
Abstract
Background
Myeloid cell leukemia-1 (Mcl-1) is a member of the Bcl-2 family, which inhibits cell apoptosis by sequestering pro-apoptotic proteins Bim and Bid. Mcl-1 overexpression has been associated with progression in leukemia and some solid tumors including prostate cancer (PCa). However, the regulatory mechanism for Mcl-1 expression in PCa cells remains elusive.
Results
Immunohistochemical analyses revealed that Mcl-1 expression was elevated in PCa specimens with high Gleason grades and further significantly increased in bone metastasis, suggesting a pivotal role of Mcl-1 in PCa metastasis. We further found that vascular endothelial growth factor (VEGF) is a novel regulator of Mcl-1 expression in PCa cells. Inhibition of endogenous Mcl-1 induced apoptosis, indicating that Mcl-1 is an important survival factor in PCa cells. Neuropilin-1 (NRP1), the "co-receptor" for VEGF165 isoform, was found to be highly expressed in PCa cells, and indispensible in the regulation of Mcl-1. Intriguingly, VEGF165 promoted physical interaction between NRP1 and hepatocyte growth factor (HGF) receptor c-MET, and facilitated c-MET phosphorylation via a NRP1-dependent mechanism. VEGF165 induction of Mcl-1 may involve rapid activation of Src kinases and signal transducers and activators of transcription 3 (Stat3). Importantly, NRP1 overexpression and c-MET activation were positively associated with progression and bone metastasis in human PCa specimens and xenograft tissues.
Conclusions
This study demonstrated that Mcl-1 overexpression is associated with PCa bone metastasis. Activation of VEGF165-NRP1-c-MET signaling could confer PCa cells survival advantages by up-regulating Mcl-1, contributing to PCa progression.
Similar content being viewed by others


Background
Acquisition of apoptosis resistance is characteristic of invasive tumor cells. Elevated expression of anti-apoptotic proteins is associated with tumor progression clinically and experimentally [1]. Myeloid cell leukemia-1 (Mcl-1), a member of the Bcl-2 family, sequesters pro-apoptotic proteins Bim and Bid, thereby inhibiting mitochondrial outer membrane permeabilization, a central control point of apoptosis [2, 3]. Mcl-1 overexpression is associated with progression in leukemia [4] and some solid tumors including prostate cancer (PCa) [5–7]. Mcl-1 was elevated in primary PCa with high Gleason grades and metastatic tumors compared to that in prostatic intraepithelial neoplasia (PIN) or lower grade tumors, suggesting a pivotal role of Mcl-1 in PCa progression [5].
Angiogenesis favors tumor cell survival, thereby contributing to progression [1]. Vascular endothelial growth factor (VEGF) is a critical pro-angiogenic factor that induces proliferation and migration of endothelial cells within tumor vasculature [8]. VEGF is expressed as several alternately spliced isoforms. VEGF165 is pre dominant, with optimal bioavailability, and responsible for VEGF biological potency, whereas VEGF121 is less potent but freely diffusible. VEGF binds two highly-related receptor tyrosine kinases, VEGF-R1 and VEGF-R2 [8]. Neuropilin-1 (NRP1) was originally identified as a receptor for the semaphorin 3 subfamily mediating neuronal guidance and axonal growth [9]. It was subsequently found to specifically bind VEGF165 but not VEGF121 on endothelial cells and tumor cells [9, 10]. NRP1 lacks a typical kinase domain, primarily functioning as a "co-receptor" to form ligand-specific receptor complexes. In response to VEGF165, NRP1 couples with VEGF-Rs to signal in endothelial cells. Though VEGF-R1 and VEGF-R2 are usually absent or expressed at very low levels in PCa cells [11], aberrant upregulation of NRP1 has been frequently observed in high grade and metastatic PCa and other solid tumors [9, 12–16]. Ectopic expression of NRP1 in PCa cells induced cell migration, increased tumor size and microvessel density, and inhibited apoptosis [17]. These observations suggested that NRP1 may be critical for PCa progression. Nonetheless, the mechanism by which NRP1 transmits VEGF signaling in PCa cells lacking VEGF-Rs remains unclear.
Previously we reported that serum VEGF levels correlate to bone metastatic status in PCa patients, and activation of VEGF signaling in PCa cells is associated with invasive phenotypes in experimental models [18]. In this study, we correlate Mcl-1 overexpression to PCa progression towards bone metastasis, and provide evidence that VEGF regulates Mcl-1 expression through NRP1-dependent activation of c-MET in PCa cells.
Results
Elevated Mcl-1 expression is associated with PCa progress and bone metastasis
To investigate the clinical significance of Mcl-1 in PCa progression, immunohistochemical (IHC) analyses were performed to determine the expression of Mcl-1 in a human PCa tissue microarray with matched normal adjacent tissue and bone metastatic bone specimens (Figure 1a). We defined tumors with Gleason score 2-6 as well-differentiated (n = 2), Gleason score 7 as intermediately-differentiated (n = 26) and Gleason score 8-10 as poorly-differentiated (n = 43). Mcl-1 immunointensity was increased from normal tissues to well-differentiated cancer and further elevated in high grade PCa, although the difference between Mcl-1 intensity in intermediate- and poorly-differentiated cancers was not statistically significant (p = 0.93). Intriguingly, Mcl-1 staining in bone metastatic tumors (n = 6) was remarkably increased compared to that in either intermediate- or poorly-differentiated PCa (p < 0.002). These data correlated elevated Mcl-1 expression to clinical PCa progression, particularly bone metastasis.

Mcl-1 is a survival factor in human PCa cells. (a) IHC staining of Mcl-1 in human PCa tissue microarray consisting of normal adjacent tissues, primary PCa and bone metastases. (b) Intracardiac (i.c) injection of ARCaPM cells in athymic mice resulted in metastases to bone and soft tissues. The ARCaPM-C2 subclone was derived from metastatic bone tissues after two rounds of intracardiac inoculation of ARCaPM cells. (c) Endogenous Mcl-1 expression in the ARCaPM model was examined by RT-PCR and western blotting analyses. (d) Effect of Mcl-1 siRNA on ARCaPM cell viability. Subconfluent ARCaPM cells on a 6-well plate were transiently transfected with Mcl-1 siRNA (30 nM) for 72 h. Endogenous expression of Mcl-1 at the mRNA and protein levels, and cell viability of ARCaPM cells as counted with trypan blue staining, were significantly inhibited by Mcl-1 siRNA treatment compared to the control. (e) Effects of Mcl-1 siRNA on ARCaPM cell apoptosis. Subconfluent ARCaPM cells were transfected with Mcl-1 siRNA or control siRNA for 72 h, expression of annexin V was measured by FACS.
Mcl-1 is a survival factor in human PCa cells
We have established several lines of human PCa models that closely mimic the clinical progression of PCa bone metastasis, including the ARCaP model [19, 20]. When inoculated either orthotopically or intracardially, ARCaPM cells are capable of metastasizing spontaneously to bone and soft tissues, forming mixed osteoblastic and osteolytic lesions in mouse skeleton [19, 21, 22]. The ARCaPM-C2 subclone, derived from metastatic bone tissues after two rounds of intracardiac injection of ARCaPM cells in athymic mice, forms predictable metastases to bone, adrenal gland and other soft tissue with higher propensity and shorter latency [21, 22] (Figure 1b). Reverse transcription-PCR (RT-PCR) and immunoblotting analyses found that Mcl-1 was substantially expressed by ARCaPM cells, and further increased in ARCaPM-C2 cells (Figure 1c). Similarly, increased Mcl-1 expression was observed in metastatic C4-2 and C4-2B cells [23] when compared to their parental, androgen-dependent human PCa cell line LNCaP (Additional file 1, Figure S1a). These results suggested a possible association between Mcl-1 expression and invasive phenotypes of PCa cells.
To examine the function of Mcl-1 in PCa cell survival, a Mcl-1 small-interfering RNA (siRNA) was transfected into ARCaPM cells. siRNA treatme nt effectively inhibited Mcl-1 expression and significantly reduced ARCaPM cell viability by ~36% after 72 h (Figure 1d). Annexin V staining by fluorescence-activated cell sorting (FACS) analysis showed that the Mcl-1 siRNA induced apoptosis in 24.4-2.3% of ARCaPM cells (Figure 1e). These results indicated that Mcl-1 may be an important survival factor in PCa cells.
VEGF regulates Mcl-1 expression in human PCa cells
Previously we reported that ARCaPM cells express high levels of endogenous VEGF, regulated by a cyclic AMP-response element binding protein (CREB)-hypoxia-inducible factor (HIF)-dependent mechanism in normoxic conditions [18]. In the present study, RT-PCR assay that could differentiate mRNA expression of VEGF165 and VEGF121 isoforms [18] was performed in ARCaPM and ARCaPM-C2 cells, showing a significant increase in the expression of both VEGF isoforms in ARCaPM-C2 cells, as confirmed at protein level by enzyme-linked immunosorbent assay (ELISA) (Figure 2a). Similarly, C4-2 cells express higher levels of VEGF when compared to LNCaP cells (Additional file 1, Figure S1b). These data suggested that VEGF expression is elevated in metastatic PCa cells.

VEGF 165 regulates Mcl-1 expression in PCa cells. (a) VEGF expression in ARCaPM and ARCaPM-C2 cells, as determined by RT-PCR and ELISA (conditioned medium). (b) Effects of recombinant VEGF165 on cell proliferation as determined by MTS assay. Top, ARCaPM cells were seeded in 96-well plates (1 × 103cells/well) for 24 h, and serum-starved overnight. The cells were further incubated with varying concentrations of VEGF165 in serum-free T-medium for 72 h. Bottom, ARCaPM cells were incubated with PBS or VEGF165 (50 ng/ml) in serum-free T-medium for varying times. (c) VEGF165 effects on Mcl-1 expression. ARCaPM cells were treated with VEGF165 at indicated concentrations and times, and Mcl-1 mRNA expression was measured. For immunoblotting, ARCaPM cells were incubated with VEGF165 (10 ng/ml) for 72 h. (d) Effects of VEGF siRNA on Mcl-1 expression. ARCaPM cells were transfected with VEGF siRNA or control siRNA (80 nM) for 72 h.
VEGF potently stimulates endothelial cell proliferation and migration, an underlying mechanism for angiogenesis during tumor progression. However, neither exogenous human VEGF165 (Figure 2b) nor VEGF121 (Additional file 1, Figure S1c) significantly affected the proliferation of ARCaPM cells at a non-saturating range (5-100 ng/ml), suggesting that VEGF is not a potent mitogen in PCa cells. Intriguingly, VEGF165 was found to rapidly induce Mcl-1 mRNA expression in a dose- and time-dependent manner, with the maximum accumulation of Mcl-1 mRNA after 2 h-incubation at the concentration of 10 ng/ml. Western blot analysis confirmed a remarkable increase of Mcl-1 protein in ARCaPM cells (Figure 2c) and LNCaP cells (Additional file 1, Figure S1d) treated with VEGF165. Conversely, when ARCaPM cells were transfected with a VEGF siRNA nucleotide that selectively inhibited expression of VEGF165 but not VEGF121, expression of Mcl-1, but not Bcl-2, was reduced after 72 h (Figure 2d). Notably, Mcl-1 expression was not affected by VEGF121 treatment (Additional file 1, Figure S1e). These results indicated that VEGF165 may specifically regulate Mcl-1 expression in PCa cells.
Differential expression of VEGF-Rs in PCa cells
Expression of VEGF-Rs was examined in several PCa cell lines with human umbilical vein endothelial cells (HUVEC) as a positive control (Figure 3a, and Additional file 1, Figure S1f). VEGF-R1 was undetectable in PCa cells. Only very low expression of VEGF-R2 could be observed in ARCaPM-C2 cells. However, NRP1 was ubiquitously expressed in PCa cells at a level comparable to that in HUVEC, and higher in metastatic ARCaPM-C2, PC3, C4-2 and C4-2B cells. NRP2, the "co-receptor" for VEGF-C [8], was not detected in PCa cells. These data implied that NRP1 may be the major receptor mediating VEGF effects in PCa cells.

NRP1 is required for basal expression and VEGF 165 induction of Mcl-1 in ARCaP M cells. (a) Expression of VEGF-Rs in PCa cells and HUVEC. (b) Effect of ectopic expression of NRP1 on Mcl-1 basal level. Subconfluent ARCaPM cells on 6-well plates were transfected with pCMV-NRP1 or pCMV-XL4 (16 μg) for 48 h (for RT-PCR) or 72 h (for immunoblotting). (c) Effects of NRP1 siRNA on Mcl-1 basal expression. ARCaPM cells were transfected with NRP1 siRNA or control siRNA (60 nM) for 48 h (for RT-PCR) or 72 h (for immunoblotting). (d) Effects of NRP1 siRNA on VEGF165 induction of Mcl-1. ARCaPM cells were transfected with NRP1 siRNA or control siRNA for 48 h, respectively. Cells were then serum-starved overnight, and treated with VEGF165 (10 ng/ml) or PBS for 2 h.
NRP1 regulates both basal expression and VEGF induction of Mcl-1 in PCa cells
The role of NRP1 in the regulation of Mcl-1 expression was investigated. ARCaPM cells cultured in serum-containing T-medium were transiently transfected with a NRP1 expression vector. Compared to the control, ectopic expression of NRP1 resulted in increased Mcl-1 at both the mRNA and protein levels (Figure 3b). Conversely, transfection with a NRP1 siRNA specifically inhibited NRP1 and reduced endogenous Mcl-1 expression in ARCaPM cells (Figure 3c). These data indicated that NRP1 may be required and sufficient for basal expression of Mcl-1 in PCa cells. Further, ARCaPM cells were transfected with NRP1 siRNA or control siRNA, and incubated with VEGF165 in serum-free T-medium for indicated time. Figure 3d showed that expression of NRP1 siRNA, but not control siRNA, abrogated VEGF165 induction of Mcl-1 in ARCaPM cells. These data indicated an indispensible role of NRP1 in mediating VEGF165 induction of Mcl-1 in PCa cells.
c-MET signaling is required for VEGF regulation of Mcl-1 in PCa cells
Since NRP1 does not contain typical kinase receptor sequences [24], we hypothesized that NRP1 may interact with certain tyrosine kinase receptor(s) to transmit VEGF autocrine signal in PCa cells lacking VEGF-Rs. Two recent studies independently demonstrated that NRP1 physically binds c-MET, and potentiates c-MET activation in response to HGF stimulation in human glioma and pancreatic cancer cells [25, 26]. It is therefore plausible that c-MET may be involved in VEGF regulation of Mcl-1 in PCa cells. Indeed, HGF activation of c-MET signaling has been shown to transcriptionally increase Mcl-1 expression in primary human hepatocytes [27].
A c-MET siRNA construct was transfected into ARCaPM cells, which effectively inhibited endogenous c-MET (Figure 4a). c-MET siRNA treatment reduced Mcl-1 protein expression, suggesting that c-MET is involved in maintaining basal expression of Mcl-1 in PCa cells. Interestingly, however, recombinant HGF treatment did not significantly affect Mcl-1 expression at either RNA or protein levels (Figure 4b), indicating that HGF-dependent activation of c-MET signaling is not sufficient to induce Mcl-1 expression in these cells.

c-MET signaling is required for VEGF 165 induction of Mcl-1 in ARCaP M cells. (a) Effects of c-MET inhibition on Mcl-1 expression. ARCaPM cells were transfected with c-MET siRNA or control siRNA (30 nM) for 72 h. (b) Effects of recombinant HGF treatment (10 ng/ml) on Mcl-1 expression at RNA (0, 2, 4 and 6 h) and protein levels (72 h). (c) ARCaPM cells were transfected with c-MET siRNA or control siRNA for 48 h, serum-starved overnight, and treated with VEGF165 (10 ng/ml) for 2 h. (d) ARCaPM cells were treated with PHA-665752 (0.5 μM) or dimethyl sulfoxide (DMSO) for 2 h, before treatment with VEGF165 (10 ng/ml) or PBS for 2 h.
We further investigated whether c-MET signaling is required for VEGF165 induction of Mcl-1. Indeed, VEGF165 only induced Mcl-1 expression in ARCaPM cells transfected with control siRNA, not in those expressing c-MET siRNA (Figure 4c). PHA-665752, a c-MET selective inhibitor [28], was used to treat ARCaPM cells prior to addition of VEGF165. PHA-665752 significantly attenuated VEGF165 induction of Mcl-1 in ARCaPM cells (Figure 4d). These data indicated that c-MET signaling is required for VEGF regulation of Mcl-1 in PCa cells.
VEGF induces c-MET activation by a NRP1-dependent mechanism in PCa cells
c-MET activation involves phosphorylation of several tyrosine residues including those at positions 1230, 1234, and 1235 [29]. To assess whether VEGF165 could induce c-MET activation, and whether this process was mediated by NRP1, ARCaPM cells were transiently transfected with NRP1 siRNA or control siRNA before VEGF165 treatment. VEGF165 induced rapid phosphorylation of c-MET at Tyr1230/1234/1235 residues in ARCaPM cells transfected with control siRNA, but this effect was significantly attenuated by expression of NRP1 siRNA. Expression of total c-MET protein was not affected by siRNA treatment (Figure 5a). These data indicated that VEGF165 activated c-MET signaling independent of HGF, and NRP1 may be indispensible in this process.

VEGF 165 induces c-MET activation through a NRP1-dependent mechanism. (a) Effects of NRP1 depletion on VEGF165-mediated c-MET phosphorylation. ARCaPM cells were transfected with NRP1 siRNA or control siRNA for 48 h, serum-starved overnight, then treated with VEGF165 (10 ng/ml) for 60 min. (b) Immunoprecipitation assay of NRP1-c-MET interaction. Serum-starved ARCaPM cells were treated with VEGF165 (10 ng/ml) for the indicated times, and immunoprecipitated with anti-NRP1 (upper), or anti-c-MET (low) antibody. (c) Co-localization of NRP1 and c-MET or p-c-MET. Serum-starved ARCaPM cells were treated with VEGF165 (10 ng/ml) or PBS for the indicated times. Immunofluorescence staining of NRP1, c-MET or p-c-MET was performed and visualized by confocal microscopy. Arrows indicate co-localization of NRP1 and c-MET or p-c-MET.
VEGF promotes interaction between NRP1 and c-MET in PCa cells
To explore whether VEGF could induce physical interaction between NRP1 and c-MET, an immunoprecipitation assay was performed in ARCaPM cells treated with VEGF165 for varying times. First, endogenous NRP1 protein was immunoprecipitated (Figure 5b, upper). There was a constitutive association between c-MET and NRP1 in the absence of VEGF165. Upon VEGF165 treatment, presence of c-MET in the NRP1 immunoprecipitates increased at 30 min and returned to baseline at 60 min. Phosphorylated c-MET (p-c-MET) significantly increased at 15 min following VEGF165 treatment, reached a peak at 30 min and slightly decreased in 60 min. Reciprocal immunoprecipitation with anti-c-MET antibody confirmed an association of NRP1 with c-MET in the absence of VEGF165. The presence of NRP1 and p-c-MET in the protein complex exhibited a similar time course following VEGF165 stimulation, with the peak at 30 min (Figure 5b, low).
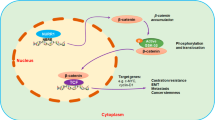
Confocal microscopy was performed to determine whether VEGF165 promotes NRP1 interaction with c-MET and activation of c-MET in ARCaPM cells. NRP1 and c-MET were found to be constitutively associated on plasma membrane, with the intensity of co-localization further increased at 30 min upon VEGF165 treatment (Figure 5c, left panel). Notably, there was a more significant increase in the intensity of co-localization of NRP1 and p-c-MET following VEGF165 stimulation (Figure 5c, right panel). The data independently supported a mechanism that NRP1 may be constitutively associated with c-MET on plasma membrane. Upon VEGF165 binding, NRP1 may further recruit c-MET and facilitate its activation, subsequently transmitting VEGF165 signal (Figure 6).

A proposed model for VEGF 165 regulation of Mcl-1 in PCa cells. NRP1 may be constitutively associated with c-MET on plasma membrane. VEGF165 engagement recruits c-MET into the protein complex and promotes its interaction with NRP1, thereby enhancing phosphorylation of c-MET. Src kinase-Stat3 signaling is subsequently activated, resulting in nuclear translocation of p-Stat3 and activation of Mcl-1 expression. Increased intracellular Mcl-1 protects PCa cells from apoptosis.
Role of Src kinases and signal transducers and activators of transcription 3 (Stat3) in VEGF induction of Mcl-1 in PCa cells
Activation of the Src kinase-Stat3 pathway is an important downstream event in c-MET signaling [30, 31] (refer to Figure 6). Recently, a Stat3 cis-element was identified in human Mcl-1 promoter [32]. We investigated whether the Src kinase-Stat3 pathway is a downstream component in NRP1 signaling in ARCaPM cells (Figure 7a). Indeed, expression of NRP1 siRNA in ARCaPM cells significantly inhibited phosphorylation of Src kinases at Tyr416 (p-Src), as well as activation of Stat3 at Tyr705 (p-Stat3), without altering expression of endogenous Src kinases and Stat3.

Src kinase-Stat3 signaling mediates VEGF 165 induction of Mcl-1 expression in ARCaP M cells. (a) Effects of NRP1 inhibition on phosphorylation of Src kinases and Stat3. ARCaPM cells were transfected with NRP1 siRNA or control siRNA for 72 h. (b) Effects of VEGF165 on the phosphorylation of Src kinases and Stat3. ARCaPM cells were treated with VEGF165 (10 ng/ml) for the indicated time, and immunoblotting was performed on total lysates (left) and nuclear extracts and cytoplasmic proteins (right). (c) Effects of Src kinases on VEGF165 regulation of Mcl-1. ARCaPM cells were treated with PP2 (10 μM) or DMSO for 2 h before treatment with VEGF165 or PBS. (d) Effects of Stat3 depletion on VEGF165 induction of Mcl-1. ARCaPM cells were transfected with Stat3 siRNA or control siRNA (80 nM) for 48 h, serum-starved overnight, then treated with VEGF165 (10 ng/ml) or PBS for 2 h.
Next we examined whether VEGF induces activation of Src kinase-Stat3 signaling in ARCaPM cells (Figure 7b). VEGF165 rapidly induced expression of both p-Src (Tyr416) and p-Stat3 (Tyr705) in a time-dependent manner in ARCaPM cells, with the peak at 60 min. Significantly, VEGF165 promoted rapid intracellular translocation of p-Stat3 (Tyr705) from the cytoplasm to the nucleus, indicating activation of Stat3-dependent gene expression. By contrast, nuclear presence of p-Stat3 (Ser727), which has been associated with HGF-induced Mcl-1 expression in primary human hepatocytes [27], was not affected. These data indicated that Src kinase-Stat3 pathway activation may be an important event following VEGF165 stimulation.
Finally, we assessed the role of Src kinase-Stat3 signaling in VEGF165 regulation of Mcl-1. PP2, a selective inhibitor of Src kinases [33], was used to treat ARCaPM cells before VEGF165 stimulation. PP2 treatment effectively abrogated VEGF165 induction of Mcl-1 (Figure 7c). Similarly, Mcl-1 mRNA expression was rapidly induced by VEGF165 in ARCaPM cells transfected with control siRNA, but not in the cells expressing Stat3 siRNA (Figure 7d). Collectively these data suggested that Src kinase-Stat3 signaling may be required for VEGF induction of Mcl-1 in PCa cells (Figure 6).
NRP1 overexpression and c-MET activation are positively associated with human PCa progression and bone metastasis
To validate the clinical significance of NRP1-c-MET signaling in PCa progression, and avoid the potential bias from using human PCa cell lines, IHC analyses were performed to determine the expression of NRP1 and p-c-MET in human PCa tissue specimens. Prostatic tissue specimens of normal/benign glands, primary and bone metastatic tumors were analyzed. NRP1 expression was increased from normal/benign glands (0/5) or well-differentiated cancer (1/5) to poorly-differentiated cancers (5/5) and bone metastatic tissues (5/5) (Figure 8a). NRP1 staining was also determined in tumor specimens from the ARCaPM xenograft model in which ARCaPM cells were inoculated into athymic mice orthotopically, resulting in skeletal metastases with a short latency [21]. Consistently, NRP1 expression was significantly greater in bone metastatic tumors than in primary tumors (Figure 8b).

Expression of NRP1 and p-c-MET is associated with bone metastatic status of human PCa specimens and the ARCaP M model. IHC analyses of NRP1 expression in human normal/benign, cancerous and metastatic prostatic tissue specimens (a) and primary and bone metastatic tissue specimens from ARCaPM model (b), and of p-c-MET expression in human PCa progression (c) and ARCaPM xenografts (d). Arrows indicate positively-stained cells.
We and others have reported that c-MET overexpression is positively associated with PCa progression [34, 35]. As shown in Figure 8c, p-c-MET was expressed at a low level in normal human prostatic tissue, but increased significantly from well-differentiated and intermediate to poorly-differentiated primary PCa. Importantly, bone metastatic PCa specimens displayed a higher expression of p-c-MET than primary PCa. p-c-MET expression was also remarkably increased in bone metastatic ARCaPM tumors (Figure 8d).
Discussion
Aberrant overexpression of Mcl-1 has been associated with poor prognosis and resistance to chemotherapy in a variety of human cancers [36]. Sensitizing tumor cells to apoptosis induction by selectively targeting Mcl-1, in combination with conventional chemotherapy, has emerged as an attractive therapeutic strategy [3, 37]. In this study, we presented evidence that elevated Mcl-1 expression is associated with clinical PCa progression, particularly bone metastasis. We further showed that activation of VEGF-NRP1-c-MET signaling is responsible for Mcl-1 expression, which may confer survival advantages allowing PCa cells to evade apoptosis and progress towards invasive states (Figure 6).
Only limited information are available on Mcl-1 expression profile in PCa. An early study found that Mcl-1 expression was increased at tumors (81%) compared with only 38% in PIN. The percentage of Mcl-1-positive cells was typically higher in Gleason grade 8-10 tumors and metastasis than lower grade tumors, but there was no significant difference in Mcl-1 immunointensity between high grade tumors (47%) and metastasis in lymph node (38%) and bone (50) [5]. Our present study confirmed elevated Mcl-1 expression in high grade (≥ 7) PCa, though the difference between Gleason score 7 and 8-10 tumors is not statistically significant. Intriguingly, our results revealed a remarkable increase in Mcl-1 immunointensity in bone metastasis compared to primary tumors, indicating that Mcl-1 overexpression is positively correlated to PCa progression towards metastatic status in clinical situation.
Accumulating evidence suggests that the function of VEGF in tumor progression may not be limited to angiogenesis [38]. Numerous tumor cells express significant levels of VEGF-Rs, which could engage VEGF and initiate multiple signaling responses involved in cell proliferation, survival and migration. VEGF autocrine signaling confers a degree of self-sufficiency that may be crucial to metastasis as the microenvironment becomes increasingly hostile. Nonetheless, it remains controversial whether VEGF has significant autocrine effects in PCa cells, since the "classical" VEGF-Rs, i.e., VEGF-R1 and -R2, are undetectable in most established PCa cell lines [11, 39, 40]. Previously we reported that serum VEGF is positively associated with bone metastatic status in PCa patients, and recapitulated this close association in the ARCaP model [18]. In this study, we investigated whether VEGF could affect PCa cell behavior in an autocrine manner. Intriguingly, VEGF165 was found to be capable of inducing Mcl-1 expression within a non-saturating range, suggesting it may act as a survival factor in PCa cells. Moreover, NRP1 was found to be highly expressed by PCa cell lines and displayed a positive association with invasiveness, suggesting that it may be the primary receptor responsible for VEGF autocrine effects in PCa cells. Gene transfer experiments supported an indispensible role of NRP1 in mediating VEGF165 regulation of Mcl-1 in metastatic PCa cells. Importantly, a positive association between NRP1 expression and in vivo bone metastatic potential was found in ARCaPM xenografts and further confirmed in clinical PCa specimens. These data collectively support a novel role of NRP1 in PCa progression and metastasis, presumably by transmitting the VEGF autocrine survival signal and enabling PCa cells to evade apoptosis.
NRP1 is not a typical "signaling receptor" since it lacks sequences predicted to have kinase receptor activities [24]. Direct interaction between NRP1 and VEGF-Rs or plexin A is essential to its function in prioritizing differential signals from VEGF165 or semaphorin 3 in endothelial cells and nerve cells [24]. Recent studies found that the NRP1 intracellular domain, especially the C-terminal three amino acids (SEA-COOH), may be required for the interaction between NRP1 and its binding partners, including NRP1-interacting protein (NIP; or RGS-GAIP-interacting protein, GIPC) and VEGF-R2 [41, 42]. However, the molecular effects and mechanism for NRP1-mediated signaling in tumor cells, especially in the absence of VEGF-Rs, remain elusive. It has been proposed that NRP1 may store or sequester VEGF165 and attract endothelial cells towards tumor, contributing to angiogenesis via juxtacrine or paracrine mechanisms [9, 17]. In this study, we investigated whether NRP1 is required for VEGF165 autocrine signaling in PCa cells lacking VEGF-Rs. We presented evidence that NRP1 and c-MET are physically associated on plasma membrane, and in response to VEGF165 stimulation, their interaction may significantly facilitate further recruitment and activation of c-MET. Though the structural and molecular basis for interaction between the two receptors needs to be further investigated, this study indicates for the first time that c-MET activity can be modulated by VEGF, and the NRP1-c-MET interaction may be a critical component in transmitting the VEGF survival signal in PCa cells. It may be plausible to assign c-MET phosphorylation as an indicator for activation of NRP1 autocrine signaling in tumor cells. Using human PCa tissue specimens as the "gold standard", we observed that NRP1 and p-c-MET were both significantly increased with progression of primary PCa, and further elevated in bone metastatic PCa, suggesting that activation of NRP1-c-MET signaling may be positively associated with clinical PCa progression. It is worth noting that these results need cautious interpretation since a limited number of patient specimens was examined, and the polyclonal antibody against p-c-MET has not been fully validated for immunohistochemical analysis in tissue sections.
In human primary hepatocytes, HGF transcriptionally induced Mcl-1 expression, but not Bcl-2 or Bcl-x(L) [27]. Our studies found that, however, HGF treatment did not significantly affect Mcl-1 protein expression in ARCaPM cells, suggesting HGF activation of c-MET signaling may not be sufficient to increase Mcl-1 expression. We postulated that VEGF165 may utilize a different mechanism to regulate Mcl-1 expression in PCa cells. An important observation supporting this hypothesis is that VEGF165 only rapidly induced Stat3 phosphorylation at Tyr705, but not altering Ser727 phosphorylation in ARCaPM cells. In hepatocytes, however, HGF triggered Ser727, but not Tyr705, phosphorylation of Stat3 [27]. It will be intriguing to investigate the role of NRP1-c-MET interaction in differentiating extracellular ligands, i.e., VEGF165 or HGF, and activating distinct downstream cascades leading to Mcl-1 expression in a highly cell context-dependent fashion.
Mcl-1 is a protein with very short half-life and is highly regulated at multiple levels in response to survival and differentiation signals. Rapid turnover of Mcl-1 protein can be initiated by stress (such as serum withdrawal) through caspase-mediated and proteasome-dependent degradation in tumor cells [3]. Previous study found that VEGF protects multiple myeloma cells against apoptosis by up-regulating Mcl-1 protein in a time-dependent manner, peaking at 6 h and returning to baseline after 24 h; change in Mcl-1 mRNA expression was not reported [43]. In ARCaPM cells, however, VEGF165 treatment rapidly induced Mcl-1 mRNA expression and significantly increased Mcl-1 protein level at 72 h. Though whether VEGF165 may also regulate Mcl-1 expression at other levels needs to be further investigated, the data presented here suggest that transcriptional activation, presumably mediated by Stat3, is an important mechanism for VEGF165 induction of Mcl-1 in PCa cells.
Interrupting NRP1 functions with mimetic peptides and monoclonal antibodies is being developed in xenograft models of human cancers [44, 45]. NRP1 monoclonal antibody has been shown to effectively inhibit tumor vascular remodeling, rendering vessels more susceptible to anti-VEGF therapy [45]. If the VEGF165-NRP1-c-MET pathway is required for Mcl-1 expression and survival in PCa cells, it would be intriguing to evaluate whether targeting NRP1 could interrupt both NRP1-c-MET signaling in tumor cells (survival) and NRP1-VEGF-Rs signaling in endothelial cells (angiogenesis). Inhibition of NRP1 signaling may be a promising strategy alone or in combination with other therapeutic approaches for treating PCa.
Methods
Cell culture
Human PCa cell lines ARCaPM, ARCaPM-C2, PC3, LNCaP, C4-2 and C4-2B, were routinely maintained in T-medium (Invitrogen, Carlsbad, CA) with 5% fetal bovine serum (FBS). ARCaPM-C2 subclone was derived from ARCaPM bone metastatic tissues as described previously [21, 22]. Where specified, ARCaPM cells were serum-starved overnight, and treated with recombinant human VEGF165, VEGF121, HGF (R&D Systems, Minneapolis, MN), c-MET inhibitor PHA-665752 (Calbiochem, San Diego, CA), or Src kinase inhibitor (PP2) (Calbiochem) in serum-free T-medium. HUVEC (The American Type Culture Collection, Manassas, VA) were maintained in endothelial basal growth medium (EBM-2) with 2% FBS. Cell proliferation was measured using the CellTiter 96 AQ proliferation assay according to the manufacturer's instructions (Promega, Madison, WI). Viable cells were counted in triplicate using a hemacytometer and trypan blue staining.
Immunohistochemical analysis
IHC staining of NRP1 and p-c-MET on human normal/benign prostatic glands, well- and poorly-differentiated primary PCa and bone metastatic PCa tissue specimens was performed as described previously [18] using goat anti-NRP1 antibody (C-19, Santa Cruz Biotechnology, Santa Cruz, CA; 1:100) and rabbit anti-p-c-MET (Tyr1230/1234/1235) antibody (44888G, Invitrogen; 1:100). IHC analysis of Mcl-1 expression was performed on a human PCa progression tissue microarray specimen (US BioMax, Inc., Rockville, MD) using rabbit anti-Mcl-1 antibody (S-19, Santa Cruz Biotechnology, 1:150). Matching normal serum was used as negative control. All IHC common reagents were obtained from Dako (Carpinteria, CA). Positive expression of NRP1, p-c-MET and Mcl-1 was defined as >15% positive staining in cell population.
Transfection
The vector harboring NRP1 cDNA (pCMV-NRP1) and the control (pCMV-XL4) (Origene, Rockville, MD) were transfected into ARCaPM cells for 48-72 h using lipofectamine 2000 (Invitrogen). Small interfering RNA (siRNA) nucleotides were transfected into ARCaPM cells according to the manufacturer's instructions. VEGF siRNA, Mcl-1 siRNA, Stat3 siRNA and control siRNA-A were purchased from Santa Cruz Biotechnology. NRP1 silencer® select validated siRNA and silencer® select control siRNA#1 were obtained from Ambion, Inc., (Austin, TX). c-MET ON-TARGET plus siRNA and ON-TARGET plus siRNA control were purchased from Dharmacon, Inc, (Chicago, IL).
Western blot analysis
Total cell lysates were prepared using radioimmunoprecipitation (RIPA) buffer (Santa Cruz Biotechnology). Nuclear proteins were extracted using a Novagen kit (EMD Biosciences, San Diego, CA). Immunoblotting analysis followed standard procedure [18] with anti-Stat3, anti-p-Stat3 (Ser705), anti-Src and anti-p-Src (Tyr416) (Cell Signaling, Danvers, MA); anti-p-Stat3 (Tyr705) (Upstate, Charlottesville, VA); anti-Mcl-1, anti-NRP1 (C-19), anti-NRP2, anti-VEGF-R2, and anti-c-MET (Santa Cruz Biotechnology); anti-p-c-MET (Tyr1230/1234/1235) (Invitrogen); and anti-β-actin (Sigma, St. Louis, MO).
RT-PCR
Total RNA was prepared with Qiagen RNeasy Kit (Valencia, CA), and RT-PCR was performed using the SuperScriptIII® One-Step RT-PCR kit (Invitrogen) following the manufacturer's protocol. The specific primer pairs are: 5'-GAGGAGGAGGAGGACGAGTT-3' (forward) and 5'-GTCCCGTTTTGTCCTTACGA-3' (reverse) (for Mcl-1); 5'-AGGACAGAGACTGCAAGTATGAC-3' (forward) and 5'-AACATTCAGGACCTCTCTTGA-3' (reverse) (for NRP1). The primers for VEGF, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [18] and Stat3 [46] were described previously. The primer pairs for human VEGF-R1, VEGF-R2 and NRP2 were purchased from R&D Systems.
ELISA
Subconfluent PCa cells were cultured in serum-free T-medium for 72 h before conditioned medium was collected. VEGF concentrations were analyzed using a Quantikine ELISA kit (R&D Systems) and normalized with total protein concentrations in CM.
Immunofluorescence and confocal imaging
Immunofluorescence was performed as described previously [47]. Goat anti-NRP1 antibody (C-19), rabbit anti-c-MET antibody (C-12, Santa Cruz Biotechnology), or rabbit anti-p-c-MET (Tyr1230/1234/1235) antibody was incubated with subconfluent ARCaPM cells at 4°C overnight. Either anti-goat Alexa Fluor® 546 or anti-rabbit Alexa Fluor® 488 secondary antibody (Invitrogen) was used at a dilution of 1:500. Cells were imaged on a Zeiss LSM 510 META [47]. In all cases, either a 63× or 100× Zeiss Plan-Apo oil objective was used (numerical aperture of 1.3 and 1.4, respectively). All images had contrast expansion performed in Adobe Photoshop.
Immunoprecipitation
The Immunoprecipitation Starter Pack (GE healthcare Bio-Sciences Corp., Piscataway, NJ) was used according to the manufacturer's instructions. Total lysates (1 mg) were immunoprecipitated with rabbit anti-c-MET antibody (C-12), rabbit anti-NRP1 antibody (H-286, Santa Cruz Biotechnology), or normal rabbit IgG (R&D Systems). Protein A/G Sepharose 4 Fast Flow beads were added to precipitate proteins, then washed and eluted. The samples were further processed for western blot analysis.
Apoptosis analysis
Cells were stained with an Annexin V-PE apoptosis detection kit (BD Biosciences, San Jose, CA) following the manufacturer's protocol, and measured using a fluorescence-activated cell sorting (FACS) caliber bench-top flow cytometer (Becton Dickinson, Franklin Lakes, NJ). The data were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Data analysis
Significance levels for comparisons of Mcl-1 expression in different Gleason score PCa were calculated by using the 2-sample t test. Treatment effects were evaluated using a two-sided Student's t test. All data represent three or more experiments. Errors are S.E. values of averaged results, and values of p < 0.05 were taken as a significant difference between means.
References
Mehlen P, Puisieux A: Metastasis: a question of life or death. Nat Rev Cancer. 2006, 6: 449-458. 10.1038/nrc1886
Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A: Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006, 9: 351-365. 10.1016/j.ccr.2006.03.027
Akgul C: Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2008
Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S, Wang HG, Zhang X, Bullrich F, Croce CM, Rai K, Hines J, Reed JC: Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with In vitro and In vivo chemoresponses. Blood. 1998, 91: 3379-3389.
Krajewska M, Krajewski S, Epstein JI, Shabaik A, Sauvageot J, Song K, Kitada S, Reed JC: Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am J Pathol. 1996, 148: 1567-1576.
Zhang B, Gojo I, Fenton RG: Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002, 99: 1885-1893. 10.1182/blood.V99.6.1885
Maeta Y, Tsujitani S, Matsumoto S, Yamaguchi K, Tatebe S, Kondo A, Ikeguchi M, Kaibara N: Expression of Mcl-1 and p53 proteins predicts the survival of patients with T3 gastric carcinoma. Gastric Cancer. 2004, 7: 78-84. 10.1007/s10120-004-0272-9
Ferrara N, Gerber HP, LeCouter J: The biology of VEGF and its receptors. Nat Med. 2003, 9: 669-676. 10.1038/nm0603-669
Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M: Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998, 92: 735-745. 10.1016/S0092-8674(00)81402-6
Whitaker GB, Limberg BJ, Rosenbaum JS: Vascular endothelial growth factor receptor-2 and neuropilin-1 form a receptor complex that is responsible for the differential signaling potency of VEGF(165) and VEGF(121). J Biol Chem. 2001, 276: 25520-25531. 10.1074/jbc.M102315200
Kitagawa Y, Dai J, Zhang J, Keller JM, Nor J, Yao Z, Keller ET: Vascular endothelial growth factor contributes to prostate cancer-mediated osteoblastic activity. Cancer Res. 2005, 65: 10921-10929. 10.1158/0008-5472.CAN-05-1809
Stephenson JM, Banerjee S, Saxena NK, Cherian R, Banerjee SK: Neuropilin-1 is differentially expressed in myoepithelial cells and vascular smooth muscle cells in preneoplastic and neoplastic human breast: a possible marker for the progression of breast cancer. Int J Cancer. 2002, 101: 409-414. 10.1002/ijc.10611
Parikh AA, Liu WB, Fan F, Stoeltzing O, Reinmuth N, Bruns CJ, Bucana CD, Evans DB, Ellis LM: Expression and regulation of the novel vascular endothelial growth factor receptor neuropilin-1 by epidermal growth factor in human pancreatic carcinoma. Cancer. 2003, 98: 720-729. 10.1002/cncr.11560
Parikh AA, Fan F, Liu WB, Ahmad SA, Stoeltzing O, Reinmuth N, Bielenberg D, Bucana CD, Klagsbrun M, Ellis LM: Neuropilin-1 in human colon cancer: expression, regulation, and role in induction of angiogenesis. Am J Pathol. 2004, 164: 2139-2151.
Latil A, Bieche I, Pesche S, Valeri A, Fournier G, Cussenot O, Lidereau R: VEGF overexpression in clinically localized prostate tumors and neuropilin-1 overexpression in metastatic forms. Int J Cancer. 2000, 89: 167-171. 10.1002/(SICI)1097-0215(20000320)89:2<167::AID-IJC11>3.0.CO;2-9
Kawakami T, Tokunaga T, Hatanaka H, Kijima H, Yamazaki H, Abe Y, Osamura Y, Inoue H, Ueyama Y, Nakamura M: Neuropilin 1 and neuropilin 2 co-expression is significantly correlated with increased vascularity and poor prognosis in nonsmall cell lung carcinoma. Cancer. 2002, 95: 2196-2201. 10.1002/cncr.10936
Miao HQ, Lee P, Lin H, Soker S, Klagsbrun M: Neuropilin-1 expression by tumor cells promotes tumor angiogenesis and progression. Faseb J. 2000, 14: 2532-2539. 10.1096/fj.00-0250com
Wu D, Zhau HE, Huang WC, Iqbal S, Habib FK, Sartor O, Cvitanovic L, Marshall FF, Xu Z, Chung LW: cAMP-responsive element-binding protein regulates vascular endothelial growth factor expression: implication in human prostate cancer bone metastasis. Oncogene. 2007, 26: 5070-5077. 10.1038/sj.onc.1210316
Zhau HY, Chang SM, Chen BQ, Wang Y, Zhang H, Kao C, Sang QA, Pathak SJ, Chung LW: Androgen-repressed phenotype in human prostate cancer. Proc Natl Acad Sci USA. 1996, 93: 15152-15157. 10.1073/pnas.93.26.15152
Zhau HE, Li CL, Chung LW: Establishment of human prostate carcinoma skeletal metastasis models. Cancer. 2000, 88: 2995-3001. 10.1002/1097-0142(20000615)88:12+<2995::AID-CNCR15>3.0.CO;2-Y
Xu J, Wang R, Xie ZH, Odero-Marah V, Pathak S, Multani A, Chung LW, Zhau HE: Prostate cancer metastasis: role of the host microenvironment in promoting epithelial to mesenchymal transition and increased bone and adrenal gland metastasis. Prostate. 2006, 66: 1664-1673. 10.1002/pros.20488
Xu J, Odero-Marah V, Wang R, Chung LW, Zhau HE: Epithelial-mesenchymal transition and bone-specific microenvironment contribute to the rapid skeletal metastasis in human prostate cancer. J Urol. 2005, 173: 125-
Wu TT, Sikes RA, Cui Q, Thalmann GN, Kao C, Murphy CF, Yang H, Zhau HE, Balian G, Chung LW: Establishing human prostate cancer cell xenografts in bone: induction of osteoblastic reaction by prostate-specific antigen-producing tumors in athymic and SCID/bg mice using LNCaP and lineage-derived metastatic sublines. Int J Cancer. 1998, 77: 887-894. 10.1002/(SICI)1097-0215(19980911)77:6<887::AID-IJC15>3.0.CO;2-Z
Fujisawa H, Kitsukawa T: Receptors for collapsin/semaphorins. Curr Opin Neurobiol. 1998, 8: 587-592. 10.1016/S0959-4388(98)80085-8
Matsushita A, Gotze T, Korc M: Hepatocyte growth factor-mediated cell invasion in pancreatic cancer cells is dependent on neuropilin-1. Cancer Res. 2007, 67: 10309-10316. 10.1158/0008-5472.CAN-07-3256
Hu B, Guo P, Bar-Joseph I, Imanishi Y, Jarzynka MJ, Bogler O, Mikkelsen T, Hirose T, Nishikawa R, Cheng SY: Neuropilin-1 promotes human glioma progression through potentiating the activity of the HGF/SF autocrine pathway. Oncogene. 2007, 26: 5577-5586. 10.1038/sj.onc.1210348
Schulze-Bergkamen H, Brenner D, Krueger A, Suess D, Fas SC, Frey CR, Dax A, Zink D, Buchler P, Muller M, Krammer PH: Hepatocyte growth factor induces Mcl-1 in primary human hepatocytes and inhibits CD95-mediated apoptosis via Akt. Hepatology. 2004, 39: 645-654. 10.1002/hep.20138
Christensen JG, Schreck R, Burrows J, Kuruganti P, Chan E, Le P, Chen J, Wang X, Ruslim L, Blake R, Lipson KE, Ramphal J, Do S, Cui JJ, Cherrington JM, Mendel DB: A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003, 63: 7345-7355.
Longati P, Comoglio PM, Bardelli A: Receptor tyrosine kinases as therapeutic targets: the model of the MET oncogene. Curr Drug Targets. 2001, 2: 41-55. 10.2174/1389450013348920
Boccaccio C, Ando M, Tamagnone L, Bardelli A, Michieli P, Battistini C, Comoglio PM: Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature. 1998, 391: 285-288. 10.1038/34657
Ponzetto C, Bardelli A, Zhen Z, Maina F, dalla Zonca P, Giordano S, Graziani A, Panayotou G, Comoglio PM: A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. 1994, 77: 261-271. 10.1016/0092-8674(94)90318-2
Isomoto H, Kobayashi S, Werneburg NW, Bronk SF, Guicciardi ME, Frank DA, Gores GJ: Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005, 42: 1329-1338. 10.1002/hep.20966
Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA: Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996, 271: 695-701. 10.1074/jbc.271.2.695
Pisters LL, Troncoso P, Zhau HE, Li W, von Eschenbach AC, Chung LW: c-met proto-oncogene expression in benign and malignant human prostate tissues. J Urol. 1995, 154: 293-298. 10.1016/S0022-5347(01)67297-5
Knudsen BS, Gmyrek GA, Inra J, Scherr DS, Vaughan ED, Nanus DM, Kattan MW, Gerald WL, Woude Vande GF: High expression of the Met receptor in prostate cancer metastasis to bone. Urology. 2002, 60: 1113-1117. 10.1016/S0090-4295(02)01954-4
Mandelin AM, Pope RM: Myeloid cell leukemia-1 as a therapeutic target. Expert Opin Ther Targets. 2007, 11: 363-373. 10.1517/14728222.11.3.363
Chen S, Dai Y, Harada H, Dent P, Grant S: Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007, 67: 782-791. 10.1158/0008-5472.CAN-06-3964
Mercurio AM, Lipscomb EA, Bachelder RE: Non-angiogenic functions of VEGF in breast cancer. J Mammary Gland Biol Neoplasia. 2005, 10: 283-290. 10.1007/s10911-006-9001-9
Sokoloff MH, Chung LW: Targeting angiogenic pathways involving tumor-stromal interaction to treat advanced human prostate cancer. Cancer Metastasis Rev. 1998, 17: 307-315. 10.1023/A:1006170612253
Soker S, Kaefer M, Johnson M, Klagsbrun M, Atala A, Freeman MR: Vascular endothelial growth factor-mediated autocrine stimulation of prostate tumor cells coincides with progression to a malignant phenotype. Am J Pathol. 2001, 159: 651-659.
Wang L, Mukhopadhyay D, Xu X: C terminus of RGS-GAIP-interacting protein conveys neuropilin-1-mediated signaling during angiogenesis. FASEB J. 2006, 20: 1513-1515. 10.1096/fj.05-5504fje
Prahst C, Heroult M, Lanahan AA, Uziel N, Kessler O, Shraga-Heled N, Simons M, Neufeld G, Augustin HG: Neuropilin-1-VEGFR-2 complexing requires the PDZ-binding domain of neuropilin-1. J Biol Chem. 2008, 283: 25110-25114. 10.1074/jbc.C800137200
Le Gouill S, Podar K, Amiot M, Hideshima T, Chauhan D, Ishitsuka K, Kumar S, Raje N, Richardson PG, Harousseau JL, Anderson KC: VEGF induces Mcl-1 up-regulation and protects multiple myeloma cells against apoptosis. Blood. 2004, 104: 2886-2892. 10.1182/blood-2004-05-1760
Hong TM, Chen YL, Wu YY, Yuan A, Chao YC, Chung YC, Wu MH, Yang SC, Pan SH, Shih JY, Chan WK, Yang PC: Targeting neuropilin 1 as an antitumor strategy in lung cancer. Clin Cancer Res. 2007, 13: 4759-4768. 10.1158/1078-0432.CCR-07-0001
Pan Q, Chanthery Y, Liang WC, Stawicki S, Mak J, Rathore N, Tong RK, Kowalski J, Yee SF, Pacheco G, Ross S, Cheng Z, Le Couter J, Plowman G, Peale F, Koch AW, Wu Y, Bagri A, Tessier-Lavigne M, Watts RJ: Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell. 2007, 11: 53-67. 10.1016/j.ccr.2006.10.018
Rivat C, Rodrigues S, Bruyneel E, Pietu G, Robert A, Redeuilh G, Bracke M, Gespach C, Attoub S: Implication of STAT3 signaling in human colonic cancer cells during intestinal trefoil factor 3 (TFF3) -- and vascular endothelial growth factor-mediated cellular invasion and tumor growth. Cancer Res. 2005, 65: 195-202.
Zhang S, Schafer-Hales K, Khuri FR, Zhou W, Vertino PM, Marcus AI: The tumor suppressor LKB1 regulates lung cancer cell polarity by mediating cdc42 recruitment and activity. Cancer Res. 2008, 68: 740-748. 10.1158/0008-5472.CAN-07-2989
Acknowledgements
This study was supported by Department of Defense PC060566, Georgia Cancer Coalition Cancer Research Award and National Cancer Institute grant 1R43CA141870 (DW), National Cancer Institute grants P01 CA98912, R01 CA122602, and Department of Defense PC060866 (LWKC).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SZ performed western blotting, immunoprecipitation, immunofluorescence confocal microscopy, and gene transfer experiments (siRNA and cDNA expression). HEZ established the ARCaP PCa progression model, provided human PCa tissue specimens, designed and performed the immunohistochemical staining of NRP1 and p-c-MET in ARCaP tumors and human PCa specimens. AOO provided human PCa tissue specimens and evaluated expression of NRP1 and p-c-MET. SF evaluated expression of Mcl-1 in PCa tissue microarray specimens. ZC performed statistical analyses. XY and SI performed cell culture and preparation of proteins and RNA samples. RW contributed to the establishment of the ARCaP animal models. FFM and LWKC participated in discussion and manuscript preparation. LWKC provided grant supports for this study. DW designed experiments and drafted manuscript. All authors read and approved the final version of this manuscript.
Electronic supplementary material
12943_2009_524_MOESM1_ESM.TIFF
Additional file 1: Figure S1. (a) Endogenous Mcl-1 expression in the lineage-related LNCaP, C4-2 and C4-2B cells, as determined by RT-PCR and western blotting analyses. (b) ELISA of VEGF levels in conditioned media of LNCaP and C4-2 cells, shown as relative VEGF concentrations normalized by total protein concentrations of the CM. (c) Effects of recombinant VEGF121 on the proliferation of ARCaPM cells. 1 × 103cells were seeded in 96-well plates for 24 h, serum-starved overnight, and cultured in the absence or presence of varying concentrations of rVEGF121 for 72 h. MTS assay was then performed. (d) Effects of VEGF165 on Mcl-1 expression in LNCaP cells. Subconfluent LNCaP cells were serum-starved overnight, and incubated for 72 h in the presence of VEGF165 (10 ng/ml) or PBS. Western blotting was performed. (e) Effects of VEGF121 on Mcl-1 expression in ARCaPM cells. Subconfluent ARCaPM cells were serum-starved overnight, and incubated for 72 h in the presence of VEGF121 (10 ng/ml) or PBS. Western blotting was performed. (f) Expression of endogenous NRP1 in LNCaP, C4-2 and C4-2B cells, as determined by RT-PCR and western blotting analyses. (g) Nuclear expression of c-MET and p-c-MET in ARCaPM and ARCaPM-C2 cells. TATA binding protein (TBP) was used as internal control of nuclear proteins. (TIFF 341 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhang, S., Zhau, H.E., Osunkoya, A.O. et al. Vascular endothelial growth factor regulates myeloid cell leukemia-1 expression through neuropilin-1-dependent activation of c-MET signaling in human prostate cancer cells. Mol Cancer 9, 9 (2010). https://doi.org/10.1186/1476-4598-9-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-9-9