
Abstract
Background
This study aimed at the identification of prognostic gene expression markers in early primary colorectal carcinomas without metastasis at the time point of surgery by analyzing genome-wide gene expression profiles using oligonucleotide microarrays.
Methods
Cryo-conserved tumor specimens from 45 patients with early colorectal cancers were examined, with the majority of them being UICC stage II or earlier and with a follow-up time of 41–115 months. Gene expression profiling was performed using Whole Human Genome 4x44K Oligonucleotide Microarrays. Validation of microarray data was performed on five of the genes in a smaller cohort.
Results
Using a novel algorithm based on the recursive application of support vector machines (SVMs), we selected a signature of 44 probes that discriminated between patients developing later metastasis and patients with a good prognosis. Interestingly, almost half of the genes was related to the patients’ immune response and showed reduced expression in the metastatic cases.
Conclusions
Whereas up to now gene signatures containing genes with various biological functions have been described for prediction of metastasis in CRC, in this study metastasis could be well predicted by a set of gene expression markers consisting exclusively of genes related to the MHC class II complex involved in immune response. Thus, our data emphasize that the proper function of a comprehensive network of immune response genes is of vital importance for the survival of colorectal cancer patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Colorectal cancer (CRC) is one of the most common cancers with an annual incidence of more than 400,000 new cases and 212,000 deaths in Europe alone [1]. Treatment decisions are based on histopathological staging of the primary tumor, considering depth of tumor infiltration and metastatic spread to lymph nodes or distant organs. Therapy consists of surgical resection of the tumor and additional chemo- or/and radiotherapy in the case of lymph node or distant metastases. Approximately 40% of patients with CRC die within 5 years due to local recurrence or distant metastases. However, histopathological staging of the primary tumor does not allow for outcome prediction, resulting in under- as well as over-treatment of particular groups of patients.
The heterogeneity of patients with complex diseases where the phenotype can have multiple genetic and environmental components [2, 3] usually prevents one from finding differentially expressed genes between different groups of patients. Therefore, recent studies aimed at outcome prediction based on gene expression [4–10], often resulting in a diverse set of genes without a common biological function. In such a situation, specialized methods of analysis for differential gene expression are required. In particular, Zhang et al. [11] developed a method appropriate for clinical data that iteratively selects groups of genes that are optimized for discriminating between different groups of patients, with patient numbers similar to those in this study. Unlike methods that attempt to select individual genes by their significant differential expression between classes of samples (e.g., SAM [12]), the gene set is selected as a set of markers that discriminate when used collectively but not necessarily when each gene is used individually. Using this methodology we were able to identify a predictive gene signature in an unbiased manner that shows a functional relationship to tumor biology. Surprisingly, most of the genes that were found to be informative for patients’ metastasis were related to the immune system but not to common tumor cell characteristics such as angiogenesis, adhesion or invasion.
Methods
Patients
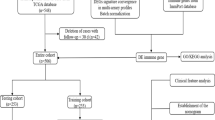
This study comprises only patients with early CRC of UICC stages I-III, with the majority of them (76%) being UICC stage II or earlier (Table 1). Inclusion criteria were: CRC, R0 resection and follow-up of at least three years or until disease recurrence. Exclusion criteria were: presence of a secondary carcinoma, patient age less than 40 years at the time of surgery, local recurrence, preoperative treatment and radiotherapy.
Specimen characteristics
Cryo-conserved tumor specimens from 45 patients and follow-up data were obtained from the Tumorbank of the Charité Comprehensive Cancer Center (CCCC), Charité Campus Buch, Berlin. Tissue samples from patients that had undergone resection for sporadic colorectal adenocarcinoma at Charité Robert Roessle Hospital (1995–2006) were enrolled for this study (after informed consent). The study was approved by the local ethics committee (Charité Universitätsmedizin Berlin). Archived primary tumor tissues were collected immediately after surgical removal and snap-frozen in liquid nitrogen according to internal protocols. In addition to routine pathological examination of the tumor tissue, histopathology of each sample used for experimental analysis was reviewed by an experienced pathologist to confirm diagnosis, tissue composition and tumor content. Only non-necrotic tissue samples lacking fatty or connective tissue and composed of at least 60% tumor cells (mean: 73%) were processed. Disease-free time was defined as the time period from the date of surgery to confirmed distant tumor metastasis date for metastatic cases and from the date of surgery to the date of last follow-up for non-recurrent patients.
Gene expression analysis
Total RNA was extracted from each frozen tumor specimen using the RNeasy Mini Prep Kit (Qiagen, Hilden, Germany). RNA quality was checked by Bioanalyzer (Agilent, Santa Clara, CA, USA). Only RNA samples showing an RNA Integrity Number (RIN) of at least 7.0 were used for labelling. Total RNA (1 μg) was labeled with Cy3 using the Low Input RNA Amplification Kit (Agilent, Santa Clara, CA, USA). Labeled cRNAs were hybridized to Whole Human Genome 4x44K Oligonucleotide Microarrays (Agilent, Santa Clara, CA, USA) following the manufacturer´s protocols. Arrays were scanned by using standard Agilent protocols and a G2565AA Microarray Scanner (Agilent, Santa Clara, CA, USA). Raw expression values were determined using Feature Extraction 8.0 software (Agilent, Santa Clara, CA, USA). Data were uploaded to GEODataSets with accession: GSE31905 and ID: 200031905 under the title: Gene expression profiling of colorectal carcinomas.
Data analysis
Microarray data analysis was performed using R/Bioconductor [13]. Raw expression values were normalized using variance stabilization normalization [14], and array quality was evaluated with the arrayQualityMetrics library [15]. Univariate differential expression analysis was performed using methods from the limma library [13], and multiple testing was controlled using the Benjamini and Hochberg method [16]. Individual probes were considered differentially expressed if they had a fold change greater than 2.0 and a false discovery rate (q-value) less than 0.05. No significant differentially expressed genes between recurrent and non-recurrent samples were observed. Principal component analysis of the data indicated that the full vectors of gene expression could not be used to discriminate between sample classes (Additional file 1: Figure S1).
Multivariate feature selection was performed using the R-SVM algorithm [11]. The algorithm was configured to perform 100 bootstrapping cross-validation steps and the best 42 probes were saved from each step. Probes that were selected at least once were kept, yielding 44 probes that were considered for further analysis. We then filtered out any probes that did not have an associated gene symbol (Table 2). The resulting 30 probes were analyzed using the Bioconductor package GOstats [17], which identified the GO term “immune system process” (GO:0002376) associated to 14 of the genes as being significantly (p-value 4.6E-12) enriched (Table 3). ROC curves, Kaplan-Meier analysis and clinical measure statistics (Table 1) were calculated using SPSS 14 (SPSS, Chicago, IL, USA).
Quantitative real-time PCR
Reverse transcription was performed using standard protocols. Quantitative Real-Time PCR (TaqMan) was carried out as described previously [18] using the following predesigned Assays-on-demand (Applied Biosystems) with beta-Actin as housekeeping gene: ACTB (Hs 030023880_g1), CD74 (Hs 00959496_m1), HLA-DMA (Hs 00157941_m1), CXCL11 (Hs 00171138_m1), CXCL9 (Hs 00970538_m1), IDO1 (Hs 00158032_m1). RT-PCR was run in a 7000 Sequence Detection System (Applied Biosystems) under the following conditions: 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Data analysis was performed according to the ΔΔCt method wherein β-Actin was used as reference gene and colorectal carcinoma cells HT29 as calibrator [19]. Significance of the differences in gene expression between samples of patients with or without later metastasis as measured by qPCR were analyzed by Mann–Whitney test using SPSS 14 (SPSS, Chicago, IL, USA).
Study design
This study includes retrospective cases, which were stratified as to contain mainly UICC II cases. Samples were taken from 1996 to 2004. Follow-up data were obtained for at least 41 months or until metastasis of disease with a follow-up time of 41–115 months (median: 77 months) for non-recurrent patients. The clinical endpoint examined was distant metastasis of disease. The number of samples initially available for this study was largely reduced because of the application of stringent quality criteria regarding sample characteristics, RNA and microarray quality (see above) and stringent criteria for the inclusion of patients.
Results
Microarray data analysis
Data analysis of gene expression profiles obtained by a comprehensive genome-wide gene expression study using oligonucleotide microarrays of 45 patients with CRC (Table 1) led to the identification of 44 probes that enabled us to discriminate between patients with distant metastasis during a follow-up time of 41 to 115 months after surgery and those who did not (Table 2). Of the 44 probes, 30 were associated with genes that had functional annotations including a gene symbol (using the hgug4112a annotation package for Bioconductor, version 2.2.11). GO-term enrichment analysis showed that the most significantly enriched terms were “immune system process”, associated with 14 genes, and “immune response”, associated with 13 genes, with the latter group being a subgroup of the former (Table 3). Only 11 of the top 47 most enriched GO terms were not directly related to immune system processes, and some of those 11 GO terms such as “response to stimulus”, “cell-cell signals”, and “taxis” might be also related to immune response mechanisms.
Predictive value of the immune response signature
The 14 genes associated to immune system processes were used for further analysis. Hierarchical clustering of the hybridization values of the corresponding 14 probes produced a branch that consisted of a majority of patients with metachronous metastases (Figure 1). The analysis suggests that the metastasized cases have more of these 14 genes expressed at low levels than non-recurrent patients, although not always the same genes. In fact, all of the 14 genes in the immune related signature have average values of expression that are lower in the set of metastasized cases than in the set of non-recurrent patients (Figure 2). According to these observations, we devised a simple classifier using the hybridization values of the probes associated to the 14 immune system related genes. If at least a certain number of the 14 probes are below their median hybridization values in all other samples, then that sample is predicted to be metastatic. By ROC-curve analysis of this gene signature (Figure 3), a cut-off of 8.5 probes was selected. This means that if at least 9 of the putative marker probes show a value below their median expression value, then that sample is classified as a case that would suffer later from metachronous metastases. This cut-off yields a specificity of 79% and a sensitivity of 75%.

Clustering of the cohort using 14 selected immune response genes. Hierarchical clustering of 45 colorectal carcinomas, using a gene expression signature composed of 14 genes related to the immune system. Each row represents a probe set for a gene and each column a sample. Samples marked in blue are metastatic cases, samples marked in bright green are cases showing no metastasis at least 3 years after surgery. The length and the subdivision of branches display the relation of the samples based on their similarity in the expression of the 14 genes. Most of the metastatic cases are grouped in the branch marked with the asterisk (*). Metastatic cases have distinctly more of these 14 genes expressed at low expression values (blue, low expression) than non-recurrent patients (red, high expression). The probe hybridization values have been scaled and centered row-wise.

Normalized expression values. The expression values of the 14 immune system-related genes were normalized and median centered. Depicted are the normalized expression values for each of the immune response genes in non-recurrent (n = 33) and metastatic (n = 12) cases.

Predictive value of the immune response signature. (A) ROC-curve of a classifier for metastasis based on number of immune system related probes (brown curve, 14 genes) expressed below the median of the whole set. A cut-off of 8.5 probes (marked with ) was selected based on the ROC-curve shown in this figure (AUC = 0.817). This cut-off yields a specificity of 79% and a sensitivity of 75% (asymptotic significance = 0.001, asymptotic 95% confidence interval = 0.678 – 0.956). For comparison, ROC-curves using one probe for each of the 30 selected genes (related or not to immune response; green curve, 30 genes), or all their associated probes (one gene may have more than one associated probe; blue curve, 44 genes), were not very different. (B) Kaplan Maier-Curve showing cumulated disease-free survival vs. disease-free survival time using the cut-off of 8.5 as described in Figure 3; p = 0.002 (logRank Mantel-Cox), n = 45.
Validation of the microarray results
For validation of microarray results qPCR was performed employing the five randomly chosen genes CD74, CXCL9, CXCL11, HLA-DMA and IDO1 from the 14 immune system related genes found by microarray analysis. Here, we used a carefully selected collection of the previously samples encompassing 11 patients which showed later metastasis and 11 patients without metastasis during the follow-up time which are similar in age, gender and tumor staging (Table 4). The results of the qPCR analysis showed that the expression of these genes is lower in cases with later metastasis for all of the examined genes, thereby confirming the microarray results (Figure 4). Differences in gene expression between both groups are significant (less than 5%), except for CD74 which shows only a trend into this direction.

Validation of the marker gene expression by quantitative RT-PCR. Expression of five marker genes (IDO1, CXCL9, CD74, HLA-DMA, CXCL11) in primary colorectal carcinomas with (always on the right side, REC = 1, n = 11) or without later metastasis (left side, REC = 0, n = 11) as determined by qRT-PCR (TaqMan). Relative amount of expression is shown in Box – Whisker - Plots. Gray columns show a 50% range of the data surrounding the median; black lines within each column mark the median; asterisks mark outliers. Differences between REC = 0 and REC = 1 groups are significant (less than 5%), except for CD74.
Discussion
One characteristic feature of the current study is its stringency with regard to sample collection and RNA/microarray quality controls. Primary CRC can be divided into two main groups, firstly tumors with chromosome instability (CIN) showing high rates of chromosome losses and gains and secondly tumors with microsatellite instability (MSI) characterized by genome-wide changes in repetitive sequences due to defects in the DNA mismatch repair system [20]. Since these two groups of CRC also differ in many aspects including survival of the patients [21], microsatellite instability of all tumors was tested and MSI samples were excluded. This strategy thus strengthens sample homogeneity.
Using a novel way of microarray data analysis (see Data analysis section) a gene signature containing 44 probe sets was identified (Table 2), which was predictive of cancer metastasis. Almost half of the genes were related to immune system processes and immune response. Obviously, one may assume that not only the specific characteristics of the tumor cells themselves, such as angiogenesis, invasion, and proliferation or apoptosis, are relevant for metastasis but also the immune response of the host organism. In fact, it is clear that the host immune response is an important factor affecting cancer progression [22]. However, the complex interplay between the tumor’s immunogenicity, the host’s immune response and the cancer cells’ capabilities of evading immune response is still a challenging subject of current research. CRCs are immunogenic and provoke the host’s immune response, its strength being related to the patient’s prognosis [23]. High concentrations of neutrophils [24], high preoperative levels of natural killer cells [25], high numbers of mast cells [26, 27], a high percentage of CD4+ T cells [28], the infiltration of the tumor with high numbers of central memory T cells and high CD8+ T cell counts [29], for example, have been found to correlate with a better prognosis. Yet, under the selective pressure of the immune system, cancers “escape” by becoming less immunogenic e.g. by failing to express MHC class I antigens – a process called immunoediting [30]. Apart from that, there is evidence that the cancer itself has a direct immunosuppressive effect, which starts at the molecular and cellular level and may lead to a basal shift in immune function [31].
Here, our analysis showed that our prognostic gene signature of 44 genes includes 14 genes related to immune response (Figure 1, Figure 2) that are down-regulated in primary carcinomas which later metastasize. In general, we hypothesize that the reduced expression of these 14 prognostic immune response genes impedes activation of CD4 T-cells, mostly involving the MHC class II pathway. Each of the 14 genes’ roles in this process is discussed in the following section. Validation experiments using qPCR confirmed downregulation of CD74, CXCL9, CXCL11, HLA-DMA and IDO1.
Reduced expression of class II molecules might impede activation of CD4 T cells
The ability of the immune system to recognize and defend against the introduction of foreign antigens depends on the ability of the host’s MHC class II, which presents peptides degraded in intracellular vesicles to circulating CD4 T cells. Class II molecules such as HLA-DMA are immunological proteins vital to the proper loading and presentation of these peptides in macrophages, immature dendritic cells, B cells, and other antigen presenting cells. HLA-DPB, HLA-DRB4, and HLA-DRB5 also belong to this group. Here, a downregulation of components of the MH) class II was found in primary carcinomas of patients with later metastasizing tumors. In accordance, Lovig et al. [32] found that patients positive for the HLA-DR determinants showed better survival than those without HLA-DR expression. Similarly, in MSI tumors with a good prognosis an upregulation of HLA-DMA was noted [33]. Furthermore, HLA-DPB1 has been found to be downregulated in metastasizing primary tumors of different origins, including colorectum [34]. Down-regulation of four MHC class II molecules has been reported to be significant for primary tumors of hepatocellular carcinoma which show later metastases [35]. Recently, the presence of inactivating mutations in the HLA-A gene was reported for squamous lung carcinomas [36].
Reduced expression of CD74, CD86, and CCL18 might impede activation of CD4 T cells
CD74 (HLA-DR-associated invariant chain) plays a critical role in MHC class II antigen processing by stabilizing peptide-free class II alpha/beta heterodimers in a complex soon after their synthesis and directing transport of the complex from the endoplasmic reticulum to compartments where peptide loading of class II takes place. CD86 (Cluster of Differentiation 86) is expressed on antigen-presenting cells that provide co-stimulatory signals necessary for T cell activation and survival [37]. CCL18 is relevant for activation of T-cells through MHC class II, and is a marker for tumor-associated macrophages [38]. Moreover, it is a prognostic marker in gastric cancer and probably plays a role in the physiological homing of lymphocytes and dendritic cells as well as in the generation of primary immune responses [39, 40].
Reduced expression of CXCL9 and CXCL11 might impede activation of T cells
CXCL9 and CXCL11 (Chemokine (C-X-C motif) ligands 9 and 11) are small cytokines belonging to the CXC chemokine family also known as “Monokine induced by gamma interferon” [41]. They are produced by three different cell types, monocytes, endothelial cells and fibroblasts, play a role as T-cell chemoattractants and are secreted in response to IFN-γ. Chemokines CXCL9, CXCL10 and CXCL11 are closely related. All three genes are located on human chromosome 4 and they all elicit their chemotactic functions by interacting with the chemokine receptor CXCR3.
A conflicting result concerning IDO1 (Indoleamine-2,3-Dioxygenase)
Elevated tryptophan catabolism in the urine and blood of tumor-bearing patients has been recognized for many decades. For example, biopsy samples taken from CRC patients show an overexpression of IDO1 [42]. Although expression did not correlate with patient survival, increased IDO1 expression did correlate with liver metastasis. However, another study in patients with HCC showed that IDO1 expression in tumor specimens was positively correlated with progression-free survival [43]. Moreover, a significant inverse correlation between the density of IDO1-positive microvessels and the number of proliferating tumor cells in primary and metastatic renal cell carcinoma was found [44]. Increased expression of IDO1 in endothelial cells of tumors correlated positively with long-term patient survival. Thus, these data suggest that IDO1 can limit tumor growth. Similarly, in our study we found an increase of IDO1 expression in primary carcinomas with good prognosis. The opposing effects of IDO1 have been discussed recently [45].
Enrichment in immune response functions among the set of genes of prognostic relevance is in agreement with other studies. CXCL9 and CXCL11 – as well as IDO1 - are part of a published prognostic signature that predicts metastasis in CRC [46]. Using Affymetrix arrays Lin et al. aimed to develop gene classifiers to predict colorectal cancer metastasis. Eleven of 19 genes in the classifier were involved in the immune response. In agreement with our results all of the 11 immune response genes were down-regulated in metastatic cases [46]. Accordingly, a comprehensive study using different assays with the aim to elucidate the mechanisms underlying immune response in CRC showed that a high expression of CXCL9 and CXCL10 is correlated with a favorable outcome of this disease [47]. Furthermore, CXCL9 and IDO1 have been shown to be prognostic markers in breast cancer [48]. Only recently, 15 immune response genes, among them IGHA1, IGHG1 and IGL@ were found to be part of a 128 genes signature that predicted metastasis in CRC [8].
Conclusions
Whereas up to now only gene signatures containing genes of various biological functions have been described for prediction of metastasis in CRC, in this study metastasis could be well predicted by a set of gene expression markers consisting exclusively of genes related to the MHC class II complex clearly involved in immune response. From our data we cannot state whether the later recurring tumor is the cause or the beneficiary of the suppressed immune response. Nevertheless, our data show that the proper function of a comprehensive network of immune response genes is of vital importance for the survival of colorectal cancer patients. Recent results indicating that the tumor microenvironment can reduce the maturation of dendritic cells [49, 50] hint to the importance of our findings and suggest avenues for prognosis and treatment.
Abbreviations
- CRC:
-
Colorectal cancer
- GO:
-
Gene ontology
- SVM:
-
Support vector machine.
References
Union E: Europe against Colorectal Cancer, Declaration of Brussels. 2007, European Union: Brussels
Markowitz SD, Bertagnolli MM: Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med. 2009, 361: 2449-2460. 10.1056/NEJMra0804588.
Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N: Colorectal cancer. Lancet. 2010, 375: 1030-1047. 10.1016/S0140-6736(10)60353-4.
Wang Y, Jatkoe T, Zhang Y, Mutch MG, Talantov D, Jiang J, McLeod HL, Atkins D: Gene expression profiles and molecular markers to predict recurrence of Dukes' B colon cancer. J Clin Oncol. 2004, 22: 1564-1571. 10.1200/JCO.2004.08.186.
Eschrich S, Yang I, Bloom G, Kwong KY, Boulware D, Cantor A, Coppola D, Kruhoffer M, Aaltonen L, Orntoft TF, Quackenbush J, Yeatman TJ: Molecular staging for survival prediction of colorectal cancer patients. J Clin Oncol. 2005, 23: 3526-3535. 10.1200/JCO.2005.00.695.
Arango D, Laiho P, Kokko A, Alhopuro P, Sammalkorpi H, Salovaara R, Nicorici D, Hautaniemi S, Alazzouzi H, Mecklin JP, Jarvinen H, Hemminki A, Astola J, Schwartz S, Aaltonen LA: Gene-expression profiling predicts recurrence in Dukes' C colorectal cancer. Gastroenterology. 2005, 129: 874-884. 10.1053/j.gastro.2005.06.066.
Pillaire MJ, Selves J, Gordien K, Gourraud PA, Gentil C, Danjoux M, Do C, Negre V, Bieth A, Guimbaud R, Trouche D, Pasero P, Mechali M, Hoffmann JS, Cazaux C: A 'DNA replication' signature of progression and negative outcome in colorectal cancer. Oncogene. 2010, 29: 876-887. 10.1038/onc.2009.378.
Jorissen RN, Gibbs P, Christie M, Prakash S, Lipton L, Desai J, Kerr D, Aaltonen LA, Arango D, Kruhoffer M, Orntoft TF, Andersen CL, Gruidl M, Kamath VP, Eschrich S, Yeatman TJ, Sieber OM: Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clin Cancer Res. 2009, 15: 7642-7651. 10.1158/1078-0432.CCR-09-1431.
Barrier A, Lemoine A, Boelle PY, Tse C, Brault D, Chiappini F, Breittschneider J, Lacaine F, Houry S, Huguier M, Van der Laan MJ, Speed T, Debuire B, Flahault A, Dudoit S: Colon cancer prognosis prediction by gene expression profiling. Oncogene. 2005, 24: 6155-6164. 10.1038/sj.onc.1208984.
Barrier A, Boelle PY, Roser F, Gregg J, Tse C, Brault D, Lacaine F, Houry S, Huguier M, Franc B, Flahault A, Lemoine A, Dudoit S: Stage II colon cancer prognosis prediction by tumor gene expression profiling. J Clin Oncol. 2006, 24: 4685-4691. 10.1200/JCO.2005.05.0229.
Zhang X, Lu X, Shi Q, Xu XQ, Leung HC, Harris LN, Iglehart JD, Miron A, Liu JS, Wong WH: Recursive SVM feature selection and sample classification for mass-spectrometry and microarray data. BMC Bioinformatics. 2006, 7: 197-10.1186/1471-2105-7-197.
Tusher VG, Tibshirani R, Chu G: Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001, 98: 5116-5121. 10.1073/pnas.091062498.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J: Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5: R80-10.1186/gb-2004-5-10-r80.
Huber W, von Heydebreck A, Sueltmann H, Poustka A, Vingron M: Parameter estimation for the calibration and variance stabilization of microarray data. Stat Appl Genet Mol Biol. 2003, 2: Article3-
Kauffmann A, Gentleman R, Huber W: arrayQualityMetrics–a bioconductor package for quality assessment of microarray data. Bioinformatics. 2009, 25: 415-416. 10.1093/bioinformatics/btn647.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I: Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001, 125: 279-284. 10.1016/S0166-4328(01)00297-2.
Falcon S, Gentleman R: Using GOstats to test gene lists for GO term association. Bioinformatics. 2007, 23: 257-258. 10.1093/bioinformatics/btl567.
Astrosini C, Roeefzaad C, Dai YY, Dieckgraefe BK, Jons T, Kemmner W: REG1A expression is a prognostic marker in colorectal cancer and associated with peritoneal carcinomatosis. Int J Cancer. 2008, 123: 409-413. 10.1002/ijc.23466.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001, 25: 402-408. 10.1006/meth.2001.1262.
Jass JR: Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007, 50: 113-130. 10.1111/j.1365-2559.2006.02549.x.
Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S: Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003, 349: 247-257. 10.1056/NEJMoa022289.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F: Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006, 313: 1960-1964. 10.1126/science.1129139.
Salama P, Platell C: Host response to colorectal cancer. ANZ J Surg. 2008, 78: 745-753. 10.1111/j.1445-2197.2008.04642.x.
Kishimoto T, Saito K, Ishikura H: [Mechanism of blood-borne metastasis in relation to the interaction between pancreatic carcinoma and endothelial cells]. Nippon Rinsho. 1995, 53: 1765-1769.
Koda K, Saito N, Takiguchi N, Oda K, Nunomura M, Nakajima N: Preoperative natural killer cell activity: correlation with distant metastases in curatively research colorectal carcinomas. Int Surg. 1997, 82: 190-193.
Nagtegaal ID, Marijnen CA, Kranenbarg EK, Mulder-Stapel A, Hermans J, van de Velde CJ, van Krieken JH: Local and distant recurrences in rectal cancer patients are predicted by the nonspecific immune response; specific immune response has only a systemic effect–a histopathological and immunohistochemical study. BMC Cancer. 2001, 1: 7-10.1186/1471-2407-1-7.
Nielsen HJ, Hansen U, Christensen IJ, Reimert CM, Brunner N, Moesgaard F: Independent prognostic value of eosinophil and mast cell infiltration in colorectal cancer tissue. J Pathol. 1999, 189: 487-495. 10.1002/(SICI)1096-9896(199912)189:4<487::AID-PATH484>3.0.CO;2-I.
Canna K, McArdle PA, McMillan DC, McNicol AM, Smith GW, McKee RF, McArdle CS: The relationship between tumour T-lymphocyte infiltration, the systemic inflammatory response and survival in patients undergoing curative resection for colorectal cancer. Br J Cancer. 2005, 92: 651-654. 10.1038/sj.bjc.6602419.
Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, Meatchi T, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Galon J: Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005, 353: 2654-2666. 10.1056/NEJMoa051424.
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD: Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002, 3: 991-998. 10.1038/ni1102-991.
Kloor M, Michel S: Immune evasion of microsatellite unstable colorectal cancers. Int J Cancer. 2010, 127: 1001-1010. 10.1002/ijc.25283.
Lovig T, Andersen SN, Thorstensen L, Diep CB, Meling GI, Lothe RA, Rognum TO: Strong HLA-DR expression in microsatellite stable carcinomas of the large bowel is associated with good prognosis. Br J Cancer. 2002, 87: 756-762. 10.1038/sj.bjc.6600507.
Bustin SA, Li SR, Phillips S, Dorudi S: Expression of HLA class II in colorectal cancer: evidence for enhanced immunogenicity of microsatellite-instability-positive tumours. Tumour Biol. 2001, 22: 294-298. 10.1159/000050630.
Ramaswamy S, Ross KN, Lander ES, Golub TR: A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003, 33: 49-54.
Matoba K, Iizuka N, Gondo T, Ishihara T, Yamada-Okabe H, Tamesa T, Takemoto N, Hashimoto K, Sakamoto K, Miyamoto T, Uchimura S, Hamamoto Y, Oka M: Tumor HLA-DR expression linked to early intrahepatic recurrence of hepatocellular carcinoma. Int J Cancer. 2005, 115: 231-240. 10.1002/ijc.20860.
Network CGA: Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012, 489: 519-525. 10.1038/nature11404.
Van Gool SW, Vandenberghe P, De BM, Ceuppens JL: CD80, CD86 and CD40 provide accessory signals in a multiple-step T-cell activation model. Immunol Rev. 1996, 153: 47-83. 10.1111/j.1600-065X.1996.tb00920.x.
Kodelja V, Muller C, Politz O, Hakij N, Orfanos CE, Goerdt S: Alternative macrophage activation-associated CC-chemokine-1, a novel structural homologue of macrophage inflammatory protein-1 alpha with a Th2-associated expression pattern. J Immunol. 1998, 160: 1411-1418.
Leung SY, Yuen ST, Chu KM, Mathy JA, Li R, Chan AS, Law S, Wong J, Chen X, So S: Expression profiling identifies chemokine (C-C motif) ligand 18 as an independent prognostic indicator in gastric cancer. Gastroenterology. 2004, 127: 457-469. 10.1053/j.gastro.2004.05.031.
Schutyser E, Richmond A, Van-Damme J: Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J Leukoc Biol. 2005, 78: 14-26. 10.1189/jlb.1204712.
Sun B, Nishihira J, Yoshiki T, Kondo M, Sato Y, Sasaki F, Todo S: Macrophage migration inhibitory factor promotes tumor invasion and metastasis via the Rho-dependent pathway. Clin Cancer Res. 2005, 11: 1050-1058.
Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, Winkler C, Werner ER, Werner-Felmayer G, Weiss HG, Gobel G, Margreiter R, Konigsrainer A, Fuchs D, Amberger A: Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res. 2006, 12: 1144-1151. 10.1158/1078-0432.CCR-05-1966.
Ishio T, Goto S, Tahara K, Tone S, Kawano K, Kitano S: Immunoactivative role of indoleamine 2,3-dioxygenase in human hepatocellular carcinoma. J Gastroenterol Hepatol. 2004, 19: 319-326. 10.1111/j.1440-1746.2003.03259.x.
Riesenberg R, Weiler C, Spring O, Eder M, Buchner A, Popp T, Castro M, Kammerer R, Takikawa O, Hatz RA, Stief CG, Hofstetter A, Zimmermann W: Expression of indoleamine 2,3-dioxygenase in tumor endothelial cells correlates with long-term survival of patients with renal cell carcinoma. Clin Cancer Res. 2007, 13: 6993-7002. 10.1158/1078-0432.CCR-07-0942.
Lob S, Konigsrainer A, Rammensee HG, Opelz G, Terness P: Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees?. Nat Rev Cancer. 2009, 9: 445-452. 10.1038/nrc2639.
Lin YH, Friederichs J, Black MA, Mages J, Rosenberg R, Guilford PJ, Phillips V, Thompson-Fawcett M, Kasabov N, Toro T, Merrie AE, van Rij A, Yoon HS, McCall JL, Siewert JR, Holzmann B, Reeve AE: Multiple gene expression classifiers from different array platforms predict poor prognosis of colorectal cancer. Clin Cancer Res. 2007, 13: 498-507. 10.1158/1078-0432.CCR-05-2734.
Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, Camus M, Gillard M, Bruneval P, Fridman WH, Pages F, Trajanoski Z, Galon J: Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology. 2010, 138: 1429-1440. 10.1053/j.gastro.2009.10.057.
Specht K, Harbeck N, Smida J, Annecke K, Reich U, Naehrig J, Langer R, Mages J, Busch R, Kruse E, Klein-Hitpass L, Schmitt M, Kiechle M, Hoefler H: Expression profiling identifies genes that predict recurrence of breast cancer after adjuvant CMF-based chemotherapy. Breast Cancer Res Treat. 2008, 118: 45-56.
Michielsen AJ, Noonan S, Martin P, Tosetto M, Marry J, Biniecka M, Maguire AA, Hyland JM, Sheahan KD, O'Donoghue DP, Mulcahy HE, Fennelly D, Ryan EJ, O'Sullivan JN: Inhibition of dendritic cell maturation by the tumor microenvironment correlates with the survival of colorectal cancer patients following bevacizumab treatment. Mol Cancer Ther. 2012, 11: 1829-1837. 10.1158/1535-7163.MCT-12-0162.
Michielsen AJ, Hogan AE, Marry J, Tosetto M, Cox F, Hyland JM, Sheahan KD, O'Donoghue DP, Mulcahy HE, Ryan EJ, O'Sullivan JN: Tumour tissue microenvironment can inhibit dendritic cell maturation in colorectal cancer. PLoS One. 2011, 6: e27944-10.1371/journal.pone.0027944.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/14/64/prepub
Acknowledgements
The authors are particularly grateful to Prof. Dr. P.M. Schlag from the Charité Comprehensive Cancer Center (CCCC) for supplying frozen samples and for fruitful discussions. The authors thank Ms. Sabine Grigull, Ms. Bianca Kochnowsky, Ms. Christina Krüger and Ms. Carola Meier for excellent technical assistance and Dr. Wolfgang Haensch for careful pathological review of the samples. This work was supported by a grant from the BMBF “InnoRegio Gesundheitsregion Berlin-Buch”. Dr. Miguel Andrade acknowledges funding from The Helmholtz Alliance on Systems Biology (Helmholtz-Gemeinschaft Deutscher Forschungszentren). The sponsors were not involved in any decisions regarding this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conception and design: MAA and WK; data acquisition: MF and TJ; data analysis and interpretation: MRH, MF, TJ and WK; manuscript writing: MF,MAA and WK. All authors read and approved the final manuscript.
Marion Fehlker, Matthew R Huska contributed equally to this work.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fehlker, M., Huska, M.R., Jöns, T. et al. Concerted down-regulation of immune-system related genes predicts metastasis in colorectal carcinoma. BMC Cancer 14, 64 (2014). https://doi.org/10.1186/1471-2407-14-64
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-14-64