreted the pathological slides. AE-E conceptualized, designed and coordinated the study, acquired and interpreted data, drafted the manuscript and finalized it. All authors read and approved the final manuscript.
Pedro A Sanchez-Lara and Anat Erdreich-Epstein contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Davidson, T.B., Sanchez-Lara, P.A., Randolph, L.M. et al. Microdeletion del(22)(q12.2) encompassing the facial development-associated gene, MN1 (meningioma 1) in a child with Pierre-Robin sequence (including cleft palate) and neurofibromatosis 2 (NF2): a case report and review of the literature. BMC Med Genet 13, 19 (2012). https://doi.org/10.1186/1471-2350-13-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-13-19