
Abstract
Background
The prognostic value of grading in breast cancer can be increased with microarray technology, but proposed strategies are disadvantaged by the use of specific training data or parallel microscopic grading. Here, we investigate the performance of a method that uses no information outside the breast profile of interest.
Results
In 251 profiled tumours we optimised a method that achieves grading by comparing rank means for genes predictive of high and low grade biology; a simpler method that allows for truly independent estimation of accuracy. Validation was carried out in 594 patients derived from several independent data sets. We found that accuracy was good: for low grade (G1) tumors 83- 94%, for high grade (G3) tumors 74- 100%. In keeping with aim of improved grading, two groups of intermediate grade (G2) cancers with significantly different outcome could be discriminated.
Conclusion
This validates the concept of microarray-based grading in breast cancer, and provides a more practical method to achieve it. A simple R script for grading is available in an additional file. Clinical implementation could achieve better estimation of recurrence risk for 40 to 50% of breast cancer patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Decisions regarding medical treatment in early breast cancer are guided by disease stage, morphological characteristics (microscopic grading), and selected biological factors - notably expression of oestrogen and progesterone receptors and the HER2 oncogene [1]. The prognostic and therapy-predictive accuracies of these factors are limited, resulting in over- and under-treatment of patients. A decade of experience from large scale gene expression studies have underscored the relevance of some factors, as well as indicated potential improvements. Retrospective data reveal a high degree of concordance between microarray-technology and established methods for determining oestrogen and progesterone receptor status [2–5], as well as expression of the HER2 oncogene [6]. Concerning histopathological grading, two studies have indicated a potential for improvement. Here, the intermediate grade (G2) seems to lack a biological correlate. Both gene expression-wise and with regards to prognosis approximately two-thirds of G2-tumors are similar to the low grade group whereas about one third resemble the high grade group [7, 8], which means that 40-50% of breast cancer patients currently get an unnecessarily imprecise estimate of their risk of recurrence.
For research as well as clinical purposes, we have been interested to use proposed methods for microarray-based grading. The more extensively investigated method [8] - now commercially available through MapQuant Dx - involves calculation of a summary score from genes with increased expression in G1 and G3 cancers, respectively (weighted -1 and +1). To allow use across different microarray platforms, scores are transformed with scale and offset terms, calculated utilizing known microscopic grading in data to be classified, in a leave-one-out cross-validation. It is unclear if the leave-one-out approach adequately deals with the obvious risk of over-fitting, and the need for parallel microscopic grading is not appealing, rendering the method unpractical and insufficiently validated. The methods proposed by Ivshina et al [7] - prediction analysis of microarrays (PAM) and statistically weighted syndromes (SWS) - require training of the algorithm in the same original data for true reproduction, which is cumbersome and may deter potential users. Also, data to be classified may have to be normalized to training data to maintain accuracy. To validate and simplify the concept of microarray-based grading, we tested a method that only uses information contained in the single tumour profile of interest.
Implementation
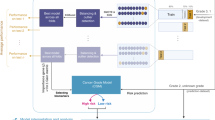
For Sotiriou's et. al. [8] set of 128 probe sets, the ranks (1 for a probe set with lowest expression in a given tumour profile, n for a gene with highest; n = number of probe sets on the microarray platform) were averaged for probe sets with increased expression in G3-tumors (n = 112), and those with increased expression in G1-tumors (n = 16), respectively. The higher average determines the genomic grade (we use Sotiriou's and co-workers term to distinguish from histopathological grade). Expression of grade-related genes is thus only related to other genes internal to the profile, resulting in a method that can classify individual profiles with no further information. Insensitivity to data set composition, no requirement for training data, and unbiased estimation of accuracy are key advantages. Sensitivity to differences in average expression between probes is a potential limitation.
Results
We tested the rank-based algorithm in a data set with 251 histopathologically graded breast cancers, here referred to as the Uppsala data set [9]. An assumption of equal average rank for G3-probe sets compared to G1-probe sets was not met: G1-probe sets had higher average expression than G3-probe sets. This resulted in bias towards low genomic grade classification calls; we therefore removed 37 lowly expressed genes from the larger group (G3-probe sets). With this optimization performed in the Uppsala cohort, we went on and validated the method in independent data (Table 1). Considerable accuracy was apparent in these published data sets - for the three series run on Affymetrix chip technology, 93%, 94%, and 83% of (histopathological) grade 1 tumours were correctly classified as low grade, and 85%, 100%, and 93% of (histopathological) grade 3 tumours were appropriately identified as high grade, translating to an overall accuracy of 88%, 97% and 88% for the Stockholm [10], Guys Hospital [11], and Oxford data [11]. The accuracy for the Netherlands Cancer Institute (NKI) data was somewhat inferior with an overall accuracy of 78%, possibly reflecting the fact that Agilent two-colour technology had been used [12]. The improvement in prognostic accuracy is illustrated in Figure 1: with array-based grading histologic G2 tumours (associated with intermediate outcome) are reclassified as either genomic G1 or G3 (associated with favourable or worse outcome, respectively). Further comparisons of original and simplified expression-based grading calls, microscopic grading calls, outcome, and unsupervised clustering results, can be found in Additional file 1.

Prognostic significance of microarray-based grading. Tumours in the respective data sets graded with either the "genomic grade" (green- red) or by a pathologist (solid, dashed and dotted). Fraction of patients alive without distant metastasis (DMFS; y-axis), years (x-axis). Genomic grade determined with the optimized signature (reduced from 128 to 91 probe sets) and mean rank. P-values = log-rank test of difference for intermediate grade tumors (hist. G2; dashed line), when stratified by microarray-based grading. Insufficient statistical power in C and D: the number of events per groups small.
Conclusions
Routine use of microarray or other RNA-profiling technology in early breast cancer is a likely future development, and projects to test this clinically are underway. Common to all these efforts is the aim to achieve improved prognostic and treatment predictive information. We take particular interest in microarray-based grading as it offers some key advantages over other proposed strategies (MammaPrint and Oncotype Dx etcetera): grade is well-known and interpretation in relation to clinical guidelines is straightforward. Change of practice - with the potential to spare patients from aggressive therapy - is likely to happen faster than with signatures introducing new taxonomies with unclear relationships to established prognostic and treatment predictive markers. Focusing on factors of known importance - like grade - also makes it possible to motivate microarray technology as an improvement in medical technology: similar or improved information (compared to that contributed by the pathology department) can be achieved at a competitive cost. Suggested methods to achieve this - in their current published form [7, 8] - are disadvantaged by methodological complexity and uncertain independent accuracy, which has motivated us to design a simpler data set-independent method and evaluate it in several tumour series. This can hopefully increase interest in the concept of microarray-based grading; an R script for the necessary calculations is available in an additional file (Additional file 2).
Availability and requirements
-
Project name: expression grade.txt
-
Project home page: available as an additional file (Additional file 2)
-
Operating system(s): any setting where R works
-
Programming language: R (http://www.r-project.org/)
-
Other requirements: none
-
License: GNU GPL (R)
-
Any restrictions to use by non-academics: none
References
Goldhirsch A, Ingle JN, Gelber RD, Coates AS, Thurlimann B, Senn HJ: Thresholds for therapies: highlights of the St Gallen International Expert Consensus on the primary therapy of early breast cancer 2009. Ann Oncol. 2009, 20: 1319-1329. 10.1093/annonc/mdp322.
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al: Molecular portraits of human breast tumours. Nature. 2000, 406: 747-752. 10.1038/35021093.
Gruvberger S, Ringner M, Chen Y, Panavally S, Saal LH, Borg A, Ferno M, Peterson C, Meltzer PS: Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res. 2001, 61: 5979-5984.
van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al: Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002, 415: 530-536. 10.1038/415530a.
Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, Haibe-Kains B, Desmedt C, Ignatiadis M, Sengstag T, Schutz F, et al: Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008, 10: R65-10.1186/bcr2124.
Bergqvist J, Ohd JF, Smeds J, Klaar S, Isola J, Nordgren H, Elmberger GP, Hellborg H, Bjohle J, Borg AL, et al: Quantitative real-time PCR analysis and microarray-based RNA expression of HER2 in relation to outcome. Ann Oncol. 2007, 18: 845-850. 10.1093/annonc/mdm059.
Ivshina AV, George J, Senko O, Mow B, Putti TC, Smeds J, Lindahl T, Pawitan Y, Hall P, Nordgren H, et al: Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006, 66: 10292-10301. 10.1158/0008-5472.CAN-05-4414.
Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, et al: Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006, 98: 262-272. 10.1093/jnci/djj052.
Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, Bergh J: An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA. 2005, 102: 13550-13555. 10.1073/pnas.0506230102.
Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S, et al: Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005, 7: R953-964. 10.1186/bcr1325.
Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, Ellis P, Harris A, Bergh J, Foekens JA, et al: Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol. 2007, 25: 1239-1246. 10.1200/JCO.2006.07.1522.
van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al: A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002, 347: 1999-2009. 10.1056/NEJMoa021967.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/11/306/prepub
Acknowledgements and funding
We thank Dr Johanna Smeds and Anna-Lena Borg for valuable technical assistance. This work was supported by grants from the Swedish Cancer Society; the Stockholm Cancer Society; the King Gustav V jubilee fund; the ALF Foundation; the Linné Foundation; the Swedish Research Council; Manchester University; the BRECT consortium at Karolinska Institutet; and the Märit & Hans Rausing initiative against breast cancer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
KW wrote the R script, performed analyses, and drafted the manuscript. JB substantially contributed to data acquisition and manuscript writing. Both authors read and approved the final manuscript.
Electronic supplementary material
12885_2011_2784_MOESM1_ESM.PDF
Additional file 1: Additional analyses and methods. comparisons of original expression-based grading calls and grade (Table S1), original and simplified calls (Table S2), outcome (Figure S1), unsupervised clustering results (Table S3), expanded materials and methods, and R script instructions. (PDF 298 KB)
12885_2011_2784_MOESM2_ESM.TXT
Additional file 2: Expression grade.txt. An R script for calculation of the simplified expression-based grade. (TXT 3 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wennmalm, K., Bergh, J. A simple method for assigning genomic grade to individual breast tumours. BMC Cancer 11, 306 (2011). https://doi.org/10.1186/1471-2407-11-306
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-11-306