
Abstract
Parainfluenza virus is an important pathogen threatening the health of animals and human, which brings human many kinds of disease, especially lower respiratory tract infection involving infants and young children. In order to control the virus, it is necessary to fully understand the molecular basis resulting in the genetic diversity of the virus. Homologous recombination is one of mechanisms for the rapid change of genetic diversity. However, as a negative-strand virus, it is unknown whether the recombination can naturally take place in human PIV. In this study, we isolated and identified a mosaic serotype 3 human PIV (HPIV3) from in China, and also provided several putative PIV mosaics from previous reports to reveal that the recombination can naturally occur in the virus. In addition, two swine PIV3 isolates transferred from cattle to pigs were found to have mosaic genomes. These results suggest that homologous recombination can promote the genetic diversity and potentially bring some novel biologic characteristics of HPIV.
Similar content being viewed by others

Introduction
Human parainfluenza virus (HPIV) is known to induce acute respiratory infections (ARI) including lower respiratory tract infection, which is a leading cause of morbidity and mortality in infants and young children [1, 2]. Until now, four serotypes of parainfluenza virus (PIV) infecting human being have been found. Especially, human PIV (HPIV) 1, 2 and 3 are the second leading causative agents of pediatric hospitalizations due to respiratory disease following respiratory syncytial virus (RSV) [3]. It is important to know the mechanism resulting in genetic and antigenic diversity of HPIV for controlling the pathogen.
As one member of the Respirovirus ge nus of the family Paramyxoviridae, HPIV is an enveloped non-segmented negative single-stranded RNA virus [4]. RNA viruses usually exhibit genetic variation, which can be attributed to their high rate of mutation during their replication process and the large population size [5]. In addition, homologous recombination has been recognized increasingly as a potentially important means of generating and shaping genetic diversity in positive strand RNA virus [6]. In several other members of the Paramyxoviridae family, Newcastle disease virus (NDV) [7–11] and human respiratory syncytial virus [12], natural recombinants have been detected. Moreover, attenuated vaccines were found to be able to influence the evolution process of NDV through exchanging their genetic material with circulating virus [7, 9, 11]. For HPIV, it is unknown whether there is natural recombinant virus circulating in the field.
In this study, we isolated and identified a natural type 3 HPIV mosaic isolate LZ22/FJ455842 (with a mosaic N gene) to show that homologous recombination can occur in HPIV3. Additionally, three HPIV1 isolates (HT88/U01082, HT89a/U01083 and HT89c/U01085) were found to be deposited in previous report. Interestingly, we also found that there were the two Swine PIV3 recombinants with mosaic L protein were thought to be associated with an cross-species infection in previous report [13, 14]. Collectively, these recombination events suggested that homologous recombination played a role in HPIV genetic diversity and rapid evolution.
Results and Discussion
The sequence of LZ22 complete genome has been deposited in GenBank (Access Number, FJ455842). And the PIV3 complete genome sequence alignment dataset was analyzed employing RDP3 software package for scanning the recombinant sequence. And the isolate LZ22 was found to have greatly strong recombination signal: RDP, p-value < 10-21; GENECOVY, p-value < 10-21, Bootscan, p-value < 10-20, MaxChi p-value < 10-3; Chimaera p-value < 10-7; Siscan, p-value < 10-8, 3Seq, p-value < 10-13. And a breakpoint was located at position 485. Two strains GP and ZHYMgz01 were suggested as representatives of its putative parent lineages.
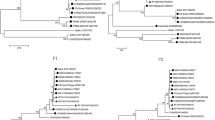
And then, Simplot software package was used to determine the recombination event [15]. Employing the Findsites subprogram of SimPlot, one potential breakpoint was located at parsimonious regions with the maximization of χ2, from positions 485 to 615 (χ2 = 122.3, P < 0.0001 of Fisher's exact test). A similarity plot (Figure 1A) which was constructed by using all sites, revealed that the sequence of LZ22 showed greater affinity with one putative parent lineage of GP in the region from position 1 to 485 than the other putative parent ZHYMgz01 (100% versus 94%). However, sequence from positions 486 to 15536, ZHYMgz01 shared greater similarity with LZ22 than GP (98% versus 95%). P value (Fisher's Exact Test) and χ2 value of the breakpoint were shown on the vertical line in Figure 1. The identical evidence also appeared in BootScanning result (Figure 1B). The region from GP lineage spanned the amino terminal 1/3 of the N protein approximately.

(A, B) Results of Similarity and BootScanning analysis of the genome of LZ22_FJ455842. The y-axis in Similarity plot (A) gives the percentage of sequence identity within a sliding window of 400 bp wide centered on the position plotted, with a step size between plots of 20 bp, while in BootScanning plot (B) represents the percentage of permuted trees. The χ2 of maximization and P value of Fisher's Exact test are shown near (or on) the vertical line. GP and ZHYMgz01 are used as two parental lineage sequences and JS_U51116 an outgroup sequence. The breakpoint is identified and located at position 485, with χ2 value maximized. The query sequence LZ22_FJ455842 demonstrates greater sequence identity and BootScanning support with GP in the beginning region while otherwise with ZHYMgz01_EU326526 in the complementary regions.
At last, The phylogenic trees were also constructed using Mega 4 to determine the recombination events [16]. From positions 1 to 485, LZ22 and GP were clustered into the same sublineage with 98% bootstrap value, while ZHYMgz01 was grouped into distinct sublineage (Figure 2A). But, in the other portion, the arrangement of the phylogenetic tree reflecting the relationship of the three isolates was in contrast with the previous one (Figure 2B). The topology of the two phylogenetic trees around the breakpoint showed a significant statistic discrepancy when the mosaic was included in analyzed data (Shimodaira-Hasegawa test, P < 0.001), constituting a powerful evidence for recombination.

Phylogenetic profiles of separate regions of LZ22_FJ455842 partitioned by cross-over events. The scale corresponds to the number of nucleotide substitutions per site. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches (only N > 70% is shown). The putative recombinants were showed with "black square". The putative parent lineages were marked with "black triangle". (A) and (B) respectively represent the phylogeny of pre- (1-485) and post- (486-15536) part of complete segments delimited by the breakpoint. The pre-part of mosaics demonstrated higher level of congruence with the GP lineage, while the post-part converge with ZHYMgz01_EU326526.
The minor putative parent of LZ22 was isolated from Japan, suggesting the existence of a global reservoir of HPIV with local subreservoirs supporting extensive levels of virus circulation, which permitted co-infection and resulted in the recombination event at last. The potential breakpoint 485 presents in gene N coding nucleoprotein spanning the region from positions 56 to 1737 of genomic sequence [17]. In the previous study, viable artificial chimeric HPIV3 recombinants were constructed, which contained the nucleoprotein open reading frame (ORF) from either BPIV3 Kansas or shipping fever (SF) strain in place of the HPIV3 N ORF. The artificial recombinant (PIV3) in which the nucleocapsid N protein had been replaced by that of bovine PIV3 was found to be attenuated in primates [18]. Here, we isolated a HPIV3 with a natural mosaic in NP ORF. Interestingly, before the breakpoint, the similarity of peptide sequences was up to 98.5% (129/131) although the similarity of gene sequences was only 94% between the two putative parent lineages. It is unknown whether the recombination in N gene is also associated with their adaptation in host cell via a changed virulence.
In addition, since there has been no report to show homologous recombination can take place between PIVs before this study; we analyzed 55 isolates (Table 1) from GenBank in order to determine to what degree genetic diversity of the virus is affected by the homologous recombination. Five additional mosaic PIV sequences were detected through the analysis of sequences from isolates characterized in previous reports:, HT88 (U01082) [19], HT89a [19] (U01083), HT89c (U01085) [19], 81-19252_Texas-81 (EU439429) [13, 14] and 92-7783_ISU-92 (EU439428) [13, 14] (Table 2). Two HN gene mosaics, HT88 and HT89a shared the same recombination event (Table 2), suggesting they descended from the same mosaic ancestor. Please also refer additional files 1, 2, 3, and 4 for the detail recombination information of each mosaic strain. These results suggested that homologous recombination did play a potential role in the evolution of the virus.
Interestingly, two mosaic viruses 81-19252_Texas-81 (U439429) and 92-7783_ISU-92 (EU439428) were reported to be involved in a cross infection between swine and bovine [13, 14]. Both of the isolates had a mosaic L gene. The transcription and replication functions of the parainfluenza virus are associated with the large RNA polymerase protein. Additionally, polyadenylation, and RNA editing activities have to do with L protein [20]. The two putative mosaics were isolated from pigs in the United States [13, 14] while both of their putative parent lineages (Shipping fever and 910N lineages) belonged to BPIV3. Viruses are largely species-specific with respect to their host and usually do not cross species boundaries [5]. Recombination processes will allow some viruses to acquire many of the key adaptive mutations in a single step, and thus make a major leap in fitness, which might result in a change of host tropism [21]. It might be necessary to further study whether the recombination event is relative to the BPIV3 cross-species infection.
In conclusion, this study provides the potential evidence that there is mosaic PIV in the field. Our observations show that homologous recombination is a molecular mechanism of PIV genetic diversity and evolution. Therefore, this study might be important for knowing the genetic basis resulting in the rapid change of PIV biologic characteristics.
Material and methods
Virus and sequencing
The virus LZ22 was isolated from lower respiratory tract of a patient infant with pneumonia in Lanzhou, Gansu Province of China in 2003. The virus was identified using previously described protocols [22]. After isolated, LZ22 was also purified 3 times by the plaque forming method in Vero cells. Before sequencing, LZ22 was passaged 13 times and amplified in Vero cells for RT-PCR. Viral RNA was extracted from Vero cell virus cultures using RNAeasy mini kit (Qiagen, Netherlands) following the manufacturer's instructions. Reverse transcription was performed using SuperScriptΠ one-step RT-PCR platinum Taq HiFi kit (Invitrogen, USA). 3' and 5' RACE were performed using 5'-full RACE CORE kit and 3'-RACE kit (Takala Dalian) to analyze the 3' and 5' UTR sequences. The primers of PCR and 3' and 5' RACE used in this study were listed in Table 3. The full genome of LZ22 was amplified and sequenced referring to previous report [22]. All PCR products were cloned into pGEM-T-vector (Promega USA) and sequenced by Takara Biotechnology (Dalian, China).
Recombination analysis
Compete genome and HN gene of PIV were retrieved from GenBank and aligned with CLUSTALW [23]. PIV3 compete genome and HPIV1 HN genes sequences analyzed in the study were listed in Table 1. Phylogenetic Neighbor-Joining (NJ) trees were set up by MEGA4 [16]. The nucleotide substitution models were optimized for Maximum-Likelihood (ML) trees employing jmodeltest (version 0.1.1) [24]. ML trees were constructed employing Phyml software with nucleotide substitution model of general time reversible model (GTR) and gamma distributed 4 (G4) [25], and displayed as graphics by using MEGA4 to determine the topology of each tree. Identification methods of homologous recombination were described as previous report [26, 27]. Briefly, the sequence alignment files were sought for potential mosaic isolates using RDP software package [24, 28]. The gene sequence similarity of mosaics and their putative parents were compared and displayed as graphics with Simplot software [15]. At last, incongruent phylogenetic relations of different gene regions delimited by crossover point were determined by phylogenetic trees.
References
Jennings LC, Anderson TP, Werno AM, Beynon KA, Murdoch DR: Viral etiology of acute respiratory tract infections in children presenting to hospital: role of polymerase chain reaction and demonstration of multiple infections. Pediatr Infect Dis J 2004, 23: 1003-1007. 10.1097/01.inf.0000143648.04673.6c
Juven T, Mertsola J, Waris M, Leinonen M, Meurman O, Roivainen M, Eskola J, Saikku P, Ruuskanen O: Etiology of community-acquired pneumonia in 254 hospitalized children. Pediatr Infect Dis J 2000, 19: 293-298. 10.1097/00006454-200004000-00006
Murphy BR, Prince GA, Collins PL, Van Wyke Coelingh K, Olmsted RA, Spriggs MK, Parrott RH, Kim HW, Brandt CD, Chanock RM: Current approaches to the development of vaccines effective against parainfluenza and respiratory syncytial viruses. Virus Res 1988, 11: 1-15. 10.1016/0168-1702(88)90063-9
Lau SK, To WK, Tse PW, Chan AK, Woo PC, Tsoi HW, Leung AF, Li KS, Chan PK, Lim WW, et al.: Human parainfluenza virus 4 outbreak and the role of diagnostic tests. J Clin Microbiol 2005, 43: 4515-4521. 10.1128/JCM.43.9.4515-4521.2005
Domingo E, Holland JJ: RNA virus mutations and fitness for survival. Annu Rev Microbiol 1997, 51: 151-178. 10.1146/annurev.micro.51.1.151
Domingo E: Virus Evolution. In Fields Virology. 5th edition. Edited by: Knipe DM, Howley PM. Lippincott/The Williams & Wilkins Co., Philadelphia, PA; 2006:391-422.
Zhang R, Wang X, Su J, Zhao J, Zhang G: Isolation and analysis of two naturally-occurring multi-recombination Newcastle disease viruses in China. Virus Res 2010, 151: 45-53. 10.1016/j.virusres.2010.03.015
Qin Z, Sun L, Ma B, Cui Z, Zhu Y, Kitamura Y, Liu W: F gene recombination between genotype II and VII Newcastle disease virus. Virus Res 2008, 131: 299-303. 10.1016/j.virusres.2007.10.001
Han GZ, He CQ, Ding NZ, Ma LY: Identification of a natural multi-recombinant of Newcastle disease virus. Virology 2008, 371: 54-60. 10.1016/j.virol.2007.09.038
Rui Z, Juan P, Jingliang S, Jixun Z, Xiaoting W, Shouping Z, Xiaojiao L, Guozhong Z: Phylogenetic characterization of Newcastle disease virus isolated in the mainland of China during 2001-2009. Vet Microbiol 2010, 141: 246-257. 10.1016/j.vetmic.2009.09.020
Chong YL, Padhi A, Hudson PJ, Poss M: The effect of vaccination on the evolution and population dynamics of avian paramyxovirus-1. PLoS Pathog 2010, 6: e1000872. 10.1371/journal.ppat.1000872
Spann KM, Collins PL, Teng MN: Genetic recombination during coinfection of two mutants of human respiratory syncytial virus. J Virol 2003, 77: 11201-11211. 10.1128/JVI.77.20.11201-11211.2003
Qiao D, Janke BH, Elankumaran S: Molecular characterization of glycoprotein genes and phylogenetic analysis of two swine paramyxoviruses isolated from United States. Virus Genes 2009, 39: 53-65. 10.1007/s11262-009-0353-2
Qiao D, Janke BH, Elankumaran S: Complete genome sequence and pathogenicity of two swine parainfluenzavirus 3 isolates from pigs in the United States. J Virol 2010, 84: 686-694. 10.1128/JVI.00847-09
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC: Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 1999, 73: 152-160.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 2007, 24: 1596-1599. 10.1093/molbev/msm092
Newman JT, Surman SR, Riggs JM, Hansen CT, Collins PL, Murphy BR, Skiadopoulos MH: Sequence analysis of the Washington/1964 strain of human parainfluenza virus type 1 (HPIV1) and recovery and characterization of wild-type recombinant HPIV1 produced by reverse genetics. Virus Genes 2002, 24: 77-92. 10.1023/A:1014042221888
Bailly JE, McAuliffe JM, Durbin AP, Elkins WR, Collins PL, Murphy BR: A recombinant human parainfluenza virus type 3 (PIV3) in which the nucleocapsid N protein has been replaced by that of bovine PIV3 is attenuated in primates. J Virol 2000, 74: 3188-3195. 10.1128/JVI.74.7.3188-3195.2000
Hetherington SV, Watson AS, Scroggs RA, Portner A: Human parainfluenza virus type 1 evolution combines cocirculation of strains and development of geographically restricted lineages. J Infect Dis 1994, 169: 248-252.
Poch O, Blumberg BM, Bougueleret L, Tordo N: Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J Gen Virol 1990,71(Pt 5):1153-1162. 10.1099/0022-1317-71-5-1153
Kuiken T, Holmes EC, McCauley J, Rimmelzwaan GF, Williams CS, Grenfell BT: Host species barriers to influenza virus infections. Science 2006, 312: 394-397. 10.1126/science.1122818
Bai MQ, An H, Bao H, Zhou X: Detection of the RSV and HPIV-3 in the low respiratory infection among infants in Lanzhou region by Nest RT-PCR. Progress in Microbiology and Immunology 2007, 35: 40-42. (In Chinese)
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997, 25: 4876-4882. 10.1093/nar/25.24.4876
Martin DP, Williamson C, Posada D: RDP2: recombination detection and analysis from sequence alignments. Bioinformatics 2005, 21: 260-262. 10.1093/bioinformatics/bth490
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 2003, 52: 696-704. 10.1080/10635150390235520
He CQ, Ding NZ, He M, Li SN, Wang XM, He HB, Liu XF, Guo HS: Intragenic recombination as a mechanism of genetic diversity in bluetongue virus. J Virol 2010, 84: 11487-11495. 10.1128/JVI.00889-10
He CQ, Xie ZX, Han GZ, Dong JB, Wang D, Liu JB, Ma LY, Tang XF, Liu XP, Pang YS, Li GR: Homologous recombination as an evolutionary force in the avian influenza A virus. Mol Biol Evol 2009, 26: 177-187. 10.1093/molbev/msn238
Gibbs MJ, Armstrong JS, Gibbs AJ: Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16: 573-582. 10.1093/bioinformatics/16.7.573
Durbin AP, McAuliffe JM, Collins PL, Murphy BR: Mutations in the C, D, and V open reading frames of human parainfluenza virus type 3 attenuate replication in rodents and primates. Virology 1999, 261: 319-330. 10.1006/viro.1999.9878
Ohsawa K, Yamada A, Takeuchi K, Watanabe Y, Miyata H, Sato H: Genetic characterization of parainfluenza virus 3 derived from guinea pigs. J Vet Med Sci 1998, 60: 919-922. 10.1292/jvms.60.919
Bailly JE, McAuliffe JM, Skiadopoulos MH, Collins PL, Murphy BR: Sequence determination and molecular analysis of two strains of bovine parainfluenza virus type 3 that are attenuated for primates. Virus Genes 2000, 20: 173-182. 10.1023/A:1008130917204
Sakai Y, Suzu S, Shioda T, Shibuta H: Nucleotide sequence of the bovine parainfluenza 3 virus genome: its 3' end and the genes of NP, P, C and M proteins. Nucleic Acids Res 1987, 15: 2927-2944. 10.1093/nar/15.7.2927
Stokes A, Tierney EL, Murphy BR, Hall SL: The complete nucleotide sequence of the JS strain of human parainfluenza virus type 3: comparison with the Wash/47885/57 prototype strain. Virus Res 1992, 25: 91-103. 10.1016/0168-1702(92)90102-F
Stokes A, Tierney EL, Sarris CM, Murphy BR, Hall SL: The complete nucleotide sequence of two cold-adapted, temperature-sensitive attenuated mutant vaccine viruses (cp12 and cp45) derived from the JS strain of human parainfluenza virus type 3 (PIV3). Virus Res 1993, 30: 43-52. 10.1016/0168-1702(93)90014-E
Horwood PF, Gravel JL, Mahony TJ: Identification of two distinct bovine parainfluenza virus type 3 genotypes. J Gen Virol 2008, 89: 1643-1648. 10.1099/vir.0.2008/000026-0
Henrickson KJ, Savatski LL: Two distinct human parainfluenza virus type 1 genotypes detected during the 1991 Milwaukee epidemic. J Clin Microbiol 1996, 34: 695-700.
Henrickson KJ, Savatski LL: Genetic variation and evolution of human parainfluenza virus type 1 hemagglutinin neuraminidase: analysis of 12 clinical isolates. J Infect Dis 1992, 166: 995-1005.
Murphy BR, Richman DD, Chalhub EG, Uhlendorf CP, Baron S, Chanock RM: Failure of attenuated temperature-sensitive influenza A (H3N2) virus to induce heterologous interference in humans to parainfluenza type 1 virus. Infect Immun 1975, 12: 62-68.
Gorman WL, Gill DS, Scroggs RA, Portner A: The hemagglutinin-neuraminidase glycoproteins of human parainfluenza virus type 1 and Sendai virus have high structure-function similarity with limited antigenic cross-reactivity. Virology 1990, 175: 211-221. 10.1016/0042-6822(90)90201-2
Matsuoka Y, Ray R, Compans RW: Sequence of the hemagglutinin-neuraminidase gene of human parainfluenza virus type 1. Virus Res 1990, 16: 107-113. 10.1016/0168-1702(90)90047-F
Acknowledgements
We would like to thank Prof. Elankumaran S for the detailed information of the two mosaic isolates 81-19252_Texas-81 (EU439429) and 92-7783_ISU-92 (EU439428); and the anonymous reviewers for helpful comments. This work was supported by a Project of Shandong Province Higher Educational Science and Technology Program (J09LCD209-12) and a Shangdong Province Young and Middle-Aged Scientists Research Awards Fund (BS2009NY011) to H.C.Q. State Scientist in Industrial Technology System of Dairy Cattle (H.H.B), Taishan Scholar and Distinguished Experts from overseas (H.H.B), A Major Application of Technological Innovations of Agriculture of Shandong Province (H.H.B).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YHT, and JQ carried out sequence collection, alignment and recombination analysis and drafted the manuscript. ZX and BMQ provided LZ22 viral sequence information. SHL participated in sequence collection. WXJ, LY, ZH participated in revision of manuscript. HHB participated in its design and coordination. And HCQ designed the study and wrote the manuscript. All authors read and approved the final manuscript.
Hui-Ting Yang, Qing Jiang contributed equally to this work.
Electronic supplementary material
12985_2010_1173_MOESM1_ESM.DOC
Additional file 1:The detail recombination information of mosaic strain 81-19252_Texas-81_EU439429. (A, B) Results of Similarity and Bootscanning analysis of 81-19252_Texas-81_EU439429. The y-axis in Similarity plot (A) gives the percentage of identity within a sliding window of 500 bp wide centered on the position plotted, with a step size between plots of 20 bp, while in Bootscanning plot (B) represents the percentage of permuted trees. Shipping_Fever_AF178655 and 910N_D84095 were used as two parental sequences and Q5592_EU277658 an outgroup sequence. Four breakpoints were identified and located by GARD at position 8688, 12595,13619 and 14175, respectively, with value maximized. The query sequence 81-19252_Texas-81_EU439429 demonstrated greater sequence identity and Bootscanning support with 910N_D84095 in the second and fourth regions while otherwise with Q5592_EU277658 in the complementary regions. (C-G) Neighbor-Jointing Phylogenetic profiles of separate regions of 81-19252_Texas-81_EU439429 partitioned by cross-over events. The scale corresponds to the number of nucleotide substitutions per site. The putative recombinants were showed with "black square". C-G) represent the phylogeny of fir-(1-8688), sec-(8689-12595), thi-(12596-13619), fou-(13620-14175) and fin-(14176-15536) part of full length segment, respectively. The sec-and fou-part of mosaics demonstrated higher level of congruence with the 910N_D84095 lineage, while the otherwise converge with Shipping_Fever_AF178655. (DOC 1006 KB)
12985_2010_1173_MOESM2_ESM.DOC
Additional file 2:The detail recombination information of mosaic strain 92-7783_ISU-92_EU439428. (A, B) Results of Similarity and Bootscanning analysis of 92-7783_ISU-92_EU439428. The y-axis in Similarity plot (A) gives the percentage of identity within a sliding window of 500 bp wide centered on the position plotted, with a step size between plots of 20 bp, while in Bootscanning plot (B) represents the percentage of permuted trees. Shipping_Fever_AF178655 and 910N_D84095 were used as two parental sequences and Q5592_EU277658 an outgroup sequence. Two breakpoints were identified and located by GARD at position 14137 and 14989, respectively, with value maximized. The query sequence 92-7783_ISU-92_EU439428 demonstrated greater sequence identity and Bootscanning support with Shipping_Fever_AF178655 in the middle region while otherwise with Q5592_EU277658 in the complementary regions. (C-E) Neighbor-Jointing Phylogenetic profiles of separate regions of 92-7783_ISU-92_EU439428 partitioned by cross-over events. The scale corresponds to the number of nucleotide substitutions per site. The putative recombinants were showed with "black square". C-E) represent the phylogeny of pre-(1-14137), mid-(14138-14989) and post-(14990-15536) part of full length segment, respectively. The pre-and post-part of mosaics demonstrated higher level of congruence with the 910N_D84095 lineage, while the mid-part converge with Shipping_Fever_AF178655. (DOC 534 KB)
12985_2010_1173_MOESM3_ESM.DOC
Additional file 3:The detail recombination information of mosaic strains HT88_U01082 and HT89a_U01083. (A, B, C, D) Results of Similarity and Bootscanning analysis of HT88_U01082 and HT89a_U01083. The y-axis in Similarity plot (A) gives the percentage of identity within sliding windows of 400 or 300 bp wide centered on the position plotted, with a step size between plots of 20 bp, while in Bootscanning plot (B, D) represents the percentage of permuted trees. Mil-49/91_U70937 and HT89b_U01084 were used as two parental sequences and CH-A-81A_M86782 and CH-A-81B_M86783 outgroup sequences. Two breakpoints were identified and located by GARD at position 392 and 839, respectively, with value maximized. The query sequences HT88_U01082 and HT89a_U01083 demonstrated greater sequence identity and Bootscanning support with HT89b_U01084 in the middle region while otherwise with Mil-49/91_U70937 in the complementary regions. (C-E) Maximum-Likelihood Phylogenetic profiles of separate regions of HT88_U01082 and HT89a_U01083 partitioned by cross-over events. The scale corresponds to the number of nucleotide substitutions per site. The putative recombinants were showed with "black square" (HT88_U01082) and "black triangle" (HT89a_U01083). E-G) represent the phylogeny of pre-(1-392), mid-(393-839) and post-(840-1728) part of complete segment HN, respectively. The pre-and post-part of mosaics demonstrated higher level of congruence with the HT89b_U01084 lineage, while the mid-part converge with Mil-49/91_U70937. (DOC 564 KB)
12985_2010_1173_MOESM4_ESM.DOC
Additional file 4:The detail recombination information of mosaic strain HT89c_U01085. (A, B,) Results of Similarity and Bootscanning analysis of HT89c_U01085. The y-axis in Similarity plot (A) gives the percentage of identity within sliding windows of 400 bp wide centered on the position plotted, with a step size between plots of 20 bp, while in Bootscanning plot (B) represents the percentage of permuted trees. Mil-58/91_U70943 and Mil-51/91_U70938 were used as two parental sequence and CH-A-81A_M86782 outgroup sequence. Two breakpoints were identified and located by GARD at position 351 and 767, respectively, with value maximized. The query sequence HT89c_U01085 demonstrated greater sequence identity and Bootscanning support with Mil-51/91_U70938 in the middle region while otherwise with Mil-58/91_U70943 in the complementary regions. (C-E) Maximum-Likelihood Phylogenetic profiles of separate regions of HT89c_U01085 partitioned by cross-over events. The scale corresponds to the number of nucleotide substitutions per site. The putative recombinants were showed with "black square". C-E) represent the phylogeny of pre-(1-351), mid-(352-767) and post-(768-1728) part of complete segment HN, respectively. The pre-and post-part of mosaics demonstrated higher level of congruence with the Mil-58/91_U70943 lineage, while the mid-part converges with Mil-51/91_U70938. (DOC 594 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yang, HT., Jiang, Q., Zhou, X. et al. Identification of a natural human serotype 3 parainfluenza virus. Virol J 8, 58 (2011). https://doi.org/10.1186/1743-422X-8-58
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-8-58