
Abstract
Background
Cytochrome P450 OleTJE from Jeotgalicoccus sp. ATCC 8456, a new member of the CYP152 peroxygenase family, was recently found to catalyze the unusual decarboxylation of long-chain fatty acids to form α-alkenes using H2O2 as the sole electron and oxygen donor. Because aliphatic α-alkenes are important chemicals that can be used as biofuels to replace fossil fuels, or for making lubricants, polymers and detergents, studies on OleTJE fatty acid decarboxylase are significant and may lead to commercial production of biogenic α-alkenes in the future, which are renewable and more environmentally friendly than petroleum-derived equivalents.
Results
We report the H2O2-independent activity of OleTJE for the first time. In the presence of NADPH and O2, this P450 enzyme efficiently decarboxylates long-chain fatty acids (C12 to C20) in vitro when partnering with either the fused P450 reductase domain RhFRED from Rhodococcus sp. or the separate flavodoxin/flavodoxin reductase from Escherichia coli. In vivo, expression of OleTJE or OleTJE-RhFRED in different E. coli strains overproducing free fatty acids resulted in production of variant levels of multiple α-alkenes, with a highest total hydrocarbon titer of 97.6 mg·l-1.
Conclusions
The discovery of the H2O2-independent activity of OleTJE not only raises a number of fundamental questions on the monooxygenase-like mechanism of this peroxygenase, but also will direct the future metabolic engineering work toward improvement of O2/redox partner(s)/NADPH for overproduction of α-alkenes by OleTJE.
Similar content being viewed by others

Background
The urgency to develop sustainable fossil fuel alternatives is driven by rapidly increasing global consumption, irreversibly diminishing reserves, unpredictable geopolitical factors, fluctuating price of crude oil, growing concerns about national energy security, and serious environmental concerns surrounding immense greenhouse gas emissions mainly resulting from combustion of fossil fuels [1–3]. Biofuels produced from biological resources represent a compelling alternative to fossil fuels because they are renewable and more environmentally friendly [4–7]. Among various biofuels, bioethanol and biodiesel are dominating the current global market. However, it is widely accepted that the ideal biofuels are bio-hydrocarbons, especially the medium- to long-chain fatty alkanes or alkenes, because they highly mimic the chemical composition and physical characteristics of petroleum-based fuels [8–10]. Thus, the biosynthetic pathways for aliphatic hydrocarbons from diverse organisms have been attracting great attentions in recent years [11–13]. Particularly, the scalable and cost-effective microbial biosynthesis of fatty alkanes or alkenes is considered as one of the most promising ways to produce ‘drop-in compatible’ biofuels [5, 8].
To date, four microbial biosynthetic pathways that convert free fatty acids or fatty acid thioesters into bio-hydrocarbons have been identified, including the cyanobacterial pathways consisting of an acyl-acyl carrier protein (acyl-ACP) reductase and an aldehyde decarbonylase, which together convert fatty acyl-ACPs into alkanes [14]; a three-gene cluster responsible for generating alkenes with internal double bonds through the head-to-head condensation of two fatty acyl-coenzyme A (acyl-CoA) molecules in Micrococcus luteus[15]; a unique P450 decarboxylase OleTJE from Jeotgalicoccus sp. ATCC 8456, which directly decarboxylates long-chain fatty acids to form α-olefins in presence of H2O2[16, 17]; and a type I polyketide synthase from Synechococcus sp. PCC 7002 [18, 19], which is capable of transforming fatty acyl-ACPs into α-olefins via sequential polyketide synthase chain elongation, keto reduction, sulfonation mediated by its sulfotransferase domain, and the coupled hydrolysis and decarboxylation catalyzed by the thioesterase domain.
Among these pathways, the single-step decarboxylation of fatty acids catalyzed by OleTJE P450 enzyme [16] apparently represents the simplest one. Moreover, it directly uses free fatty acids instead of fatty acid thioesters as substrates, which is believed to be advantageous for metabolic engineering because fatty acids are more abundant and their abundance and composition can be well manipulated in E. coli[20–22], one of the most developed microbial cell factories. Thus, this P450 fatty acid decarboxylative machinery may hold great potential to be engineered into a biological α-alkene-producing system. Notably, in addition to being biofuel molecules, α-alkenes are also used broadly for making lubricants, polymers and detergents [17, 23, 24].
The superfamily of cytochrome P450 enzymes (CYPs) are considered among the most versatile biocatalysts in nature [25]. Typically, P450 enzymes employ one or more redox partner proteins to transfer two electrons from NAD(P)H to the heme iron reactive center for dioxygen activation, and then insert one atom of O2 into their substrates [25–27]. Therefore, these oxidative biocatalysts are often termed as P450 monooxygenases. However, the CYP152 family members such as P450SPα[28], P450BSβ[29] and OleTJE[16] have been identified to exclusively use H2O2 as the sole electron and oxygen donor, and are thus classified into a unique P450 peroxygenase category.
Practically, the peroxygenase activity of P450 enzymes is often treated as an advantageous feature because H2O2 is much cheaper than NADPH and redox proteins in many P450 applications such as biocatalysts for in vitro synthetic reactions and enzyme additives in laundry detergents [30, 31]. However, the large-scale production of low-cost α-alkene biofuels by OleTJE P450 decarboxylase cannot rely on the H2O2-dependent enzymatic system because the use of large amounts of peroxide is cost prohibitive, and high concentration of H2O2 can quickly deactivate biocatalysts [32].
Therefore, the H2O2-independent activity of OleTJE, if it exists, is preferred for cost-effective microbial production of α-alkenes. In the present study, we demonstrated such activity for the first time. Through engineering a self-sufficient version of OleTJE by fusing it to the Rh odococcus fusion reductase (RhFRED) domain from Rhodococcus sp. NCIMB 9784 [33], the catalytic activity of the resultant fusion P450 enzyme can be solely driven by NADPH. We also found that E. coli flavodoxin (Fld) and flavodoxin reductase (FdR) are capable of supporting OleTJE activity as well. Guided by these new findings, our initial metabolic engineering efforts based on the H2O2-independent decarboxylation of fatty acids by OleTJEin vivo led to a group of α-alkene overproducers with the best one producing 97.6 mg·l-1 total α-alkenes.
Results
In vitro fatty acid decarboxylation by OleTJEand OleTJE-RhFRED
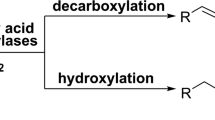
The unusual activity of the first P450 fatty acid decarboxylase OleTJE was determined by reconstituted in vitro reactions [16]. Specifically, OleTJE was able to decarboxylate stearic acid and palmitic acid to generate 1-heptadecene and 1-pentadecene, respectively, in the presence of H2O2 (Figure 1A). It also catalyzed the α- and β-hydroxylation of fatty acids as side reactions. Using the purified P450 enzymes (Additional file 1: Figure S1 and Additional file 2: Figure S2), we first confirmed these three types of activity of OleTJE using myristic acid (C14) as substrate. Consistent with the previous report [16], fatty acid decarboxylation was the dominant reaction, while the α- and β-hydroxymyristic acid only accounted for 0.2% and 6.1% of total products, respectively (Figure 1B).

Decarboxylation and hydroxylation reactions catalyzed by OleT JE. (A) The decarboxylation and hydroxylation of fatty acids (C12 to C20) catalyzed by OleTJE. (B) Product distribution of the decarboxylation and hydroxylation reactions catalyzed by purified OleTJE and OleTJE-RhFRED, respectively. Myristic acid was used as substrate. 1-TE, 1-tridecene; α-OH MA, α-hydroxy myristic acid; β-OH MA, β-hydroxy myristic acid.
According to previous amino acid sequence analysis, OleTJE was assigned to the CYP152 family, with the well-studied P450BSβ[29, 34, 35] from Bacillus subtilis and P450SPα[28, 36] from Sphingomonas paucimobilis as family members. Essentially, this family of P450 enzymes utilizes H2O2 as the sole electron and oxygen donor to oxidize their substrates, in contrast to most other P450 monooxygenases, whose catalytic activity depends on dioxygen, NAD(P)H and redox partner protein(s) [37]. Thus, these P450 enzymes are functionally referred to as peroxygenases [38]. Indeed, it was reported that P450 reductase systems such as ferredoxin and ferredoxin reductase did not support the activity of P450BSβ and P450SPα[36, 39]. This is consistent with the substitution of Pro246 and Arg245 (numbering in OleTJE) in CYP152 peroxygenases for the highly conserved threonine and an acidic residue (for example, Glu or Asp) in most P450 monooxygenases (Additional file 3: Figure S3), which are involved in a specific H-bond network responsible for delivery of protons during the P450 catalytic cycle [34, 40].
However, in the study of another CYP152-member P450CLA, from the anaerobic microorganism Clostridium acetobutylicum[41], it was demonstrated that both P450CLA and P450BSβ functioned when provided with O2, NADPH and flavodoxin/flavodoxin reductase from E. coli or the diflavin reductase domain of P450BM3 from Bacillus megaterium, suggesting an alternative monooxygenase-like mechanism of these P450 peroxygenases.
To investigate whether OleTJE peroxygenase can also function as a monooxygenase like P450CLA and P450BSβ, the RhFRED reductase domain from Rhodococcus sp. NCIMB 9784 [33] was fused to the C-terminus of OleTJE. In the presence of the electron donor NADPH, the fusion protein OleTJE-RhFRED successfully converted lauric acid (C12) into 1-undecene with the conversion ratio of 51.1 ±0.3%, approximately half that of OleTJE plus H2O2 (93.0 ±4.3% conversion; Figure 2). Notably, the product distribution (decarboxylation versus hydroxylation) of the OleTJE-RhFRED catalyzed reaction was similar to that of the OleTJE reaction (Figure 1B), indicating the fused reductase domain has no significant impact on the catalytic mechanism of P450 domain. As control, OleTJE cannot directly use NADPH as electron donor. OleTJE-RhFRED was not active by itself. Interestingly, OleTJE-RhFRED retained its ability to use H2O2 as a cofactor, but there was not a significant additive effect between NADPH and H2O2 for supporting the activity of OleTJE-RhFRED (Figure 2).

Gas chromatography-mass spectroscopy analysis of decarboxylation reactions (10 min) catalyzed by OleT JE or OleT JE -RhFRED under different reaction systems. (A) OleTJE + H2O2; (B) OleTJE + NADPH; (C) OleTJE-RhFRED + NADPH; (D) OleTJE-RhFRED + H2O2;(E) OleTJE-RhFRED + NADPH + H2O2;(F) OleTJE-RhFRED in absence of NADPH; (G) authentic standards of 1-undecene (U) lauric acid (LA) and heptadecanoic acid (IS: internal standard).
To prevent the spontaneous generation of H2O2 during the OleTJE-RhFRED reaction, which could complicate interpretation of the results, dithiothreitol (DTT: the reducing agent often used in the storage and reaction buffer of P450 proteins) was omitted from all buffers used during protein purification, storage and reaction because it has previously been reported that DTT, dioxygen and the heme iron center of P450 can react to generate H2O2[16, 42]. To further exclude the possibility that H2O2 could be produced from the peroxide shunt pathway during the monooxygenase catalytic cycle [27], bovine liver catalase that is able to efficiently eliminate H2O2 was added to the fatty acid decarboxylation reactions (in the DTT-free buffer) catalyzed by OleTJE and OleTJE-RhFRED. In the OleTJE plus H2O2 system, pre-addition of catalase completely abolished the reaction (Figure 3). By contrast, in the OleTJE-RhFRED plus NADPH reaction, pre-added catalase did not significantly change (indeed, it slightly improved) the conversion of myristic acid to 1-tridecene (Figure 3). This clearly indicates that the decarboxylation activity of OleTJE-RhFRED can be solely supported by NADPH.

Effects of catalase on the in vitro decarboxylation activity of three OleT JE (1 μM) reaction systems. Systems comprised OleT JE plus H 2 O 2 , OleT JE -RhFRED plus NADPH, and OleT JE plus flavodoxin and flavodoxin reductase plus NADPH. The percentage conversion of myristic acid is shown beside each bar.
Substrate specificity of OleTJE and OleTJE-RhFRED
The substrate preference of OleTJE and OleTJE-RhFRED provides useful information to help understand the in vivo behavior of these fatty acid decarboxylases and to guide the metabolic engineering of fatty acid biosynthesis. Thus, we determined their substrate specificity (Figure 4) using a number of straight-chain saturated fatty acids with even-numbered chain length ranging from C8 to C20. In the case of OleTJE (Figure 4A), myristic acid (C14) turned out to be the best substrate for olefin production among the tested fatty acids with the conversion ratio of 97.0 ± 2.8%. Lauric acid (C12) and palmitic acid (C16) were sub-optimal. OleTJE was only able to convert a minority of stearic acid (C18) and arachidic acid (C20), but could not decarboxylate capric acid (C10) or caprylic acid (C8).

Substrate preference spectrum of (A) OleT JE and (B) OleT JE -RhFRED. The substrate preference was determined by calculating the percentage conversion of each fatty acid substrate into corresponding α-alkene product. In these assays, 0.2 μM enzymes were used.
In comparison, the substrate preference spectrum of OleTJE-RhFRED slightly shifted toward shorter fatty acids (Figure 4B), which suggests that the P450-reductase interaction might induce small conformational change of the OleTJE active site. Specifically, OleTJE-RhFRED showed the highest activity against lauric acid (83.8 ±0.1% conversion), but was inactive toward arachidic acid. Similar to OleTJE, OleTJE-RhFRED could not decarboxylate capric acid or caprylic acid. It is also worth noting that the system of OleTJE-RhFRED plus NADPH plus O2 was less active than OleTJE plus H2O2 when reacting with most tested fatty acids except for lauric acid, suggesting OleTJE might have evolved to become a better peroxygenase than a monooxygenase.
In vivo production of α-alkenes in Escherichia coli
Motivated by the efficient in vitro conversion of free fatty acids (Cn) to corresponding α-alkenes (Cn-1) by two related but mechanistically distinct decarboxylation systems including OleTJE plus H2O2 and OleTJE-RhFRED plus NADPH plus O2, we sought to engineer hydrocarbon-producing strains of E. coli. To guarantee sufficient substrate supply of free fatty acids for OleTJE P450 decarboxylase, two previously engineered fatty acid-overproducing strains including XL100 (BL21:ΔfadD) and XL100/(pMSD8 + pMSD15) [43] were used as hosts for the decarboxylases. Transformation of these two strains with pET28b-oleT JE or pET28b-oleT JE -RhFRED resulted in YL5 (XL100 with pET28b-oleT JE ), YL6 (XL100 with pET28b-oleT JE -RhFRED), YL7 (XL100 with pET28b-oleT JE , pMSD8 and pMSD15), and YL8 (XL100 with pET28b-oleT JE -RhFRED, pMSD8 and pMSD15) (Table 1).
As expected, all these engineered strains successfully accumulated multiple α-alkenes, including 1-tridecene, trideca-1,6-diene, 1-pentadecene, pentadecene-1,8-diene and heptadeca-1,10-diene (Figure 5A and Additional file 4: Figure S4), which correspond to the decarboxylation products of the 14:0, 14:1, 16:0, 16:1 and 18:1 fatty acids, respectively. This fatty acid composition is consistent with previous reports [43]. In all cases, the major alkene product was heptadeca-1,10-diene with the highest yield of 7.7 mg·l-1 in YL7. Since XL100/(pMSD8 + pMSD15) is able to produce more free fatty acids than XL100 [43], the total alkene titers of YL7 and YL8 were significantly greater than those of YL5 and YL6. Notably, expression of P450 decarboxylases did not significantly affect the cell growth of all strains (Additional file 5: Figure S5).

Production of α -alkenes by heterologous expression of OleT JE or OleT JE -RhFRED in fatty acid-overproducing Escherichia coli strains. (A) Production of α-alkenes by OleTJE and OleTJE-RhFRED in two fatty acid-overproducing E. coli strains. The line chart denotes the alkene productivity. (B) The α-alkene production and fatty acid conversion profiles of strain YL7 in the defined mineral medium. The histograms demonstrate the titer of each detected α-alkene titers. The line chart demonstrates the conversion ratio of corresponding fatty acids. FFA, free fatty acid.
According to a previous report [44], H2O2 levels in growing E. coli are less than 20 nM, and 2 μM H2O2 could cause substantial growth inhibition. Therefore, the physiological concentration of H2O2 is far below the optimal H2O2 concentration (200 to 500 μM) [16] for supporting OleTJE catalysis. Thus, we had predicted that OleTJE-RhFRED using endogenous NADPH, whose physiological concentration is around 120 μM [45], might catalyze the decarboxylation more efficiently than OleTJE using H2O2 as a cofactor. Unexpectedly, the highest titer (11.8 mg·l-1) and productivity (4.5 mg·g dcw-1) of total alkenes was achieved by YL7 with OleTJE instead of YL8 with OleTJE-RhFRED. This contradiction has led to a hypothesis that OleTJE is able to employ the native redox system of E. coli to drive catalysis, such as P450CLA and P450BSβ[41].
To test this hypothesis, we expressed and purified the E. coli Fld and FdR. In the in vitro reaction containing OleTJE, Fld, FdR and NADPH, myristic acid was substantially converted into 1-tridecene (Figure 3). Therefore, Fld and FdR probably better serve OleTJE than the fused RhFRED in vivo, explaining why YL7 was a better alkene producer than YL8. However, the supportive role of H2O2 cannot be entirely neglected.
Inspired by these results, we attempted to enhance the level of dissolving dioxygen for improving OleTJE catalysis during the cultivation of the best alkene producer, YL7, by applying a higher rotation rate of 250 rpm. Moreover, the defined mineral medium [14] containing 3% glucose was used to standardize the culture condition for the future metabolic engineering, and the culture time was extended to 40 h. Significantly, the total alkene titer of YL7 under these conditions was 97.6 mg·l-1 (Figure 5B), which is almost seven-fold higher than that in lysogeny broth (LB) medium at 220 rpm for 20 h (Figure 5A). The productivity of total alkenes was also improved from 4.5 mg·g dcw-1 (in LB over 20 h) to 24.9 mg·g dcw-1 (in the defined mineral medium over 40 h, data not shown). Again, heptadeca-1,10-diene was the major alkene product (41.4 mg·l-1). As control, the production of free fatty acids by the strain XL100/(pMSD8 + pMSD15) without OleTJE expression was evaluated. Octadec-11-enoic acid was the most abundant fatty acid with a titer of 96.1 mg·l-1 (Additional file 6: Figure S6). This well explains why its decarboxylated product, heptadeca-1,10-diene, had the highest yield among produced alkenes (Figure 5). With respect to fatty acid conversion, octadec-11-enoic acid had the highest conversion ratio (85.9%) followed by hexadec-9-enoic acid (85.7%), myristic acid (75.7%), palmitic acid (74.0%) and lauric acid (24.4%). The low conversion of lauric acid seems to be inconsistent with the in vitro result (Figure 4A), which is likely due to the low intracellular concentration of the C12 fatty acid.
Discussion
The cytochrome P450 enzymes are a superfamily of b-type heme proteins, capable of catalyzing more than 20 different types of reactions [25, 46, 47]. The H2O2-assisted decarboxylation of long-chain fatty acids catalyzed by P450 OleTJE represents a novel activity of this highly versatile superfamily. This activity may be mechanistically similar to that of P450Rm (CYP53B) from the yeast Rhodotorula minuta[48], which decarboxylates isovalerate to form isobutene.
According to protein sequence alignment (Additional file 3: Figure S3), OleTJE belongs to the CYP152 peroxygenase family together with the well-studied P450BSβ and P450SPα[28, 29], and other members. Functionally, these P450 peroxygenases were previously thought to have no ability of utilizing dioxygen to drive catalysis as typical P450 monooxygenases do [39]. However, we have unambiguously demonstrated that OleTJE can perform H2O2-independent catalysis when partnering with the fused RhFRED reductase or the E. coli Fld/FdR system to transfer electrons from NADPH to the heme iron reactive center. This strongly suggests that OleTJE can undergo the monooxygenase catalytic cycle to generate the highly reactive ferryl-oxo cation radical species (Compound I) for catalysis. In the well-accepted mechanism for dioxygen activation in P450 monooxygenases (Figure 6), two protons (and two electrons from NADPH) are required for generation of Compound I [27]. However, the conserved threonine and an acidic residue involved in proton delivery in normal P450 monooxygenases [34, 40] are replaced by Pro and Arg, respectively, which are absolutely conserved in OleTJE and all other known CYP152 family members (Additional file 3: Figure S3). This strongly suggests an unknown proton transfer pathway. To elucidate this hypothetical pathway, we are currently seeking to solve the crystal structure of OleTJE.

Proposed two alternative catalytic mechanisms of OleT JE with (monooxygenase-like) or without (peroxygenase-like) redox systems. The Arg245 is proposed to be required for substrate anchoring via the strong electrostatic interactions with the carboxyl group of fatty acid substrate. The supply of two protons in the putative monooxygenase catalytic cycle remains unclear. The dashed arrow indicates the peroxide shunt pathway.
Evolutionarily, because the early Earth’s environment probably had more H2O2 and peroxygenated organic chemicals than O2, P450 peroxygenases are presumed to have emerged ahead of P450 monooxygenases [38]. Thus, the ability of most P450 monooxygenases to use H2O2 as a surrogate for the O2/2e-/2H+ system (peroxide shunt pathway, Figure 6) could be understood as a remnant function inherited from their peroxygenase ancestors, whereas the OleTJE P450 peroxygenase bearing the monooxygenase property might represent a transition species during the evolving process from peroxygenase to monooxygenase.
Under certain circumstances, such as in vitro synthetic reactions using purified P450s and application of P450 enzymes in laundry detergents, the peroxygenase activity supported by H2O2 is advantageous because expensive redox partner proteins and NAD(P)H are not needed [30, 31]. However, the peroxygenase nature is not a good feature if attempting to construct a biofuel-producing microorganism by taking advantage of the fatty acid decarboxylation activity of OleTJE. Essentially, the intracellular level of H2O2 cannot be increased to the concentration (101 to 102 μM range) required for efficiently supporting OleTJE because at this level, H2O2 is toxic or fatal to all organisms including E. coli. Thus, that the activity of P450 OleTJE can be supported by O2/redox partner(s)/NADPH besides H2O2 is a significant discovery because the former three factors are more viable targets for metabolic engineering [49–51]. Being aware of this, future metabolic engineering work aiming to improve the in vivo productivity of α-alkenes by OleTJE probably should be directed to improvement of the intracellular level of dioxygen, redox partner protein(s) and/or NADPH. For example, among numerous metabolic strategies for in vivo up-regulation of NADPH [49], the NADPH regenerating system could be used for maintaining the high intracellular level of this reducing cofactor to better support the activity of OleTJE and the overproduction of fatty acids [52, 53], both of which require a sufficient supply of NADPH.
In the mutant E. coli strains with up-regulated fatty acid biosynthesis (XL100 and XL100/(pMSD8 + pMSD15)), both OleTJE and OleTJE-RhFRED were functional, therefore significantly converting fatty acids into α-alkenes. Consistent with the previous report [16], the common major product of four engineered alkene producers (YL5-8) turned out to be heptadeca-1,10-diene, but the second most abundant alkene varied (Figure 5A). This is likely because octadec-11-enoic acid (the precursor of heptadeca-1,10-diene) was the predominant component of the fatty acid pool of the tested strains (Additional file 6: Figure S6). However, there might be an additional reason that octadec-11-enoic acid is a preferred substrate in vivo. Interestingly, in previous studies [43, 54, 55], palmitic acid rather than octadec-11-enoic acid was the major component in the fatty acid pool of E. coli. This inconsistency could be due to different culture conditions.
After 20 h cultures of YL7 in LB medium, significant amounts of fatty acids remained unreacted with OleTJE (Additional file 4: Figure S4). By contrast, the majority of fatty acids in YL7 were consumed by OleTJE over the longer and more oxygenated cultivation in the defined mineral medium (Figure 5B). This suggests that, at the current stage, the yield of fatty acids might be the major limiting factor for further improvement of α-alkene titers. Thus, the overproduction of fatty acids needs to be significantly optimized prior to other engineering efforts on up-regulating the level of OleTJE, redox partners, O2 and NADPH.
In this report, the best bio-hydrocarbon-producing strain YL7 accumulated 97.6 mg·l-1 of total alkenes. This yield is comparable to or better than a majority of engineered alkane and alkene biosynthetic pathways with reported yields. These include the artificial alkane biosynthetic pathway in E. coli consisting of the carboxylic acid reductase from Mycobacterium marinum that catalyzes formation of fatty aldehydes directly from fatty acids, and the aldehyde decarbonylase from Synechocystis sp. PCC 6803 to produce alkanes (yield: approximately 2 mg·l-1) [56]; the hybrid system using the fatty acid reductase complex (LuxC, LuxE and LuxD) to provide fatty aldehyde as the substrate for downstream aldehyde decarbonylase to generate alkanes (yield: approximately 2 to 5 mg·l-1) [20]; and the ATP required decarboxylation of 3-hydroxy-3-methylbutyrate catalyzed by the R74H mutant of mevalonate diphosphate decarboxylase from Saccharomyces cerevisiae (productivity: 5,888 pmol·h-1·g cells-1) [57]. It is only lower than the 300 mg·l-1 of total alkane titer produced when the cyanobacterial pathway consisting of the acyl-ACP reductase Orf1594 from Synechococcus elongates PCC7942 and the aldehyde decarbonylase from Nostoc punctiforme PCC73102 was heterologously expressed in E. coli[14]. However, it is expected that more metabolic engineering efforts and optimization of fermentation will further increase the total alkene titers of the OleTJE/E. coli system, which is currently ongoing in our laboratory.
Conclusions
The H2O2 independence of OleTJE described in this work not only raises a number of fundamental questions regarding its monooxygenase-like mechanism, but also could direct future metabolic engineering work toward improvement of O2/redox partner(s)/NADPH for optimal activity of OleTJEin vivo. Considering its high conversion rate in vitro and H2O2-independent functionality in vivo, it is of great potential for engineering a hyper-producer of α-alkenes on the basis of OleTJE.
Methods
Materials
Fatty acid substrates, terminal alkene authentic standards, and derivatizing reagents were purchased from TCI (Shanghai, China). Antibiotics were obtained from SolarBio (Beijing, China). Other chemicals were from Sigma Aldrich (St. Louis, MO, USA) or Ameresco (Solon, OH, USA). Oligonucleotides were synthesized by Sangon Biotech (Shanghai, China), and their sequences are shown in Additional file 7: Table S1. The Pfu DNA polymerases and all restriction endonucleases were obtained from Fermentas (Vilnius, Lithuania) or Takara (Dalian, China). The kits used for molecular cloning were from OMEGA Bio-Tek (Jinan, China) or Promega (Madison, WI, USA). Protein purification used Qiagen Ni-NTA resin (Valencia, CA, USA), Millipore Amicon Ultra centrifugal fliters (Billerica, MA, USA) and PD-10 desalting columns from GE Healthcare (Piscataway, NJ, USA). Bovine liver catalase was purchased from Sigma Aldrich.
Molecular cloning
Strains and plasmids constructed and used in this study are listed in Table 1. The gene of oleT JE was amplified from the genomic DNA of Jeotgalicoccus sp. ATCC 8456 using the primer pair of OleT-NdeI/OleT-Hin dIII (Table 1). The gel-cleaned PCR fragment was double digested by Nde I and Hin dIII and subsequently ligated into the Nde I/Hin dIII pre-treated pET28b to afford pET28b-oleT JE (Additional file 8: Figure S7A). For pET28b-oleT JE -RhFRED, the genes of oleT JE and RhFRED were first fused by overlap extension PCR [58]. Briefly, the gene encoding RhFRED reductase was amplified from the previously constructed pET28b-pikC-RhFRED[59, 60] with a pair of primers including RhFRED-F and RhFRED-R. The oleT JE gene was amplified from pET28b-oleT JE , using the primers OleT-F and OleT-RhFRED-OE. Then the two PCR fragments with overlap sequence were mixed, annealed, extended and finally amplified with the OleT-F/RhFRED-R primer pair, giving rise to the fused gene of oleT JE -RhFRED. This fusion product was digested by Nde I/Hin dIII and inserted into the Nde I/Hin dIII-digested pET28b to generate pET28b-oleT JE -RhFRED (Additional file 8: Figure S7B).
Using the genomic DNA of E. coli DH5α as template, the two genes encoding Fld and FdR were amplified with the primer pairs of Fld-BamHI-F/Fld-SalI-R and FdR-BamHI-F/FdR-SalI-R, respectively. Next, the fld and fdR genes were sub-cloned into pACYCDuet-1 and pCDFDuet-1 respectively, resulting in pACYCDuet-fld and pCDFDuet-fdR. All sub-cloned sequences were confirmed by DNA sequencing at Sangon Biotech, Shanghai.
Protein overexpression and purification
The E. coli BL21(DE3) cells carrying the recombinant expression vector were grown at 37°C for 16 to 20 h in LB medium containing certain selective antibiotics (50 μg·ml-1 kanamycin, 34 μg·ml-1 chloramphenicol, or 50 μg·ml-1 streptomycin), which were used to inoculate (1:100 ratio) Terrific Broth medium containing corresponding antibiotics, thiamine (1 mM), 10% glycerol and a rare salt solution [61]. Cells were grown at 37°C for 3 to 4 h until the optical density at 600 nm (OD600) reached 0.6 to 0.8, at which isopropyl-β-D-thiogalactopyranoside (IPTG, 0.2 mM as final concentration) and δ-aminolevulinic acid (0.5 mM, only for P450 expression) were added, followed by 18 h of cultivation at 18°C.
Protein purification was carried out as described elsewhere [61] with slight modifications. Specifically, the cell pellets harvested by centrifugation (5,000 × g, 4°C, 15 min) were stored at -80°C and melted at ambient temperature immediately before use. Then the cell pellets were resuspended in 40 ml of pre-chilled lysis buffer (pH 8.0, 50 mM NaH2PO4, 300 mM NaCl, 10% glycerol and 10 mM imidazole) through vortexing. Following a sonication step, the cell lysate was centrifuged at 12,000 × g for 30 min to remove the insoluble fraction. To the supernatant, 1 ml of Ni-NTA resin was added and gently mixed at 4°C for 1 h. The slurry was loaded onto an empty column, and washed with approximately 100 ml of wash buffer (pH 8.0, 50 mM NaH2PO4, 300 mM NaCl, 10% glycerol and 20 mM imidazole) until no proteins were detectable in flow-through. The bound target proteins were eluted with elution buffer (pH 8.0, 50 mM NaH2PO4, 300 mM NaCl, 10% glycerol and 250 mM imidazole). The eluent was concentrated with an Amicon Ultra centrifugal filter, and buffer exchanged on a PD-10 desalting column. Finally, the desalted purified proteins (Additional file 1: Figure S1) in storage buffer (pH 7.4, 50 mM NaH2PO4, 10% glycerol) were flash-frozen by liquid nitrogen and stored at -80°C for later use.
Determination of protein concentration
Following the method described by Omura and Sato [62], the CO-bound reduced difference spectrum (Additional file 2: Figure S2) of each P450 enzyme was recorded on a UV-visible spectrophotometer DU 800 (Beckman Coulter, Fullerton, CA, USA). The functional P450 concentration was calculated using the extinction coefficient (ϵ450–490) of 91,000 M-1·cm-1. The concentration of Fld and FdR from E. coli was determined using ϵ579 = 4,570 M-1·cm-1[63] and ϵ456 = 7,100 M-1·cm-1[64], respectively.
In vitro enzymatic assays with purified proteins
The fatty acid decarboxylation assays containing 0.2 to 1.0 μM OleTJE or OleTJE-RhFRED, 200 μM fatty acid substrate (from C8 to C20), 500 μM H2O2 (for OleTJE) or 500 μM NADPH (for OleTJE-RhFRED) in 200 μl of storage buffer were carried out at 28°C for 2 h. In the assay to test whether E. coli Fld and FdR are able to support the activity of OleTJE, 5 μM Fld and 5 μM FdR were mixed with 200 μM myristic acid, 500 μM NADPH and 1 μM OleTJE in 200 μl of storage buffer. To remove spontaneously generated H2O2, bovine liver catalase was added to the final concentration of 20 U·ml-1.
All above described reactions were quenched by addition of 20 μl of 10 M HCl. Heptadecanoic acid was added as internal standard and the mixture was extracted by 200 μl ethyl acetate. Samples were then analyzed by gas chromatography-mass spectroscopy (GC-MS; see below).
In vivo production of α-alkenes
The E. coli fadD deletion mutant strain XL100 [43] that overproduces fatty acids was selected as the starting host for construction of alkene-producing strains. Plasmids pMSD8 and pMSD15 were gifts from Dr. John Cronan and were used in the strain XL100 to construct fatty acid-overproducing strains. Bacterial cells transformed with certain plasmid(s) (Table 1) were either grown in 50 ml of LB medium or in 50 ml of defined mineral medium [14] containing 3% glucose as the carbon source, supplemented with appropriate selective antibiotics, thiamine (1 mM) and a rare salt solution. All cultivations were performed at 37°C and induced at an OD600 of 0.9 to 1.0 with 0.4% arabinose (for pMSD15) followed by 0.2 mM IPTG (for pMSD8, and pET28b-oleT JE or pET28b-oleT JE -RhFRED) after 0.5 h. Next, cells were grown at 28°C (the optimal temperature for OleTJE expression) for an additional 20 h at 220 rpm (in LB medium) or 40 h at 250 rpm (in the defined mineral medium). For analysis of hydrocarbon production, 20 ml of culture with 10 μl heptadecanoic acid added as internal standard was sonicated for 10 min and then thoroughly mixed with an equal volume of chloroform-methanol (2:1, vol/vol). The aqueous-organic mixture was centrifuged (8,000 × g for 15 min) for phase separation. The organic phase was transferred into a clean tube, evaporated under a nitrogen flow, and re-dissolved in 500 μl of n-hexane as the testing sample. Prior to GC-MS analysis, 5 μl eicosane was added as calibration standard. All experiments were repeated two to four times.
Analytical methods
The GC-MS analytical method for hydrocarbon and fatty acid samples was adapted from Guan et al.[65]. The analyses were performed on an Agilent 7890A gas chromatograph equipped with a capillary column HP-INNOWAX (Agilent Technologies, Santa Clara, CA, USA; cross-linked polyethylene glycerol, i.d. 0.25 μm film thickness, 30 m by 0.25 mm) coupled to an Agilent 5975C MSD single quadrupole mass spectrometer operated under electron ionization mode at 70 eV in the scan range of 50 to 500 m/z. The helium flow rate was set to 1 ml·min-1. The oven temperature was controlled initially at 40°C for 4 min, then increased at the rate of 10°C per min to 250°C, and held for 15 min. The injecting temperature was set to 280°C with the injection volume of 1 μl under splitless injection conditions. Under these conditions, the previously reported thermal degradations of α- and β-hydroxy fatty acids to form alkenes (the minor products of OleTJE catalyzed reaction) in the GC inlet [16] were not observed (Additional file 9: Figure S8), making the silylating protection of the hydroxyl group unnecessary if only caring about the α-alkene products. To detect α- and β-hydroxy fatty acids, samples were derivatized with an equal volume of N, O-bis(trimethylsilyl)trifluoroacetamide with 1% trimethylchlorosilane at 70°C for 15 min. GC-MS analysis followed the previous protocol developed by Rude et al.[16] except for using the Agilent J&W DB-5 MS column (i.d. 0.25 μm film thickness, 50 m by 0.25 mm). During GC-MS analysis, peak identity was determined by comparison of retention time and fragmentation pattern with authentic standard compounds where available and to the National Institute of Standards and Technology, USA mass spectral database. The location of double bond(s) in α-olefins was deduced by derivatization with dimethyl disulfide as described previously [66]. Quantification was achieved by comparison of integrated peak areas with calibration curves of authentic standards. The conversion percentages of free fatty acids to corresponding α-alkenes were estimated using the equation: [total alkenes]/([total alkenes] + [total free fatty acids]).
Abbreviations
- ACP:
-
acyl carrier protein
- CoA:
-
coenzyme A
- CYP:
-
cytochrome P450 enzyme
- DTT:
-
dithiothreitol
- FdR:
-
flavodoxin reductase
- Fld:
-
flavodoxin
- GC-MS:
-
gas chromatography-mass spectrometry
- IPTG:
-
Isopropyl-β-D-thiogalactopyranoside
- LB:
-
lysogeny broth
- OD:
-
optical density
- PCR:
-
polymerase chain reaction
- RhFRED:
-
Rhodococcus fusion reductase.
References
Shafiee S, Topal E: When will fossil fuel reserves be diminished? Energy Policy 2009, 37: 181-189. 10.1016/j.enpol.2008.08.016
Stephanopoulos G: Challenges in engineering microbes for biofuels production. Science 2007, 315: 801-804. 10.1126/science.1139612
Kerr RA: Global warming is changing the world. Science 2007, 316: 188-190. 10.1126/science.316.5822.188
Peralta-Yahya PP, Zhang F, del Cardayre SB, Keasling JD: Microbial engineering for the production of advanced biofuels. Nature 2012, 488: 320-328. 10.1038/nature11478
Lee SK, Chou H, Ham TS, Lee TS, Keasling JD: Metabolic engineering of microorganisms for biofuels production: from bugs to synthetic biology to fuels. Curr Opin Biotechnol 2008, 19: 556-563. 10.1016/j.copbio.2008.10.014
Savage DF, Way J, Silver PA: Defossiling fuel: how synthetic biology can transform biofuel production. ACS Chem Biol 2008, 3: 13-16. 10.1021/cb700259j
Demain AL: Biosolutions to the energy problem. J Ind Microbiol Biotechnol 2009, 36: 319-332. 10.1007/s10295-008-0521-8
Fortman JL, Chhabra S, Mukhopadhyay A, Chou H, Lee TS, Steen E, Keasling JD: Biofuel alternatives to ethanol: pumping the microbial well. Trends Biotechnol 2008, 26: 375-381. 10.1016/j.tibtech.2008.03.008
Li H, Cann AF, Liao JC: Biofuels: biomolecular engineering fundamentals and advances. Annu Rev Chem Biomol Eng 2010, 1: 19-36. 10.1146/annurev-chembioeng-073009-100938
Wackett LP: Biomass to fuels via microbial transformations. Curr Opin Chem Biol 2008, 12: 187-193. 10.1016/j.cbpa.2008.01.025
Zhang F, Rodriguez S, Keasling JD: Metabolic engineering of microbial pathways for advanced biofuel production. Curr Opin Biotechnol 2011, 22: 775-783. 10.1016/j.copbio.2011.04.024
Qiu Y, Tittiger C, Wicker-Thomas C, Le Goff G, Young S, Wajnberg E, Fricaux T, Taquet N, Blomquist GJ, Feyereisen R: An insect-specific P450 oxidative decarbonylase for cuticular hydrocarbon biosynthesis. Proc Natl Acad Sci USA 2012, 109: 14858-14863. 10.1073/pnas.1208650109
Niehaus TD, Okada S, Devarenne TP, Watt DS, Sviripa V, Chappell J: Identification of unique mechanisms for triterpene biosynthesis in Botryococcus braunii . Proc Natl Acad Sci USA 2011, 108: 12260-12265. 10.1073/pnas.1106222108
Schirmer A, Rude MA, Li X, Popova E, del Cardayre SB: Microbial biosynthesis of alkanes. Science 2010, 329: 559-562. 10.1126/science.1187936
Beller HR, Goh EB, Keasling JD: Genes involved in long-chain alkene biosynthesis in Micrococcus luteus . Appl Environ Microbiol 2010, 76: 1212-1223. 10.1128/AEM.02312-09
Rude MA, Baron TS, Brubaker S, Alibhai M, del Cardayre SB, Schirmer A: Terminal olefin (1-alkene) biosynthesis by a novel P450 fatty acid decarboxylase from Jeotgalicoccus species. Appl Environ Microbiol 2011, 77: 1718-1727. 10.1128/AEM.02580-10
Alibhai MF, Rude MA, Schirmer A: Methods and compositions for producing olefins. 20110196180 A1 (Patent) 2012
Mendez-Perez D, Begemann MB, Pfleger BF: Modular synthase-encoding gene involved in α -olefin biosynthesis in Synechococcus sp. strain PCC 7002. Appl Environ Microbiol 2011, 77: 4264-4267. 10.1128/AEM.00467-11
Gu L, Wang B, Kulkarni A, Gehret JJ, Lloyd KR, Gerwick L, Gerwick WH, Wipf P, Håkansson K, Smith JL, Sherman DH: Polyketide decarboxylative chain termination preceded by O -sulfonation in curacin A biosynthesis. J Am Chem Soc 2009, 131: 16033-16035. 10.1021/ja9071578
Howard TP, Middelhaufe S, Moore K, Edner C, Kolak DM, Taylor GN, Parker DA, Lee R, Smirnoff N, Aves SJ, Love J: Synthesis of customized petroleum-replica fuel molecules by targeted modification of free fatty acid pools in Escherichia coli . Proc Natl Acad Sci USA 2013, 110: 7636-7641. 10.1073/pnas.1215966110
Lennen RM, Pfleger BF: Engineering Escherichia coli to synthesize free fatty acids. Trends Biotechnol 2012, 30: 659-667. 10.1016/j.tibtech.2012.09.006
Youngquist JT, Lennen RM, Ranatunga DR, Bothfeld WH, Marner WD II, Pfleger BF: Kinetic modeling of free fatty acid production in Escherichia coli based on continuous cultivation of a plasmid free strain. Biotechnol Bioeng 2012, 109: 1518-1527. 10.1002/bit.24420
Popov YV, Uskach YL, Ledenev SM, Kroman DA, Pavlova VA: Synthesis of a sulfur-containing additive to transmission lubricants based on the С20–С26 fraction of α -olefins. Russ J Appl Chem 2013, 86: 447-449. 10.1134/S1070427213030257
Kioupis LI, Maginn EJ: Molecular simulation of poly- α -olefin synthetic lubricants: impact of molecular architecture on performance properties. J Phys Chem B 1999, 103: 10781-10790. 10.1021/jp992399n
Coon MJ: Cytochrome P450: nature’s most versatile biological catalyst. Annu Rev Pharmacol Toxicol 2005, 45: 1-25. 10.1146/annurev.pharmtox.45.120403.100030
Munro AW, Girvan HM, McLean KJ: Variations on a (t)heme-novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat Prod Rep 2007, 24: 585-609. 10.1039/b604190f
Poulos TL, Johnson EF: Cytochrome P450: Structure, Mechanism, and Biochemistry. 3rd edition. New York: Kluwer Academic/Plenum Publishers; 2005.
Matsunaga I, Sumimoto T, Ueda A, Kusunose E, Ichihara K: Fatty acid-specific, regiospecific, and stereospecific hydroxylation by cytochrome P450 (CYP152B1) from Sphingomonas paucimobilis : substrate structure required for α -hydroxylation. Lipids 2000, 35: 365-371. 10.1007/s11745-000-533-y
Matsunaga I, Ueda A, Fujiwara N, Sumimoto T, Ichihara K: Characterization of the ybdT gene product of Bacillus subtilis: novel fatty acid β -hydroxylating cytochrome P450. Lipids 1999, 34: 841-846. 10.1007/s11745-999-0431-3
Joo H, Lin Z, Arnold FH: Laboratory evolution of peroxide-mediated cytochrome P450 hydroxylation. Nature 1999, 399: 670-673. 10.1038/21395
Cirino PC, Arnold FH: A self-sufficient peroxide-driven hydroxylation biocatalyst. Angew Chem Intl Ed 2003, 42: 3299-3301. 10.1002/anie.200351434
Andre C, Kim SW, Yu X-H, Shanklin J: Fusing catalase to an alkane-producing enzyme maintains enzymatic activity by converting the inhibitory byproduct H2O2 to the cosubstrate O2. Proc Natl Acad Sci USA 2013, 110: 3191-3196. 10.1073/pnas.1218769110
Roberts GA, Grogan G, Greter A, Flitsch SL, Turner NJ: Identification of a new class of cytochrome P450 from a Rhodococcus sp. J Bacteriol 2002, 184: 3898-3908. 10.1128/JB.184.14.3898-3908.2002
Lee DS, Yamada A, Sugimoto H, Matsunaga I, Ogura H, Ichihara K, Adachi S, Park SY, Shiro Y: Substrate recognition and molecular mechanism of fatty acid hydroxylation by cytochrome P450 from Bacillus subtilis . Crystallographic, spectroscopic, and mutational studies. J Biol Chem 2003, 278: 9761-9767. 10.1074/jbc.M211575200
Shoji O, Fujishiro T, Nakajima H, Kim M, Nagano S, Shiro Y, Watanabe Y: Hydrogen peroxide dependent monooxygenations by tricking the substrate recognition of cytochrome P450BSβ. Angew Chem Intl Ed 2007, 46: 3656-3659. 10.1002/anie.200700068
Fujishiro T, Shoji O, Nagano S, Sugimoto H, Shiro Y, Watanabe Y: Crystal structure of H2O2-dependent cytochrome P450SPα with its bound fatty acid substrate: insight into the regioselective hydroxylation of fatty acids at a position. J Biol Chem 2011, 286: 29941-29950. 10.1074/jbc.M111.245225
Munro AW, Girvan HM, Mason AE, Dunford AJ, McLean KJ: What makes a P450 tick? Trends Biochem Sci 2013, 38: 140-150. 10.1016/j.tibs.2012.11.006
Hrycay EG, Bandiera SM: The monooxygenase, peroxidase, and peroxygenase properties of cytochrome P450. Arch Biochem Biophys 2012, 522: 71-89. 10.1016/j.abb.2012.01.003
Matsunaga I, Sumimoto T, Ayata M, Ogura H: Functional modulation of a peroxygenase cytochrome P450: novel insight into the mechanisms of peroxygenase and peroxidase enzymes. FEBS Lett 2002, 528: 90-94. 10.1016/S0014-5793(02)03261-1
Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG: The catalytic pathway of cytochrome P450cam at atomic resolution. Science 2000, 287: 1615-1622. 10.1126/science.287.5458.1615
Girhard M, Schuster S, Dietrich M, Duerre P, Urlacher VB: Cytochrome P450 monooxygenase from Clostridium acetobutylicum : a new α -fatty acid hydroxylase. Biochem Biophys Res Comm 2007, 362: 114-119. 10.1016/j.bbrc.2007.07.155
Netto LE, Stadtman ER: The iron-catalyzed oxidation of dithiothreitol is a biphasic process: hydrogen peroxide is involved in the initiation of a free radical chain of reactions. Arch Biochem Biophys 1996, 333: 233-242. 10.1006/abbi.1996.0386
Lu X, Vora H, Khosla C: Overproduction of free fatty acids in E. coli : implications for biodiesel production. Metab Eng 2008, 10: 333-339. 10.1016/j.ymben.2008.08.006
Seaver LC, Imlay JA: Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli . J Bacteriol 2001, 183: 7182-7189. 10.1128/JB.183.24.7182-7189.2001
Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD: Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli . Nat Chem Biol 2009, 5: 593-599. 10.1038/nchembio.186
Guengerich FP: Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol 2001, 14: 611-650. 10.1021/tx0002583
Guengerich FP, Munro AW: Unusual cytochrome P450 enzymes and reactions. J Biol Chem 2013, 288: 17065-17073. 10.1074/jbc.R113.462275
Fujii T, Nakamura K, Shibuya K, Tanase S, Gotoh O, Ogawa T, Fukuka H: Structural characterization of the gene and corresponding cDNA for the cytochrome P450rm from Rhodotorula minuta which catalyzes formation of isobutene and 4-hydroxylation of benzoate. Mol Gen Genet 1997, 256: 115-120. 10.1007/s004380050552
Fasan R, Crook NC, Peters MW, Meinhold P, Buelter T, Landwehr M, Cirino PC, Arnold FH: Improved product-per-glucose yields in P450-dependent propane biotransformations using engineered Escherichia coli . Biotechnol Bioeng 2011, 108: 500-510. 10.1002/bit.22984
Wei X-X, Chen G-Q: Applications of the VHb gene vgb for improved microbial fermentation processes. Methods Enzymol 2008, 436: 273-287.
Bai L, Li P, Zhang H, Duan Y, Lin Z: Semi-rational engineering of cytochrome P450sca-2 in a hybrid system for enhanced catalytic activity: insights into the important role of electron transfer. Biotechnol Bioeng 2013, 110: 2815-2835. 10.1002/bit.24960
Yu X, Liu T, Zhu F, Khosla C: In vitro reconstitution and steady-state analysis of the fatty acid synthase from Escherichia coli . Proc Natl Acad Sci USA 2011, 108: 18643-18648. 10.1073/pnas.1110852108
Liu T, Vora H, Khosla C: Quantitative analysis and engineering of fatty acid biosynthesis in E. coli . Metab Eng 2010, 12: 378-386. 10.1016/j.ymben.2010.02.003
Sullivan KH, Hegeman GD, Cordes EH: Alteration of the fatty acid composition of Escherichia coli by growth in the presence of normal alcohols. J Bacteriol 1979, 138: 133-138.
Marr AG, Ingraham JL: Effect of temperature on the composition of fatty acids in Escherichia coli . J Bacteriol 1962, 84: 1260-1267.
Akhtar MK, Turner NJ, Jones PR: Carboxylic acid reductase is a versatile enzyme for the conversion of fatty acids into fuels and chemical commodities. Proc Natl Acad Sci USA 2013, 110: 87-92. 10.1073/pnas.1216516110
Gogerty DS, Bobik TA: Formation of isobutene from 3-hydroxy-3-methylbutyrate by diphosphomevalonate decarboxylase. Appl Environ Microbiol 2010, 76: 8004-8010. 10.1128/AEM.01917-10
Heckman KL, Pease LR: Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc 2007, 2: 924-932. 10.1038/nprot.2007.132
Li S, Podust LM, Sherman DH: Engineering and analysis of a self-sufficient biosynthetic cytochrome P450 PikC fused to the RhFRED reductase domain. J Am Chem Soc 2007, 129: 12940-12941. 10.1021/ja075842d
Li S, Chaulagain MR, Knauff AR, Podust LM, Montgomery J, Sherman DH: Selective oxidation of carbolide C–H bonds by an engineered macrolide P450 mono-oxygenase. Proc Natl Acad Sci USA 2009, 106: 18463-18468. 10.1073/pnas.0907203106
Anzai Y, Li S, Chaulagain MR, Kinoshita K, Kato F, Montgomery J, Sherman DH: Functional analysis of MycCI and MycG, cytochrome P450 enzymes involved in biosynthesis of mycinamicin macrolide antibiotics. Chem Biol 2008, 15: 950-959. 10.1016/j.chembiol.2008.07.014
Omura T, Sato R: The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J Biol Chem 1964, 239: 2379-2385.
Fujii K, Galivan JH, Huennekens FM: Activation of methionine synthase: further characterization of flavoprotein system. Arch Biochem Biophys 1977, 178: 662-670. 10.1016/0003-9861(77)90238-7
Jenkins CM, Waterman MR: NADPH-flavodoxin reductase and flavodoxin from Escherichia coli : characteristics as a soluble microsomal P450 reductase. Biochemistry 1998, 37: 6106-6113. 10.1021/bi973076p
Guan W, Zhao H, Lu X, Wang C, Yang M, Bai F: Quantitative analysis of fatty-acid-based biofuels produced by wild-type and genetically engineered cyanobacteria by gas chromatography–mass spectrometry. J Chromatogr A 2011, 1218: 8289-8293. 10.1016/j.chroma.2011.09.043
Bauer S, Schulte E, Thier H-P: Composition of the surface wax from tomatoes. I. Identification of the components by GC/MS. Eur Food Res Technol 2009, 219: 223-228.
Acknowledgements
This work was supported by National Natural Science Foundation of China under grant number NSFC31270855 and funding from the Recruitment Program of Global Experts, 2012. It was also carried out in the framework of the Boeing-QIBEBT Joint Research Laboratory for Sustainable Aviation Biofuels Collaboration Agreement. We are grateful to Mr Xufeng Liu, Mr Zhi Zhu and Dr Xiaoming Tan for helpful strategic and technical discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SL conceived of the study. YL, CW, XL and SL designed the experiments. YL, JY and WZ performed the experiments including plasmid construction, protein overexpression, purification, characterization and enzymatic assays. YL, CW and WG carried out GC-MS analysis. YL, CW and SL drafted the manuscript. JY, WZ, WG and XL helped to revise the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
13068_2013_427_MOESM1_ESM.tiff
Additional file 1: Figure S1: SDS-PAGE analysis of purified (A) OleTJE and (B) OleTJE-RhFRED. M, protein marker. (TIFF 7 MB)
13068_2013_427_MOESM3_ESM.tiff
Additional file 3: Figure S3: Protein sequence alignment of CYP152 family members and three selected P450 monooxygenases. Protein sequences were obtained from NCBI protein databases. CYP152A1 (P450BSβ from Bacillus subtilis); CYP152A2 (P450CLA from Clostridium acetobutylicum); CYP152B1 (P450SPα from Sphingomonas paucimobilis); CYP152B2 (from Azotobacter vinelandii); CYP152C1 (from Rhodobacter sphaeroides); CYP152C2 (from Rhodobacter sphaeroides); CYP152D1 (from Streptomyces scabies); CYP152E1 (from Cyanothece sp. CCY0110); CYP107L1 (P450PikC from Streptomyces venezuelae); CYP102A1 (P450BM3 from Bacillus megaterium); CYP101A1 (P450cam from Pseudomonas putida). Conserved amino acid residues are shaded. The Arg and Pro absolutely conserved in CYP152 family are marked by asterisks. (TIFF 2 MB)
13068_2013_427_MOESM4_ESM.tiff
Additional file 4: Figure S4: GC-MS analysis of the organic extract of the YL7 culture in LB broth. Eicosane and heptadecanoic acid are served as calibration standard (CS) and internal standard (IS), respectively. (TIFF 1 MB)
13068_2013_427_MOESM8_ESM.tiff
Additional file 8: Figure S7: Plasmid maps for (A) pET28b-oleT JE and (B) pET28b-oleT JE -RhFRED expression vectors. (TIFF 823 KB)
13068_2013_427_MOESM9_ESM.tiff
Additional file 9: Figure S8: GC-MS analysis of the α- and β-hydroxy myristic acid authentic standards on HP-INNOWAX capillary column. (A) Without derivatization, α-hydroxy myristic acid was unseen due to its high boiling point. Importantly, no thermally degraded terminal olefin (1-tridecene) was observed in the dashed box. (B) Without derivatization, β-hydroxy myristic acid was unseen due to its high boiling point. Again, no thermally degraded terminal olefin (1-tridecene) was observed in the dashed box. (C) The decarboxylation reaction of myristic acid (2 h) catalyzed by OleTJE. The peak shown in the dashed box corresponds to 1-tridecene. (TIFF 627 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Liu, Y., Wang, C., Yan, J. et al. Hydrogen peroxide-independent production of α-alkenes by OleTJE P450 fatty acid decarboxylase. Biotechnol Biofuels 7, 28 (2014). https://doi.org/10.1186/1754-6834-7-28
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1754-6834-7-28