
Abstract
Initially, the CALPHAD (Calculation of Phase Diagrams) method was established as a tool for treating thermodynamics and phase equilibria of multicomponent systems. Since then the method has been successfully applied to diffusion mobilities in multicomponent systems, creating the foundation for simulation of diffusion processes in these systems. Recently, the CALPHAD method has been expanded to other phase-based properties, including molar volumes and elastic constants, and has the potential to treat electrical and thermal conductivity and even two-phase properties, such as interfacial energies. Advances in the CALPHAD method or new information on specific systems frequently require that already assessed systems be re-assessed. Therefore, the next generation of CALPHAD necessitates data repositories so that when new models are developed or new experimental and computational information becomes available the relevant low-order (unary, binary, and ternary) systems can be re-assessed efficiently to develop the new multicomponent descriptions. The present work outlines data and infrastructure needs for efficient CALPHAD assessments and updates, highlighting the requirement for data repositories with flexible data formats that can be accessed by a variety of tools and that can evolve as data needs change. Within these repositories, the data must be stored with the appropriate metadata to enable the evaluation of the confidence of the stored data.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Review
The first efforts using computational methods to describe Gibbs energy functions to represent the phases and describe phase equilibria were made more than 60 years ago as reviewed in[1]. However, only after computers became available did these efforts become systematic. In 1970, Kaufman and Bernstein[2] presented a collection of analytical thermodynamic descriptions of the Gibbs energy of the phases of binary and ternary systems as functions of temperature and concentration. These descriptions could be used for the calculation of phase equilibria and thermochemical properties in a large number of systems. This collection established the CALPHAD method as a valuable tool for the treatment of multicomponenta phase equilibria and spawned the development of several software packages and databases with collections of thermodynamic descriptions of multicomponent systems[1]. However, it soon became obvious that more sophisticated models were needed to account for the structure (long and short range order) of these phases when describing the concentration dependence. With the development of more physics-based model descriptions[3] the reproducibility of the experimental information improved, but also required the revisions of systems that had been already described by Kaufman and Bernstein[2].
Kaufman and Bernstein also presented the concept of “lattice stabilities” to describe the thermochemical properties of the stoichiometric and often hypothetical end-member phases in solid solutions. Although it is straightforward to obtain functions for the stable forms of an element from their thermochemical properties, obtaining these quantities for end-members that are not stable proved to be a challenge. In the early days of CALPHAD these quantities were frequently estimated from the extrapolation of the properties of the solution phases to the non-stableb end-member phase. This practice resulted in different quantities being used by different authors for the same non-stable end-member form of an element or phase. This was a severe impediment to the development of descriptions for multicomponent systems since only one description of an element (phase) in a specific structure can be used, e.g., the description of end-member A must be the same in all systems A-B, A-C, etc. This problem was greatly alleviated by the publication of the lattice stabilities of the elements by Dinsdale[4]. These lattice stabilities allowed the development of descriptions that were consistent with respect to the description of the elements, but also made the re-assessment of older descriptions necessary.
The approach used by the CALPHAD method lends itself to the modeling of other phase properties. It has been successfully employed for the description of diffusion mobilities[5, 6], molar volumes[7], and elastic constants[8]. Since the descriptions of these properties are also based on the properties of the end-members and the models being used, these properties are also strongly affected by any change to the model.
The strengths of the CALPHAD method are that the data obtained from the calculation with a CALPHAD description are self-consistent and that these descriptions can be used for the extrapolation[9] of multicomponent systems. This makes the CALPHAD method attractive for the prediction of materials properties[10] and the coupling with materials simulation codes, such as solidification simulations[11] or phase field simulations[12]. With this ability to describe multicomponent materials properties as functions of composition and temperature, CALPHAD-based approaches have served as the foundation for several successful materials design programs[13, 14].
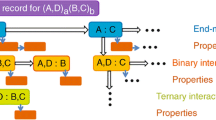
For the construction of functional databases to describe multicomponent systems it is imperative that the same models are used for the description of the temperature, pressure and the concentration dependence of the Gibbs energy of each phase in all of the constitutive binary and ternary systems. Therefore, any modification of a constitutive subsystem has a compounding effect on the description of a multicomponent system because it affects every description of systems that includes this subsystem; e.g., a change in the binary A-B system affects the description of the ternary systems A-B-C, A-B-D, etc. This compounding effect also makes re-assessments of higher component subsystems necessary. Figure 1 demonstrates this compounding effect illustrating the number of binary and ternary re-assessments needed if one unary description is changed in a six-component system. Since the inception of the CALPHAD method, models, functions for end-member phases (including the quantities of the pure elements) and the descriptions of the systems have been constantly evolving and re-assessments are continuously needed[15]. These re-assessments are needed not only as the result of the availability of improved model descriptions or lattice stabilities for end-member phases, but also due to the availability of new experimental and theoretical data that improve the knowledge of a system. Although each re-assessment is progress, it is also a setback in the development of the description of multicomponent systems because of the previously mentioned compounding effect that delays the growth of these databases and the extension to the modeling of other phase properties using the CALPHAD method. As a result of this the CALPHAD community has become increasingly reluctant to adopt changes in models and end-member phase properties, even when it is known that updates need to be executed. This clearly demonstrates the urgent need for tools and data repositories to streamline the re-assessment process and make it more efficient.

The data dependencies in the development of multicomponent CALPHAD databases are illustrated in the above figure. For the six component A-B-C-D-E-F system, if the unary description for component C is changed then all the circled binary and ternary systems must be re-evaluated.
The purpose of this article is to give a perspective of future development of the CALPHAD method within the concept of the Materials Genome Initiative[16] and to identify data repositories, data compilation and software needs.
Thermodynamics
Thermodynamics defines the state of a system when interacting with the surroundings, based on the first and second laws of thermodynamics. The first law of thermodynamics describes those interactions, and the second law of thermodynamics governs the evolution inside the system. Consequently, how the system is controlled from the surroundings dictates the state function used to describe the system. Under typical experimental conditions, temperature, pressure, and mass are the variables being controlled, thus the Gibbs energy is the characteristic state function widely used in materials science and engineering. The Gibbs energy of a system with more than one phase is the sum of the Gibbs energies of individual phases. The Gibbs energy of a stoichiometric phase, i.e., a completely ordered phase, can be simply described by its Gibbs energy of formation from its components.
For a solution phase, the chemical activity of a component, i, is defined as ai = fixi with fi being the activity coefficient and xi the mole fraction, and its value depends on the reference state where ai = 1. A commonly used reference state is the stable element reference (SER) state, i.e., its stable structure at 298 K and 105 Pa, with its chemical potential denoted by, though it is sometimes more convenient to use a pure component in the same structure of the solution as the reference state of chemical activity, with its chemical potential denoted by, where α represents a specific structure/phase. The Gibbs energy can thus be written using both reference states of chemical activities as follows.
With.
where and are the excess Gibbs energy of mixing in J/mol, and is the lattice stability of component i in structure α with respect to the SER state, T is the temperature in K and R is the gas constant in J/(mol K). It is self-evident that the lattice stability is extremely important as it dictates the value of Gibbs energy of mixing. Any change in lattice stability will result in a different excess Gibbs energy of mixing to maintain the same Gibbs energy of the phase. The excess Gibbs energy of mixing is commonly modeled using the Muggianu extension of the Redlich-Kister formalism due to its symmetrical characteristics when applied to multicomponent systems[17, 18], i.e.,
where the summation of i, j, k is over all components, and, Li, Lj, and Lk are the excess binary and ternary mixing terms and are temperature-dependent using a form similar to that shown in[4], i.e.,
where the coefficients (a,b,c,d,e, and f) are model parameters evaluated from thermochemical and phase equilibrium data, typically with m ≤ 2 and only and being used. Model parameters often need to be re-evaluated when new data become available, particularly data for metastable phases and metastable states. New data at low temperatures now accessible by first-principles calculations based on density functional theory (DFT)[19] also necessitate the revision of model parameters to extend and validate the Gibbs energy functions to 0 K. The Einstein and Debye models have been proposed to describe the heat capacity[20], and the implementation of these models into CALPHAD descriptions is being investigated by several groups[21–23]. Vřešt’ál et al.[24] developed an extension of the Dinsdale[4] Gibbs energy functions of the pure elements to 0 K employing an extended Einstein model. New model descriptions for the temperature and pressure dependence of the pure elements have been discussed at a recent workshop[25–29].
For solution phases with short-range and long-range ordering additional order-parameters are needed to fully define the state of the phase. Long-range ordering is commonly treated by sublattice models[30] with the Gibbs energy of mixing defined in the individual sublattices (This is discussed in detail in the Section “Crystallography”). The cluster site approximation[31] treats long-range and short-range ordering; however, sublattice models were shown capable of including both long-range and short-range ordering[32, 33]. Magnetic ordering has been treated by adding a magnetic contribution to the total Gibbs energy. This magnetic contribution is based on the local magnetic moment theory of Curie and Weiss[34, 35]. However, it has been recognized that current descriptions of the magnetic contributions to the Gibbs energy still cannot accurately describe all the observed properties[21, 27, 36, 37], and a concept of mixture of individual magnetic microstates has been proposed[38–41].
Modeling of a liquid remains a challenge, particularly the liquid properties below the melting temperature. A two-state model[42] has been suggested to describe the temperature dependence of the Gibbs energy but this model has been demonstrated for only a few cases[36, 43] and is partially supported by the recent DFT-based molecular dynamic simulations[44]. Short-range ordering in the liquid phase can be described using a quasi-chemical model[45], the associate model[46, 47] or the liquid ionic sublattice model[48].
Currently, CALPHAD modeling is based on the Gibbs energy, which describes a thermodynamic system at constant temperature and pressure, as most experimental data are collected under these conditions. However, describing a thermodynamic system at constant temperature and volume using the Helmholtz energy may be useful for direct implementation of data from first-principles calculations and coupling with diffusion simulations of systems with large molar volume differences. Although, Lu and Chen[49] have investigated the implementation of the Helmholtz energy within the CALPHAD approach for fcc-Cu, this approach has yet to be tested for multicomponent, multiphase systems.
Crystallography
The basis for any CALPHAD description is the model selection. The models for temperature and pressure dependence are usually selected for the entire system while the model for the concentration dependence is specific for each phase. The modeling of ordered phases with homogeneity ranges or interstitial solutions are usually described with the sublattice model. In the early days of the CALPHAD method, the specifics of the sublattice model were chosen in part out of convenience to describe the appearance of a phase in the phase diagram without much consideration of the physical properties of the phase. However, for accurate description of the phase properties and reliable extrapolation to multicomponent systems it is imperative that the crystal structure of the phase is considered when the model is chosen. The choice of model has not only implications on the thermodynamic modeling, but also on the modeling of diffusion, molar volume and other phase-based properties.
The basic premise of the sublattice model is that a sublattice is assigned for each distinct site in the crystal structure. Each of the sublattices can be occupied by one or more species, which are usually elements, ions or a vacancy forming regular type solutions. End-members of a phase represent the phase with only one species on each sublattice. In a full thermodynamic description of a multicomponent phase the number of parameters to describe the end-members and regular type interactions could become excessively large. For example, if all the elements in the system were allowed to occupy each sublattice in a phase, the number of end-member phases would be nk where n is the number of elements and k is the number of sublattices. One option is to generate these parameters from first-principles calculations, and the other option is to reduce the number of parameters by introducing constraints or decreasing the number of sublattices. Constraints are usually based on nearest neighbor and, maybe, second nearest neighbor configurations while similarities, such as co-ordination number, point symmetry etc., and the changes in site fraction upon deviation from the ideal stoichiometry are used as criterion for combining different sublattices. Therefore, the use of crystallographic information is crucial for selecting the proper model descriptions.
The selection of the model to describe the concentration dependence of the Gibbs energy function of a phase is typically made during the assessment of a binary or ternary system. Consideration of the phase properties only in the system being assessed may result in a model description that is not suitable to describe the phase correctly in a multicomponent system. For example, the substitutional behavior on two or more distinct but similar sites could be very similar in the A-B system while in the A-C or A-B-C system the substitutional behavior may be quite different. If in the assessment of the A-B system the model is simplified by combining the similar sites into one sublattice, this model would not be capable of describing the substitutional behavior in the A-C or A-B-C systems. Therefore, it is essential to examine all the mechanisms of deviation from stoichiometry of a phase in all systems where this phase occurs before any combinations of sublattices are used for its model description. Therefore, site occupation data are also important data for the refinement of the model parameters.
The crystallographic information needed to correctly determine a phase can be found in classic compilations, such as Pearson’s Handbook[50], or in electronic databases, such as ICSD[51], Pauling File[52] or Pearson’s Crystal Data[53]. The advantage of electronic databases is that they are easily searchable and allow with relatively little effort the extraction of phase information for all elemental combinations for which data for this phase are available. The electronic databases also offer the advantage that they can be combined with other software that could be developed to assist in model selection. Since the model of the crystal structure is critical to the development of functional databases for multicomponent systems, unique identifiers for the crystal structure that are linked to the description of the individual phases are needed. Such an identifier could be the space group and Wyckoff sequence as recommended by Brown et al.[54] and adopted by the International Union of Pure and Applied Chemistry (IUPAC).
Phase diagram data
Important phase diagram data for the development and verification of a CALPHAD type description are phase boundary data. These data are either determined from isothermal measurements or scans of phase changes with heating and cooling experiments. In most of these experimental measurements the pressure is kept constant. Isothermal measurements are usually carried out on samples that have been heat treated and quenched; only in rare cases is an isothermal measurement made in situ. These measurements can be either quantitative phase analysis, such as electron probe microanalysis (EPMA) or energy dispersive x-ray analysis (EDX), or semi-quantitative observations of phase changes with slightly different compositions in metallographic or x-ray diffraction samples. Scans with temperature can be simple thermal analysis (TA), differential thermal analysis (DTA) or differential scanning calorimetry (DSC). With these latter methods the signal caused by the enthalpy change in the material resulting from the phase change is recorded. Zhao[55] provides an overview of the experimental methods available for the determination of phase diagrams.
For an assessment of the thermodynamic description using an optimization procedure the experimental error associated with these data needs to be input. In addition to the experimental error itself other factors that affect accuracy must also be considered, such as material purity and heat treatment history of the sample. Therefore, it is imperative that a repository of experimental phase diagram data contains all this information. Unfortunately not all experimental work reported in the literature contains these data in sufficient detail and some of these accuracies need to be estimated during an assessment. When auxiliary estimates on data accuracy have been made in an assessment this also needs to be documented.
Thermochemical data
The availability of thermochemical data is extremely valuable for the development of a thermodynamic description of a system. However, in general the only quantities that can be directly measured are enthalpies and chemical potentials while all other quantities, such as heat capacity or entropies, are derived from these measurements. The majority of enthalpies are determined with a variety of calorimetric methods[56, 57]. The chemical potentials are usually determined from electromotive force (emf)[58] or vapor pressure measurements[59, 60]. Depending on the specific method the values are obtained either through direct or indirect measurements. In cases where the values stem from indirect measurement methods, the values may need to be used in their converted form; however, in all other cases the directly measured value should be used. Unfortunately, the originally measured data are not always reported and the information given may be inadequate to reconstruct the originally measured data. The reported values may carry errors that have accumulated from measurement to measurement, and also may be affected by the choice of reference, which is used to correct for temperature differences at different stages of the experiment. Even though it is desirable to use only data from direct measurements these data may not be available. For particular systems, the melting points of the components or other thermochemical properties of the system may limit the use of some direct measurement techniques. Thus, when using indirectly obtained data, the way these data are derived must be documented and taken into account when the accuracy of these data is evaluated.
A data repository of experimental phase diagram data and thermochemical data and all the relevant metadata, including documentation on the error estimation, is needed to improve the accuracy, efficiency, and reproducibility of the CALPHAD assessments.
Diffusion
CALPHAD-based diffusion mobility databases have been developed based on the phenomenological theory of diffusion for multicomponent systems developed by Onsager[5, 6]. Assuming irreversible thermodynamics and an exchange mechanism in a crystalline material, the diffusional flux, Jk, of a component is assumed to be proportional to a thermodynamic force, for an isothermal isobaric system,
where Dki is the diffusivity (m2/s), ci is the concentration of component i (Note: where xi is the mole fraction of component i, and Vm is the molar volume of the phase), and z is the diffusion distance. Rewriting Eq 5 in terms of the more convenient quantities, the chemical potentials μι, and using the chain rule, the flux and diffusivity can be expressed as
where is a proportionality factor which is dependent of the mobility of individual components, and ∂μi/∂cj are the thermodynamic factors. Thus, the diffusivities are defined by a pure thermodynamic factor and pure kinetic factor (). The kinetic factor, , is defined by the atomic mobilities as follows:
Where δik represents the Kronecker delta and =1 when i = k and = 0 when i ≠ k, and Vi is the partial volume of element i, and yVa is the fraction of vacant lattice sites on the sublattice where i is dissolved. The atomic mobility, Mi, is defined by an Arrhenius function:
where Qi and, the activation energy and frequency factor, respectively, are expanded using Redlich-Kister polynomials to express the composition and temperature dependence (similar to the excess Gibbs energy in Eq 3.) of the mobility and are the functions stored in the diffusion mobility databases[5, 6]. It should be noted by assuming the mobility matrix is diagonal in a lattice fixed frame of reference, as followed in the Darken approximation, the coupling between fluxes, otherwise known as the vacancy wind (Manning-effect) is neglected. For additional information on the inclusion of these factors see the following work[61, 62].
When radioactive tracers are used in a homogeneous material, the concentration gradient approaches zero and tracer diffusivity is defined as a function of the kinetic factors, which are proportional to the atomic mobility. Thus, the tracer diffusivity is independent of a concentration gradient,. While the temperature dependence of the diffusivity of many species can be described by an Arrhenius relation, there are some materials that have anomalous behavior at low temperatures, such as β-Ti and β-Zr. These diffusivities could be described using a two-exponential fit[63].
Tracer diffusivity data from single crystal material are the preferred experimental data on which to build a diffusion mobility database, as these data are not dependent on a thermodynamic factor or influenced by the Gibbs energy. However, for a given system, it is often difficult to find enough tracer diffusivity data to complete a diffusion mobility description (some data sources of diffusivity data, mostly tracer and impurity data, found in[64–66]). Thus, chemical and intrinsic diffusivity data are used to complete the diffusion assessments. Using these data requires a thermodynamic database to calculate the concentration gradients and to determine the diffusion mobilities[67–69]. In the volume-fixed frame of reference for substitutional elements the chemical diffusivity, VDkj, is defined as
The derivatives of the chemical potential can be calculated using a CALPHAD-based description of the thermodynamics. The intrinsic diffusivity, LDkj, is defined in the lattice-fixed frame of reference, where the sum of the diffusion fluxes equals the vacancy flux, as:
Similar to the thermodynamic and phase diagram data, it is essential when reporting diffusivity data to include not only the measurement error, but also the type of material used, the initial composition/s and relative errors, and the methods used to analyze the data.
Just as the CALPHAD thermodynamic databases require a consistent set of reference lattice stabilities, the diffusion mobility databases also require a consistent set of reference tracer-diffusion mobilities for the pure elements. However, unlike the lattice stabilities, which have been established, the reference tracer-diffusion mobilities have yet to be established. A reference tracer-diffusion mobility database for the pure elements is in the process of being developed in conjunction with NIST and a group of international researchers[70]. The reference mobilities are determined by reviewing all of the available experimental data and assessments for a given element. When multiple assessments exist for the same element, weight mean statistics are used to determine which assessment best represents the critically reviewed experimental data[71]. The reference mobility database also requires values for the non-stable end-members. These quantities may be estimated using diffusion correlations[72, 73] or using data from first-principle calculations[19, 74–76] or atomistic simulations[77], when possible.
The choice of sublattice model is also important in the development of robust diffusion mobility databases. Even if only tracer diffusivity data are used to developed the diffusion mobility description, eventually the diffusion and thermodynamic descriptions will be used together to predict diffusional controlled phase transformations and thus the same phase model must be used by both the diffusion and thermodynamic database. The choice of phase model must accurately represent the crystal structure and the active diffusion mechanism. For example, in modeling the austenite (fcc) phase for a steel, carbon should not be placed on the substitutional sublattice, but rather on the interstitial sublattice.
Physical phase-based properties related to the thermodynamics
Molar volume and thermal expansion
Recently, the CALPHAD approach has been applied to more phase-based properties, such as molar volume and elastic moduli, and databases of assessments of functions for these properties are in the process of being developed. Again essential to the development of these CALPHAD-based descriptions are reference experimental data and a reference database for the pure end-member quantities in various phases. Molar volumes for the stable crystalline phases and compounds can be determined using high accuracy lattice parameter measurements, such as x-ray or neutron diffraction. For liquids, molar volumes may be measured directly, determined from measured volume changes occurring during melting or solidification, or derived from changes in the melting temperature as a function temperature. Databases can be developed assuming pressure independent volumes below 1 GPa[78]. Above pressures of 1 GPa, the pressure dependent volumes should be included by using the appropriate equation of state (EOS)[79] (This will be discussed further in the next section). Hallstedt et al.[78] recommended that metastable end points can be extrapolated from binary lattice parameter data using Vegard’s law, assuming that a linear composition dependence holds. In using this process as many binary systems as possible should be considered to determine an average extrapolated metastable end-member quantity. Low solubility in a given phase may limit the application of this extrapolation method. When this occurs, using first-principles calculations to estimate the lattice parameter is the preferred method.
The temperature dependence of the molar volume (thermal expansion) and heat capacity are related in both the Mie-Grüneisen and Einstein models. The molar volume, is defined as function of linear expansion (CLE), α, as the following:
Where the first term represents the nonmagnetic molar volume and the second term,, is the magnetic contribution to the molar volume. The variable is the molar volume at the reference temperature, T0, usually 298.15 K. Lu et al.[7, 80] applied the CALPHAD method to expand the nonmagnetic volumetric expansivity by defining:
where the constants a, b, c, and d are evaluated using available data from lattice parameter expansion data determined using diffraction methods or measured length changes determined using dilatometric and interferometric methods. (It should be noted that high temperature diffraction data are preferred as vacancy contributions can be minimized.) The magnetic contributions to the molar volume are treated by Fernández Guillermet[81]. Based on this formalism Lu et al. developed descriptions for a number of fcc, bcc and hcp elements[7]. Lu et al.[7] acknowledged that the current modeling efforts do not describe the Invar effect well in Fe-Ni alloys and additional modeling efforts are needed[41]. Lu et al.[7] demonstrated that the Debye-Grüneisen model is useful to approximate thermal expansion coefficient for metastable and unstable phases and systems where experimental data are lacking. Kim et al.[82] demonstrate the use of first-principles to develop multicomponent composition thermal expansion coefficients descriptions for Ni-Al based systems.
Alternatively, Hallstedt et al.[78] noted that the thermal expansion contribution to the volume is generally small and thus, a simple polynomial may be sufficient at this time.
Assuming.
For metastable phases, Hallstedt et al.[78] suggested using the same thermal expansion descriptions that correspond to the stable phases. If thermal expansion data for a liquid phase are not available, it has been recommended to assume that the thermal expansion is proportional to the volume of the liquid using the same proportionality constant that is defined for the solid phase. The additivity rule can be used as approximation for intermediate phases with missing thermal expansion data[78]. Hallstedt demonstrated the feasibility of this approach for Al, Mg, and Si[83].
Recently, Zhang et al.[84] demonstrated yet another approach for modeling the thermal expansion. This approach was used to evaluate 42 pure metallic elements and was based on Debye-Grüneisen model. The approached showed potential to for use to extrapolate thermal expansion coefficients when sufficient temperature data is unavailable.
Elastic properties
To apply a CALPHAD approach to modeling the elastic properties requires using an EOS, which can be expressed in the form V = V (T,P) and can be integrated, and data on the bulk moduli. The data needed to describe the EOS include the volume, Vo(T), compressibility, Ko(T), and the pressure dependence of the compressibility at a reference temperature[78]. Most of the bulk moduli for the elements and many compounds are known. When experimental data are not available first-principles calculations, using volume functions fitted to an appropriate EOS, can be employed[85, 86]. The temperature dependence can be obtained through the thermal expansion assuming that the temperature dependence of elastic stiffness coefficients mainly results from the volume change as a function of temperature. This approach has been demonstrated by Liu et al.[8] for the Mg-Al system.
As noted previously to describe the volume at pressures above 1 GPa, the pressure dependence must be included in the EOS[78]. Lu et al.[79] developed such an EOS based on the work of Jacobs and Oonk[87], which extrapolates well to at least 6000 K and 200 GPa.
Where c = c(T) is defined by the pressure data, κ is the isothermal compressibility, and κo is the isothermal compressibility at Po. The Gibbs energy is then expressed as.
The temperature dependence of Vm(T,Po) is given by Eq. 13. Other EOS have also been considered[88–90]; however, all of these EOS have had some compatibility issues with the CALPHAD method that results in the incorrect prediction of high pressure thermophysical properties. Brosh et al.[91] proposed another EOS which attempts to address some of these capability issues; however, an EOS which satisfactorily accounts for the compatibility issues with CALPHAD has yet to be identified. Hammerschmidt et al.[28] give a summary of the calculation of the elastic constants and point out that the magnetic properties of a phase also need to be taken into account for their treatment.
Physical base properties independent of thermodynamics
The phase-based properties that have been addressed thus far are either thermodynamic quantities or use the thermodynamics to fully describe the property. However, the CALPHAD method has been demonstrated to apply to other properties.
Electrical resistivity and thermal conductivity
The electrical resistivity of a phase is composed of contributions from scattering of conducting electrons and static lattice defects. The electrical conductivity, the inverse of the electrical resistivity, ρ, is related to the thermal conductivity via Wiedemann-Franz law, which states:
where λ is the thermal conductivity, L is the Lorentz number and T is the absolute temperature (K). However it should be noted when the heat is conducted by phonons, as in an insulating phase, the assumptions made in the Wiedemann-Franz law no longer apply. The temperature of these quantities can be described by linking to the heat capacity as demonstrated, by Grimvall[92], or by using a simple polynomial as shown in the work by Terada et al.[93].
Using a CALPHAD approach the composition dependence of these quantities can be expressed. For example, the electrical resistivity in a metallic binary solid solution can be expressed as a sum of the pure component contributions and a mixing term, as proposed by Nordheim for a metallic binary solid regular solution[94]:
where ρα is the electrical resistivity of phase α, ρA and ρB are the electrical resistivities of components A and B in the α phase, and k is an excess mixing term.
Terada et al.[95, 93] have reviewed the thermal conductivity in a wide range of intermetallic compounds and have found that the Wiedemann-Franz law applies for not only stoichiometric intermetallic compounds, but also for off-stoichiometric compositions, such as NiAl and CoAl. Using the work of Terada et al.[93] and others[96] should provide a starting basis for thermal conductivity and electrical resistivity CALPHAD-based databases.
For insulating materials, such as oxides and semiconductors, Gheribi and Chartrand[97] developed a model based on the Debye model to predict thermal conductivity. This CALPHAD-based model, which simultaneously optimizes the thermal expansion, heat capacity and bulk modulus, also correctly predicts the thermal conductivity as a function of temperature.
In addition to the phase-based properties described in this work, the CALPHAD approach could be used to describe the composition and temperature of optical, thermo-electric, acoustic properties of materials.
Interphase properties
The ability to simulate microstructure evolution requires not only input of many phase-based properties (thermodynamic, diffusion, volume), but also many interphase dependent properties. The physical properties of molten alloys are dependent on the surface tension (i.e., for solder and solidification applications). Using Bulter’s equation, which defines an equilibrium between the bulk and monolayer surface, Picha et al.[98] and Tanaka et al.[99] have developed CALPHAD-based descriptions for various low melting binary metal systems and molten salt systems. The interfacial energy between two solid phases is an essential input for simulating precipitation and coarsening processes; however, these values are often difficult to measure and calculate. Establishing a database of available measured interfacial energies and methods to extrapolate these quantities is critical for the use of many precipitation codes. Currently, composition independent interfacial energies between phases are used within most precipitation and coarsening simulation tools[100–102]; however, if sufficient data were collected and stored composition-dependent interfacial energies CALPHAD databases might be developed. Efforts to estimate the composition dependent interfacial energies for a given set of solid phases were reviewed by Costae Silva et al.[103], who noted that while thermodynamic approaches can provide reasonable estimates, fundamental first-principles approaches[104] should be used as the reference values. More recently, Shi and Luo[105] developed a method to estimate the interfacial energy associated with a grain boundary between two phases. Combining these and other methods with data available from coarsening experiments[106, 107] and single-sensor DTA nucleation cooling experiments[101] a CALPHAD based databases could be established, where the interfacial energies are defined by two phases.
Grain boundary diffusion models, thermal migration models[108], and nucleation models are also used to predict composition dependent microstructure evolution and require additional material dependent input, such as the grain boundary diffusion coefficients, dislocation densities, and nucleation kinetics. Eventually composition and temperature dependent databases for these quantities will also be required. Thus, experimental databases documenting these inputs should also be established; however, the specific format for these data is yet to be determined.
Data repositories and tools
Repositories
Based on the evolution of CALPHAD modeling and multicomponent database development, it is now clear that data repositories are essential for improving the efficiency of future modeling and assessment work. Without these repositories, the effort needed to improve current descriptions will continue to be burdensome and continue to slow the implementation of new models. As CALPHAD modeling expands to other phase-based properties there will be even greater need for these data repositories. For these repositories to be effective, these data need to be well documented. This documentation needs to be sufficient such that the user does not need to obtain the original work to determine the error associated with the data for the application of interest.
Although there are currently many various data compilations (such as[109, 110]), it is rare that these data can be easily manipulated into the desired format. Some of these data compilations, especially those in printed form, are rarely updated. The advantages of electronic compilations include the possibility of easier data manipulation and the ability to easily update with new information.
NIST has been charged with developing materials data infrastructure and data repositories to support the Materials Genome Initiative (MGI). NIST has initiated several different efforts in the areas of data capture, data dissemination, and data processing to develop this infrastructure, as illustrated in Figure 2. As a first step, files used in the assessment of the description of a given system should be collected and stored electronically in a public repository, such as that initiated by NIST and Kent State[111] (http://nist.matdl.org/). Figure 3 is screen shot of this repository, which is a customized version of the open source software DSpace (http://www.dspace.org/), showing the development of different materials-based data communities. Authors are asked to contribute the files from their work. These files can then be linked to the originally published work. It is advantageous that the files used to assess the description can be directly associated to the files containing the description. Figure 4 illustrates the relationship the evaluated data files and the files containing the functional descriptions. Although these files are software specific, translation tools can be used to make the data available to other software codes.

Schematic representing NIST data efforts in the areas of data capture, data dissemination, and data processing. Data capture efforts include the Thermodynamic Research Center (TRC) expansion of the Guided Data Capture program (http://trc.nist.gov/GDC.html) and developing data curation tools based on ontologies. Data dissemination efforts include the TRC ThermoML archive and the NIST interatomic potential repository (http://www.ctcms.nist.gov/potentials/). The overlapping areas show the development of workflow tools, data repositories, ontologies using natural language processing (NLP), and meta-data standards that are needed for all the data efforts.

Screen shot of the customized DSpace repository ( http://www.nist.matdl.org) to support a variety open and private data communities.

Schematic illustrating the proposed relationship between evaluated data files and CALPHAD functional descriptions to be used in developing a CALPHAD-base file repository.
Data may be collected in repositories either as single value points or as a data series. Most data can be stored as single value points; however, some data must be stored as a data series. For example, a diffusion profile is a data series where the individual data points must be stored together to be meaningful. The format for the repository, while not specific, must be flexible enough to accommodate extensions while maintaining easy data retrieval. The format must be able to define a unique material in an unambiguous way such that the same material identifier can be used by other databases and tools searching for information on that specific material. In addition, phase names must also be unique and unambiguous. Examples of currently used identifiers for chemicals include the Chemical Abstracts Registry (CAS) number[112] and International Chemical Identifier (InChI)[113]. An example for defining phase names is given by Schmid-Fetzer et al.[114]. One possibility for a flexible data format is the use of various extensible markup languages. Some of the languages to be consider for use include MatML specifically designed for the interchange of materials data[115] and ThermoML for thermochemical and thermophysical property data[116–118]. Other choices to consider are the HDF5 (Hierarchical Data Format) format used by the Materials Data Environment[119] or data-interchange formats such as, JSON (Java Script Object Notation)[120] or BSON (Binary Script Object Notation)[121]. Regardless of the choice of data format, the data stored should ideally include descriptions of the appropriate metadata. These metadata would include information describing how the individual datum was obtained. These descriptions could include information on material purity, alloy preparation, etc., for experimental data and information on the method and input used, etc., for calculated data. For phase data it is essential that the data are linked to the crystal structure of the phase. Following the recommendations of the CCN (Commission on Crystallographic Nomenclature), the crystal structure should be inputted using the format prescribed by Brown et al.[54]. A preliminary summary of the information needed for data typically used in CALPHAD assessments is shown in Table 1. If all entries in Table 1 are provided, this would represent an ideal data description case. In reality, it is accepted that information to all entries in Table 1 may not be known. For example, a diffusion couple composition profile may be entered for a given system: ideally, the profile should be entered with a corresponding micrograph of the microstructure and the compositions should be identified as single phase or multiphase. However, this information may not be available, but the composition profile still provides valuable information and should be entered.
ESPEI
In a project supported by the United States Automotive Materials Partnership (USAMP) of Department of Energy, Shang et al.[122] developed an relational infrastructure, ESPEI (Extensible Self-optimizing Phase Equilibrium Infrastructure), based on SQL (structured query language). In ESPEI, all data are organized through the phase identifiers with one for each individual phase-based on crystallographic information. There are currently about 300 phase identifiers in ESPEI covering known symmetries designated by space groups. The input data are tabulated for each phase in three groups: thermochemical data from first-principles calculations, thermochemical data from experiments, and phase equilibrium data. Each data point is divided into multiple entries with one for each element in the phase, which enables the extension to multi-component systems. In addition to the value and uncertainty, each entry details the independent variables and their uncertainties such as temperature, pressure, composition, magnetic data, and other miscellaneous information. The ESPEI SQL data are searchable by elements. An automation procedure was developed to model Gibbs energy functions of individual phases in binary and ternary systems using the three groups of data mentioned previously.
The data flow in ESPEI is illustrated in Figure 5. ESPEI heavily depends on thermochemical data from DFT-based first-principles calculations for both stoichiometric and solution phases at finite temperatures[19] due to the lack of experimental measurements of thermochemical properties of individual phases. Even though the discrepancies on lattice stability between the classic CALPHAD modeling and DFT-based first-principles calculations still exist[123] progresses have been made to narrow the differences such as bcc Ti[124] and fcc W[125], even for liquid solution phases[44]. The efficient special quasirandom structures (SQS) approach[126–128] is particularly useful in predicting the enthalpy and entropy of mixing in solid solution phases using phonon or Debye models[129, 130]. Consequently, the thermodynamic model parameters of all individual phases can be evaluated solely from DFT-based first-principles calculations. On the other hand, the uncertainty in DFT-based first principles calculations prohibits accurate predictions of phase equilibrium temperature and compositions between two or more phases, and model parameters are further refined using phase equilibrium data from experiments.

Schematic illustrating data flow in ESPEI.
Conclusions
The CALPHAD method is a powerful tool for assessing the properties of multicomponent, multiphase materials that can readily be coupled with materials simulation tools. However, the method is currently lacking strategies for straightforward implementation of new models and new data to update databases. The basis for efficient update methods is the availability of the data that are needed for determination of the functions used by the CALPHAD method.
The strengths of the CALPHAD method are that the data obtained from the calculation with a CALPHAD description are self-consistent and that descriptions of constitutive subsystems can be combined for the extrapolation[9] of multicomponent systems. However, from the assessment of the descriptions to the prediction of the properties in a multicomponent system, the confidentiality interval of the calculated data is not available from the calculations. Although several attempts[131, 132] have been made to quantify the reliability of these calculated data none of these methods has been tested for its general applicability for results from calculations of multicomponent systems. For full implementation into an Integrated Computational Materials Engineering (ICME) approach, information on the confidence of the multicomponent predictions is needed to design materials within specified property tolerances. While the CALPHAD approach only requires unary, binary, and ternary data, multicomponent data are essential for providing verification and confidence assessments of the multicomponent material property predictions. Thus, it is important that multicomponent property data are also published and stored in data repositories[133].
As outlined in the 2008 National Research Council report on ICME[134], a data repository infrastructure is needed. The report suggests following the example of the human genome project (HGP)[135]. This would require that data from government sponsored research be published in publically available databases as recently mandated by the recent US government executive order[136]. Likewise, materials-oriented research journals should require that the reported data be deposited in a publically available database before the research is accepted for publication. The development of a cyber-infrastructure is required as a first step, within the framework of the Materials Genome Initiative[16]. This infrastructure is needed to host repositories, to provide tools to enable efficient population of reference data repositories, and to integrate the variety of different repositories and tools currently being developed. NIST has initiated several projects and collaborations to develop this infrastructure and host some of these repositories (http://www.nist.matdl.org andhttp://www.materialsdata.nist.gov/dspace/xmlui).
Endnotes
aIn this article the term “multicomponent” is used to describe systems with more than 3 components.
bThe term “non-stable” is used if a distinction between metastable and unstable is not available or not needed.
References
Saunders N, Miodownik AP: CALPHAD Calculation of Phase Diagrams: A Comprehensive Guide. Pergamon Materials Series. Elsevier Science Inc, New York; 1998.
Kaufman L, Bernstein H: Computer Calculation of Phase Diagrams. Academic Press, London; 1970.
Lukas HL, Fries SG, Sundman B: Computational Thermodynamics: The CALPHAD Method. Cambridge University Press, Cambridge; 2007.
Dinsdale AT: SGTE Data for Pure Elements. CALPHAD 1991, 15(4):317–425. 10.1016/0364-5916(91)90030-N
Ågren J: Diffusion in Phases with Several Components and Sublattices. J Phys Chem Solids 1982, 43(5):421–430. 10.1016/0022-3697(82)90152-4
Andersson J-O, Ågren J: Models for numerical treatment of multicomponent diffusion in simple phases. J Appl Phys 1992, 72(4):1350–1355. 10.1063/1.351745
Lu X-G, Selleby M, Sundman B: Assessments of molar volume and thermal expansion for selected bcc, fcc, and hcp metallic elements. CALPHAD 2005, 29: 68–89. 10.1016/j.calphad.2005.05.001
Liu ZK, Zhang H, Ganeshan S, Wang Y, Mathaudhu SN: Computational modeling of effects of alloying elements on elastic coefficients. Scr Mater 2010, 63(7):686–691. doi:10.1016/j.scriptamat.2010.03.049 doi:10.1016/j.scriptamat.2010.03.049 10.1016/j.scriptamat.2010.03.049
Hillert M: Phase Equilibria, Phase Diagrams and Phase Transformations. 2nd edition. Cambridge University Press, Cambridge; 2007.
Saunders N, Kucherenko S, Li X, Miodownik AP, Schille J: A new computer program for predicting materials properties. J Phase Equil 2001, 22(4):463–469. doi:10.1361/105497101770333036 doi:10.1361/105497101770333036 10.1361/105497101770333036
Guo J, Samonds M: Alloy Thermal Physical Property Prediction Coupled Computational Thermodynamics with Back Diffusion Consideration. J Phase Equil Diffus 2007, 28(1):58–63. doi:10.1007/s11669–006–9005–6 doi:10.1007/s11669-006-9005-6 10.1007/s11669-006-9005-6
Steinbach I, Böttger B, Eiken J, Warnken N, Fries SG: CALPHAD and phase-field modeling: A successful liaison. J Phase Equil Diffus 2007, 28(1):101–106. 10.1007/s11669-006-9009-2
Olson GB: Genomic materials design: The ferrous frontier. Acta Mater 2013, 61(3):771–781. doi:10.1016/j.actamat.2012.10.045 doi:10.1016/j.actamat.2012.10.045 10.1016/j.actamat.2012.10.045
Kang Y-B, Aliravci C, Spencer P, Eriksson G, Fuerst C, Chartrand P, Pelton A: Thermodynamic and volumetric databases and software for magnesium alloys. JOM 2009, 61(5):75–82. doi:10.1007/s11837–009–0076–9 doi:10.1007/s11837-009-0076-9 10.1007/s11837-009-0076-9
Kaufman L, Ågren J: CALPHAD, first and second generation – Birth of the materials genome. Scr Mater 2014, 70(1):3–6.
National Science and Technology Council: Materials Genome Initiative for Global Competitiveness. Washington DC; 2011. http://www.whitehouse.gov/mgi
Redlich O, Kister AT: Algebraic Representations of Thermodynamic Properties and the Classification of Solutions. Ind Eng Chem 1948, 40(2):345–348. 10.1021/ie50458a036
Muggianu YM, Gambino M, Bros JP: Enthalpies Of Formation Of Liquid Alloys Bismuth-Gallium-Tin At 723 k - Choice Of An Analytical Representation Of Integral And Partial Thermodynamic Functions Of Mixing For This Ternary-System. J Chim Phys-Chim Biol 1975, 72(1):83–88.
Liu ZK: First-Principles Calculations and CALPHAD Modeling of Thermodynamics. J Phase Equil Diffus 2009, 30(5):517–534. doi:10.1007/s11669–009–9570–6 doi:10.1007/s11669-009-9570-6 10.1007/s11669-009-9570-6
Chase MW, Ansara I, Dinsdale A, Eriksson G, Grimvall G, Höglund L, Yokokawa H: Group 1: Heat capacity models for crystalline phases from 0 K to 6000 K. CALPHAD 1995, 19(4):437–447.
Xiong W, Hedstrom P, Selleby M, Odqvist J, Thuvander M, Chen Q: An improved thermodynamic modeling of the Fe-Cr system down to zero kelvin coupled with key experiments. CALPHAD 2011, 35(3):355–366. doi:10.1016/j.calphad.2011.05.002 doi:10.1016/j.calphad.2011.05.002 10.1016/j.calphad.2011.05.002
Palumbo M, Fries SG, Pasturel A: Temperature Dependence of Thermodynamic Quantities Calculated from First-Principles. Paper presented at the COST MP0602 HISOLD, Brno, Czech Republic; 2011.
Palumbo M, Fries SG, Hammerschmidt T, Körmann F, Hickel T: SAPIENS Thermophysical Database for Pure Elements: DFT and Experiments. Paper presented at the 18th Symposium on Thermophysical Properties, Boulder, CO USA; 2012.
Vřešt’ál J, Štrof J, Pavlů J: Extension of SGTE data for pure elements to zero Kelvin temperature—A case study. CALPHAD 2012, 37: 37–48. doi:10.1016/j.calphad.2012.01.003 doi:10.1016/j.calphad.2012.01.003
Rogal J, Divinski SV, Finnis MW, Glensk A, Neugebauer J, Perepezko JH, Schuwalow S, Sluiter MHF, Sundman B: Perspectives on point defect thermodynamics. physica status solidi (b) 2014, 251(1):97–129. doi:10.1002/pssb.201350155 doi:10.1002/pssb.201350155 10.1002/pssb.201350155
Palumbo M, Burton B, Costa e Silva A, Fultz B, Grabowski B, Grimvall G, Hallstedt B, Hellman O, Lindahl B, Schneider A, Turchi PEA, Xiong W: Thermodynamic modelling of crystalline unary phases. physica status solidi (b) 2014, 251(1):14–32. 10.1002/pssb.201350133 10.1002/pssb.201350133 10.1002/pssb.201350133
Körmann F, Breidi AAH, Dudarev SL, Dupin N, Ghosh G, Hickel T, Korzhavyi P, Muñoz JA, Ohnuma I: Lambda transitions in materials science: Recent advances in CALPHAD and first-principles modelling. physica status solidi (b) 2014, 251(1):53–80. doi:10.1002/pssb.201350136 doi:10.1002/pssb.201350136 10.1002/pssb.201350136
Hammerschmidt T, Abrikosov IA, Alfè D, Fries SG, Höglund L, Jacobs MHG, Koßmann J, Lu XG, Paul G: Including the effects of pressure and stress in thermodynamic functions. physica status solidi (b) 2014, 251(1):81–96. doi:10.1002/pssb.201350156 doi:10.1002/pssb.201350156 10.1002/pssb.201350156
Becker CA, Ågren J, Baricco M, Chen Q, Decterov SA, Kattner UR, Perepezko JH, Pottlacher GR, Selleby M: Thermodynamic modelling of liquids: CALPHAD approaches and contributions from statistical physics. physica status solidi (b) 2014, 251(1):33–52. doi:10.1002/pssb.201350149 doi:10.1002/pssb.201350149 10.1002/pssb.201350149
Sundman B, Ågren J: A Regular Solution Model For Phases With Several Components And Sub-Lattices, Suitable For Computer-Applications. J Phys Chem Solids 1981, 42(4):297–301. 10.1016/0022-3697(81)90144-X
Oates WA, Zhang F, Chen SL, Chang YA: Improved cluster-site approximation for the entropy of mixing in multicomponent solid solutions. Phys Rev B 1999, 59(17):11221–11225. 10.1103/PhysRevB.59.11221
Andersson JO, Fernández Guillermet A, Hillert M, Jansson B, Sundman B: A Compound-Energy Model of Ordering in a Phase with Sites of Different Coordination Numbers. Acta Metallugrica 1986, 34(3):437–445. 10.1016/0001-6160(86)90079-9
Hillert M: The compound energy formalism. J Alloy Compd 2001, 320(2):161–176. 10.1016/S0925-8388(00)01481-X
Hillert M, Jarl M: MODEL FOR ALLOYING EFFECTS IN FERROMAGNETIC METALS. CALPHAD 1978, 2(3):227–238. 10.1016/0364-5916(78)90011-1
Inden G: Determination Of Chemical And Magnetic Interchange Energies In Bcc Alloys.1. General Treatment. Z Metallkd 1975, 66(10):577–582.
Chen Q, Sundman S: Modeling of thermodynamic properties for Bcc, Fcc, liquid, and amorphous iron. J Phase Equil 2001, 22(6):631–644. doi:10.1007/s11669–001–0027–9 doi:10.1007/s11669-001-0027-9 10.1007/s11669-001-0027-9
Sundman B, Ohnuma I, Dupin N, Kattner UR, Fries SG: An assessment of the entire Al–Fe system including D03 ordering. Acta Mater 2009, 57(10):2896–2908. doi:10.1016/j.actamat.2009.02.046 doi:10.1016/j.actamat.2009.02.046 10.1016/j.actamat.2009.02.046
Wang Y, Hector LG, Zhang H, Shang SL, Chen LQ, Liu ZK: A thermodynamic framework for a system with itinerant-electron magnetism. J Physics-Condensed Matter 2009, 21(32):326003. 10.1088/0953-8984/21/32/326003
Shang SL, Wang Y, Liu ZK: Thermodynamic fluctuations between magnetic states from first-principles phonon calculations: The case of bcc Fe. Phys Rev B 2010, 82(1):014425. doi:014425 10.1103/PhysRevB.82.014425 doi:014425 10.1103/PhysRevB.82.014425
Shang SL, Saal JE, Mei ZG, Wang Y, Liu ZK: Magnetic thermodynamics of fcc Ni from first-principles partition function approach. J. Applied Physics 2010, 108(12):123514. doi:123514 10.1063/1.3524480 doi:123514 10.1063/1.3524480 10.1063/1.3524480
Wang Y, Shang SL, Zhang H, Chen LQ, Liu ZK: Thermodynamic fluctuations in magnetic states: Fe3Pt as a prototype. Philos Mag Lett 2010, 90(12):851–859. 10.1080/09500839.2010.508446
Ågren J, Cheynet B, ClavagueraMora MT, Hack K, Hertz J, Sommer F, Kattner U: GROUP 2: Extrapolation of the heat capacity in liquid and amorphous phases. CALPHAD 1995, 19(4):449–480.
Ågren J: Thermodynamics of Supercooled Liquids and their Glass Transition. Phys Chem Liq 1988, 18(2):123–139. doi:10.1080/00319108808078586 doi:10.1080/00319108808078586 10.1080/00319108808078586
Han J, Wang WY, Wang C, Wang Y, Liu X, Liu Z-K: Accurate determination of thermodynamic properties for liquid alloys based on ab initio molecular dynamics simulation. Fluid Phase Equilib 2013, 360: 44–53.
Pelton AD, Blander M: Thermodynamic Analysis of Ordered Liquid Solutions by a Modified Quasi-Chemical Approach - Application to Silicate Slags. Metallogr Trans B-Process Metallurgy 1986, 17(4):805–815. 10.1007/BF02657144
Sommer F: Association Model For The Description Of The Thermodynamic Functions Of Liquid Alloys.1. Basic Concepts. Z Metallkd 1982, 73(2):72–76.
Sommer F: Association Model For The Description Of Thermodynamic Functions Of Liquid Alloys.2. Numerical Treatment And Results. Z Metallkd 1982, 73(2):77–86.
Hillert M, Jansson B, Sundman B, Ågren J: A 2-Sublattice Model For Molten Solutions With Different Tendency For Ionization. Metal Trans A 1985, 16(2):261–266. doi:10.1007/bf02816052 doi:10.1007/bf02816052 10.1007/BF02816052
Lu XG, Chen Q: A CALPHAD Helmholtz energy approach to calculate thermodynamic and thermophysical properties of fcc Cu. Philos Mag 2009, 89(25):2167–2194. doi:10.1080/14786430903059004 doi:10.1080/14786430903059004 10.1080/14786430903059004
Villars P, Calvert LD: Pearson’s Handbook of Crystallographic Data for Intermetallic Phases. ASM International, Materials Park; 1991.
FIZ/NIST: Inorganic Crystal Structure Database (ICSD) Version 2011/2. NIST 2011. Accessed Jan 9 2012 http://www.nist.gov/srd/nist84.cfm
PAULING FILE. http://paulingfile.com/
Villars P, Cenzual K: Pearson’s Crystal Data. 2012. Available via ASM International.http://www.asminternational.org/materials-resources/online-databases/-/journal_content/56/10192/6382084/DATABASE.
Brown ID, Abrahams SC, Berndt M, Faber J, Karen VL, Motherwell WDS, Villars P, Westbrook JD, McMahon B: Report of the working group on crystal phase identifiers. Acta Crystallogr Sect A 2005, 61: 575–580. doi:10.1107/s010876730503179x doi:10.1107/s010876730503179x 10.1107/S010876730503179X
Zhao J-C: Methods for Phase Diagram Determination. Elsevier, Amsterdam; 2007.
Colinet C: Comparison of enthalpies of formation and enthalpies of mixing in transition metal based alloys. Thermochim Acta 1998, 314(1–2):229–245.
Colinet C: High temperature calorimetry: recent developments. J Alloy Compounds 1995, 220(1–2):76–87. doi:10.1016/0925–8388(94)06032–0 doi:10.1016/0925-8388(94)06032-0
Ipser H, Miikula A, Katayama I: Overview: The emf method as a source of experimental thermodynamic data. CALPHAD 2010, 34: 271–278. doi:10.1016/j.calphad.2010.05.001 doi:10.1016/j.calphad.2010.05.001 10.1016/j.calphad.2010.05.001
Predel B: Recent trends and developments of experimental methods for the determination of thermodynamic quantities of alloys. CALPHAD 1982, 6(3):199–216. doi:10.1016/0364–5916(82)90002–5 doi:10.1016/0364-5916(82)90002-5 10.1016/0364-5916(82)90002-5
Ferro R, Cacciamani G, Borzone G: Remarks about data reliability in experimental and computational alloy thermochemistry. Intermetallics 2003, 11: 1081–1094. 10.1016/S0966-9795(03)00145-6
Belova I, Murch GE: Expressions for vacancy-wind factors occurring in interdiffusion in ternary and higher-order alloys. Acta Mater 2007, 55: 627–634. 10.1016/j.actamat.2006.08.055
Swoboda B, Van der Ven A, Morgan D: Assessing Concentration Dependence of FCC Metal Alloy Diffusion Coefficients Using Kinetic Monte Carlo. J Phase Equilib Diffus 2010, 31(3):250–259. doi:10.1007/s11669–010–9706–8 doi:10.1007/s11669-010-9706-8 10.1007/s11669-010-9706-8
Herzig C, Kohler U, Divinski SV: Tracer diffusion and mechanism of non-Arrhenius diffusion behavior of Zr and Nb in body-centered cubic Zr-Nb alloys. J Appl Phys 1999, 85(12):8119–8130. doi:10.1063/1.370650 doi:10.1063/1.370650 10.1063/1.370650
Neumann G, Tuijn C: Self-Diffusion and Impurity Diffusion in Pure Metals: Handbook of Experimental Data, vol 14. Pergamon Materials Series. 1st edition. Elsevier, New York; 2009.
Bakker H, Bonzel HP, Bruff CM, Dayananda MA, Gust W, Horvath J, Kaur I, Kidson GV, LeClaire AD, Mehrer H, Murch GE, Neumann G, Stolica N, Stolwijk NA (Eds): Difusion in Solid Metals and Alloys, vol 26. Landolt-Börnstein. Numerical Data and Functional Relationships in Science and Technology Springer-Verlag, Berlin; 1990.
Gale WF, Totemeier TC: Smithells Metals Reference Book. 8th edition. Elsevier, Amsterdam; 2004.
Cui YW, Kato R, Omori T, Ohnuma I, Oikawa K, Kainuma R, Ishida K: Revisiting diffusion in Fe-Al intermetallics: Experimental determination and phenomenological treatment. Scr Mater 2010, 62(4):171–174. http://dx.doi.org/10.1016/j.scriptamat.2009.10.011
Cui YW, Jiang M, Ohnuma I, Oikawa K, Kainuma R, Ishida K: Computational study of atomic mobility in Co-Fe-Ni ternary fcc alloys. J Phase Equilib Diffus 2008, 29(4):312–321. doi:10.1007/s11669–008–9341–9 doi:10.1007/s11669-008-9341-9 10.1007/s11669-008-9341-9
Engström A, Ågren J: Assessment of diffusional mobilities in face-centered cubic Ni Cr Al alloys. Z. Metallkde 1996, 87(2):92–97.
Campbell CE: NIST Diffusion Workshop. 2009. http://www.nist.gov/mml/msed/thermodynamics_kinetics/Diffusion-Workshop-Group.cfm
Campbell CE, Rukhin AL: Evaluation of self-diffusion data using weighted means statistics. Acta Mater 2011, 59(13):5194–5201. doi:10.1016/j.actamat.2011.04.055 doi:10.1016/j.actamat.2011.04.055 10.1016/j.actamat.2011.04.055
Brown AM, Ashby MF: Correlations for Diffusion Constants. Acta Metall 1980, 28: 1085–1101. 10.1016/0001-6160(80)90092-9
Askill J: Tracer Diffusion Data for Metals. Alloys and Simple Oxides, Plenum, New York; 1970.
Mantina M, Wang Y, Arroyave R, Chen LQ, Liu ZK, Wolverton C: First-principles calculation of self-diffusion coefficients. Phys Rev Lett 2008, 100(21):215901.
Mantina M, Wang Y, Chen LQ, Liu ZK, Wolverton C: First principles impurity diffusion coefficients. Acta Mater 2009, 57(14):4102–4108. 10.1016/j.actamat.2009.05.006
Van Der Ven A, Yu H-C, Ceder G, Thornton K: Vacancy mediated substitutional diffusion in binary crystalline solids. Prog Mater Sci 2010, 55(2):61–105. doi:10.1016/j.pmatsci.2009.08.001 doi:10.1016/j.pmatsci.2009.08.001 10.1016/j.pmatsci.2009.08.001
Mendelev MI, Mishin Y: Molecular dynamics study of self-diffusion in bcc Fe. Phys Rev B 2009., 80(14): http://dx.doi.org/10.1103/PhysRevB.80.144111
Hallstedt B, Dupin N, Hillert M, Höglund L, Lukas H, Schuster JC, Solak N: Thermodynamic models for crystalline phases. Composition dependent models for volume, bulk modulus and thermal expansion. CALPHAD 2007, 31: 28–37. 10.1016/j.calphad.2006.02.008
Lu X-G, Selleby M, Sundman B: Implementation of a new model for pressure dependence of condensed phases in Thermo-Calc. CALPHAD 2005, 29(1):49–55. doi:10.1016/j.calphad.2005.04.001 doi:10.1016/j.calphad.2005.04.001 10.1016/j.calphad.2005.04.001
Lu X-G, Selleby M, Sundman B: Theoretical modeling of molar volume and thermal expansion. Acta Mater 2005, 53(8):2259–2272. doi:10.1016/j.actamat.2005.01.049 doi:10.1016/j.actamat.2005.01.049 10.1016/j.actamat.2005.01.049
Fernández Guillermet A: Critical-Evaluation Of The Thermodynamic Properties Of Cobalt. Int J Thermophys 1987, 8(4):481–510. 10.1007/BF00567107
Kim D, Shang S-L, Liu Z-K: Effects of alloying elements on thermal expansions of γ-Ni and γ’-Ni3Al by first-principles calculations. Acta Mater 2012, 60(4):1846–1856. doi:10.1016/j.actamat.2011.12.005 doi:10.1016/j.actamat.2011.12.005 10.1016/j.actamat.2011.12.005
Hallstedt B: Molar volumes of Al, Li, Mg and Si. CALPHAD 2007, 31(2):292–302. doi:10.1016/j.calphad.2006.10.006 doi:10.1016/j.calphad.2006.10.006 10.1016/j.calphad.2006.10.006
Zhang B, Li X, Li D: Assessment of thermal expansion coefficient for pure metals. Calphad 2013, 43(1):7–17. http://dx.doi.org/10.1016/j.calphad.2013.08.006
Wang Y, Wang JJ, Zhang H, Manga VR, Shang SL, Chen LQ, Liu ZK: A first-principles approach to finite temperature elastic constants. J Phys-Condens Matter 2010, 22(22):225404. doi:10.1088/0953–8984/22/22/225404 doi:10.1088/0953-8984/22/22/225404 10.1088/0953-8984/22/22/225404
Shang SL, Saengdeejing A, Mei ZG, Kim DE, Zhang H, Ganeshan S, Wang Y, Liu ZK: First-principles calculations of pure elements: Equations of state and elastic stiffness constants. Comput Mater Sci 2010, 48(4):813–826. doi:10.1016/j.commatsci.2010.03.041 doi:10.1016/j.commatsci.2010.03.041 10.1016/j.commatsci.2010.03.041
Jacobs MHG, Oonk HAJ: A realistic equation of state for solids. The high pressure and high temperature thermodynamic properties of MgO. CALPHAD 2000, 24(2):133–147. doi:10.1016/s0364–5916(00)00019–5 doi:10.1016/s0364-5916(00)00019-5 10.1016/S0364-5916(00)00019-5
Saxena SK: Pressure–volume equation of state for solids. J Phys Chem Solids 2004, 65(8–9):1561–1563. doi:10.1016/j.jpcs.2004.02.003 doi:10.1016/j.jpcs.2004.02.003
Jacobs MHG, van den Berg AP, de Jong BHWS: The derivation of thermo-physical properties and phase equilibria of silicate materials from lattice vibrations: Application to convection in the Earth’s mantle. CALPHAD 2006, 30(2):131–146. doi:10.1016/j.calphad.2005.10.001 doi:10.1016/j.calphad.2005.10.001 10.1016/j.calphad.2005.10.001
Jacobs MHG, de Jong BHWS: An investigation into thermodynamic consistency of data for the olivine, wadsleyite and ringwoodite form of (Mg, Fe)2SiO4. Geochim Cosmochim Acta 2005, 69(17):4361–4375. doi:10.1016/j.gca.2005.05.002 doi:10.1016/j.gca.2005.05.002 10.1016/j.gca.2005.05.002
Brosh E, Makov G, Shneck RZ: Application of CALPHAD to high pressures. CALPHAD 2007, 31(2):173–185. doi:10.1016/j.calphad.2006.12.008 doi:10.1016/j.calphad.2006.12.008 10.1016/j.calphad.2006.12.008
Grimvall G: Thermophysical Properties of Materials. 2nd edition. Elsevier, Amsterdam; 1999.
Terada Y, Ohkubo K, Mohri T, Suzuki T: Thermal conductivity of intermetallic compounds with metallic bonding. Mater Trans 2002, 43(12):3167–3176. doi:10.2320/matertrans.43.3167 doi:10.2320/matertrans.43.3167 10.2320/matertrans.43.3167
Nordheim L: Electron theory of metals I. Ann Physik 1931, 9: 607.
Terada Y, Ohkubo K, Mohri T, Suzuki T: A comparative study of thermal conductivity in alloys and compounds. Mater Sci Eng A 2000, 278(1–2):292–294.
Schroder K: CRC Handbook of Electrical Resistivitives of Binary Metallic Alloys. 1983.
Gheribi A, Chartrand P: Application of the CALPHAD method to predict the thermal conductivity in dielectric and semiconductor crystals. CALPHAD 2012, 39: 70–79. http://dx.doi.org/10.1016/j.calphad.2012.06.002
Picha R, Vrest IJ, Kroupa A: Prediction of alloy surface tension using a thermodynamic database. CALPHAD 2004, 28: 141–146. 10.1016/j.calphad.2004.06.002
Tanaka T, Hack K, Hara S: Use of thermodynamic data to determine surface tension and viscosity of metallic alloys. MRS Bull 1999, 24(4):45–50.
Shi PF, Engström A, Sundman B, Ågren J: Thermodynamic Calculations and Kinetic Simulations of Some Advanced Materials. In Advanced Material Science and Technology, Pts 1 and 2, vol 675–677. Edited by: Tan Y, Ju DY. Stafa-Zurich: Materials Science Forum. Trans Tech Publications Ltd; 2011:961–974. http://www.scientific.net/MSF.675–677.961
Olson GB, Jou HJ, Jung J, Sebastian JT, Misra A, Locci I, Hull D: Precipitation model validation in 3(rd) generation aeroturbine disc alloys. Superalloys 2008. Minerals, Metals & Materials Soc, Warrendale PA; 2008:923–932.
Wu K, Zhang F, Chen S, Cao W, CY A: A modeling tool for the precipitation simulations of superalloys during heat treatments. Superalloys 2008 2008, 933–939. Minerals, Metals, & Materials Soc. Warrendale, PA Minerals, Metals, & Materials Soc. Warrendale, PA
Costa e Silva A, Ågren J, Clavaguera-Mora MT, Djurovic D, Gómez-Acebo T, Lee B-J, Liu Z-K, Miodownik P, Seifert HJ: Applications of computational thermodynamics — the extension from phase equilibrium to phase transformations and other properties. CALPHAD 2007, 31(1):53–74. doi:10.1016/j.calphad.2006.02.006 doi:10.1016/j.calphad.2006.02.006 10.1016/j.calphad.2006.02.006
Turchi PEA, Abrikosov IA, Burton B, Fries SG, Grimvall G, Kaufman L, Korzhavyi P, Rao Manga V, Ohno M, Pisch A, Scott A, Zhang W: Interface between quantum-mechanical-based approaches, experiments, and CALPHAD methodology. CALPHAD 2007, 31(1):4–27. doi:10.1016/j.calphad.2006.02.009 doi:10.1016/j.calphad.2006.02.009 10.1016/j.calphad.2006.02.009
Shi X, Luo J: Developing grain boundary diagrams as a materials science tool: A case study of nickel-doped molybdenum. Phys Rev B 2011, 84(1):014105.
Ardell AJ: A1-L12 interfacial free energies from data on coarsening in five binary Ni alloys, informed by thermodynamic phase diagram assessments. J Mater Sci 2011, 46(14):4832–4849. doi:10.1007/s10853–011–5395-x doi:10.1007/s10853-011-5395-x 10.1007/s10853-011-5395-x
Sudbrack CK, Noebe RD, Seidman DN: Compositional pathways and capillary effects during isothermal precipitation in a nondilute Ni–Al–Cr alloy. Acta Mater 2007, 55(1):119–130. doi:10.1016/j.actamat.2006.08.009 doi:10.1016/j.actamat.2006.08.009 10.1016/j.actamat.2006.08.009
Höglund L, Ågren J: Simulation of Carbon Diffusion in Steel Driven by a Temperature Gradient. J Phase Equil Dif 2010, 31(3):212–215. doi:10.1007/s11669–010–9673–0 doi:10.1007/s11669-010-9673-0 10.1007/s11669-010-9673-0
MatNavi: NIMS Materials Database. 2012. . Accessed July 2012 2012 http://mits.nims.go.jp/index_en.html
Landolt-Börnstein Database, Springer Materials. 2014. http://www.springermaterials.com/navigation/
Bartolo L, Campbell CE, Kattner UR: CALPHAD File Repositories. 2013. http://nist.matdl.org/dspace/
American Chemical Society CAS REGISTRY 2014. http://www.cas.org/expertise/cascontent/registry/index.html
International Union of Pure and Applied Chemistry (IUPAC), International Chemical Identifier 2011. http://www.iupac.org/home/publications/e-resources/inchi.html
Schmid-Fetzer R, Andersson D, Chevailer PY, Eleno L, Fabrichnaya O, Kattner UR, Sundman B, Wang C, Watson A, Zabdyr L, Zinkevich M: Assessment techniques, database design and software facilities for thermodynamics and diffusion. CALPHAD 2007, 31: 38–52. 10.1016/j.calphad.2006.02.007
Kaufman JG, Begley EF: MatML: A data interchange markup language. Adv Mater Process 2003, 161(11):35–36.
Chirico RD, Frenkel M, Diky VV, Marsh KN, Wilhoit RC: ThermoML-An XML-based approach for storage and exchange of experimental and critically evaluated thermophysical and thermochemical property data. 2. Uncertainties. J Chem Eng Data 2003, 48(5):1344–1359. doi:10.1021/je034088i doi:10.1021/je034088i 10.1021/je034088i
Frenkel M, Chirico RD, Diky VV, Dong Q, Frenkel S, Franchois PR, Embry DL, Teague TL, Marsh KN, Wilhoit RC: ThermoML - An XML-based approach for storage and exchange of experimental and critically evaluated thermophysical and thermochemical property data. 1. Experimental data. J Chem Eng Data 2003, 48(1):2–13. doi:10.1021/je025645o doi:10.1021/je025645o 10.1021/je025645o
Frenkel M, Chirico RD, Oiky VV, Marsh KN, Dymond JH, Wakeham WA: ThermoML (dagger) - An XML-based approach for storage and exchange of experimental and critically evaluate thermophysical and thermochemical property data. 3. Critically evaluated data, predicted data, and equation representation. J Chem Eng Data 2004, 49(3):381–393. doi:10.1021/je049890e doi:10.1021/je049890e 10.1021/je049890e
Boyce DE, Dawson PR, Miller MP: The Design of a Software Environment for Organizing, Sharing, and Archiving Materials Data. Mater Trans A 2009, 40A(10):2301–2318. doi:10.1007/s11661–009–9889-y doi:10.1007/s11661-009-9889-y
ECMA International, ECMA-404 Standard, 1st edition: JavaScript Object Notation. 2013. http://www.json.org/
Binary JSON. 2012. http://bsonspec.org/
Shang S, Wang Y, Liu ZKESPEI: Extensible, Self-optimizing Phase Equilibrium Infrastructure for Magnesium Alloys. In Magnesium Technology 2010. Edited by: Agnew SR, Neelameggham NR, Nyberg EA, Sillekens WH. WA: Seattle; 2010:617–622.
Wang Y, Curtarolo S, Jiang C, Arroyave R, Wang T, Ceder G, Chen LQ, Liu ZK: Ab initio lattice stability in comparison with CALPHAD lattice stability. Calphad 2004, 28(1):79–90. http://dx.doi.org/10.1016/j.calphad.2004.05.002 10.1016/j.calphad.2004.05.002
Mei ZG, Shang SL, Wang Y, Liu ZK: Density-functional study of the thermodynamic properties and the pressure-temperature phase diagram of Ti. Phys Rev B 2009, 80(10):104116. doi:10.1103/PhysRevB.80.104116 doi:10.1103/PhysRevB.80.104116
Ozolins V: First-Principles Calculations of Free Energies of Unstable Phases: The Case of fcc W. Phys Rev Lett 2009, 102(6):065702. doi:10.1103/PhysRevLett.102.065702 doi:10.1103/PhysRevLett.102.065702
Zunger A, Wei SH, Ferreira LG, Bernard JE: Special quasirandom structures. Phys Rev Lett 1990, 65(3):353–356. doi:10.1103/PhysRevLett.65.353 doi:10.1103/PhysRevLett.65.353 10.1103/PhysRevLett.65.353
Jiang C, Wolverton C, Sofo J, Chen LQ, Liu ZK: First-principles study of binary bcc alloys using special quasirandom structures. Phys Rev B 2004, 69(21):214202. doi:10.1103/PhysRevB.69.214202 doi:10.1103/PhysRevB.69.214202
Shin D, Arroyave R, Liu ZK, Van de Walle A: Thermodynamic properties of binary hcp solution phases from special quasirandom structures. Phys Rev B 2006, 74(2):024204. doi:10.1103/PhysRevB.74.024204 doi:10.1103/PhysRevB.74.024204
Wang Y, Zacherl CL, Shang SL, Chen LQ, Liu Z: Phonon dispersions in random alloys: a method based on special quasi-random structure force constants. J Phys-Condens Matter 2011., 23(48): 485403 485403
Shang SL, Wang Y, Kim DE, Zacherl CL, Du Y, Liu ZK: Structural, vibrational, and thermodynamic properties of ordered and disordered Ni (1-x) Pt (x) alloys from first-principles calculations. Phys Rev B 2011., 83(14): 144204 144204
Malakhov DV: Confidence intervals of calculated phase boundaries. CALPHAD 1997, 21(3):391–400. doi:10.1016/s0364–5916(97)00039–4 doi:10.1016/s0364-5916(97)00039-4 10.1016/S0364-5916(97)00039-4
Stan M, Reardon B: A Bayesian approach to evaluating the uncertainty of thermodynamic data and phase diagrams. CALPHAD 2003, 27(3):319–323. doi:10.1016/j.calphad.2003.11.002 doi:10.1016/j.calphad.2003.11.002 10.1016/j.calphad.2003.11.002
National Research Council: Retooling Manufacturing:Bridging Design, Materials, and Production. The National Academies Press, Washington, DC; 2004. http://www.nap.edu/catalog.php?record_id=11049
Integrated Computational Materials Engineering: A Transformational Discipline for Improved Competitiveness and National Security. The National Academies Press, Washington, DC; 2008. http://www.nap.edu/catalog.php?record_id=12199
Department of Energy: Human Genome Project information. 2003. . Accessed March 3, 2012 2012 http://www.ornl.gov/sci/techresources/Human_Genome/home.shtml
Obama B: Making Open and Machine Readable the New Default for Government Information. US Government Executive Order, The White House. 2013. http://www.whitehouse.gov/the-press-office/2013/05/09/executive-order-making-open-and-machine-readable-new-default-government
Acknowledgments
The work by ZK Liu is partially supported by the National Science Foundation (NSF) through grant DMR-1006557.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
CEC reviewed the data needed to develop the diffusion mobility, molar volume and thermal expansion descriptions. CEC also contributed the discussion on expanding the CALPHAD approach to other phase-based properties that are independent of the thermodynamics, including the thermal conductivity and interphase properties. URK reviewed the development of the CALPHAD approach and the crystallographic, phase equilibria, thermochemical data needed to develop the thermodynamic descriptions. CEC reviewed the data needed to develop the diffusion mobility, molar volume and thermal expansion descriptions. Z-KL contributed the discussion on elastic properties and the development of the ESPEI. URK and CEC both contributed to the discussion on data repositories and needed data infrastructure. All the authors collaborated in presenting the motivation of this work and in the discussion of all the work presented. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article

Cite this article
Campbell, C.E., Kattner, U.R. & Liu, ZK. The development of phase-based property data using the CALPHAD method and infrastructure needs. Integr Mater Manuf Innov 3, 158–180 (2014). https://doi.org/10.1186/2193-9772-3-12
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1186/2193-9772-3-12