
Abstract
Activation of the coagulation system is a fundamental host defense mechanism. Microorganisms that have invaded the body are trapped and disposed of in clots. Monocytes/macrophages are widely accepted as the main players in the procoagulant process; however, recent evidence suggests that neutrophils also play important roles. Tissue factor, which initiates the extrinsic coagulation cascade, is reportedly expressed on the surface of neutrophils, as well as on microparticles derived from neutrophils. Neutrophil extracellular traps (NETs) are another source of tissue factor. The components of NETs, such as DNA, histones, and granule proteins, also provide procoagulant activities. For instance, DNA initiates the intrinsic pathway, histones are a strong generator of thrombin, and granule proteins such as neutrophil elastase, cathepsin G and myeloperoxidase contribute to the suppression of the anticoagulation systems. Although understanding of the mechanisms that are involved in coagulation/fibrinolysis in sepsis has gradually progressed, the impact of neutrophils on thrombogenicity during sepsis remains to be addressed. Since the importance of the connection between coagulation and inflammation is advocated nowadays, further research on neutrophils is required.
Similar content being viewed by others


Introduction
Activation of the coagulation system is a critical step in the prevention of bacterial dissemination as the coagulation system participates in host defense via fibrin deposition and thrombus formation [1]. Recently, Engelmann and Massberg [2] introduced the term ‘immunothrombosis’ , which designates an innate immune response induced by the formation of thrombi in microvessels and is supported by immune cells and specific thrombosis-related molecules to generate an intravascular scaffold (Figure 1). Immunothrombosis facilitates the recognition, containment, and destruction of pathogens at the infectious site, thereby protecting host integrity without inducing major collateral damage. Once these reactions occur systemically, however, they lead to disseminated intravascular coagulation, and subsequently multiple organ failure and death [3].

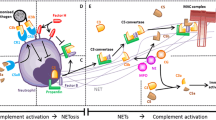
Neutrophils induce blood coagulation during sepsis. Neutrophils accumulate and adhere to the vascular endothelium in collaboration with platelets during sepsis. There, they express tissue factor (TF), release TF-bearing neutrophil microparticles (NMPs), and expel neutrophil extracellular traps (NETs) that initiate the coagulation cascade. In addition, neutrophil-derived granule proteins, especially neutrophil elastase (NE), participate in thrombus formation by inhibiting tissue factor pathway inhibitor (TFPI) and anticoagulants such as antithrombin (AT) and activated protein C (APC). Both thrombi formation and endothelial damage lead to substantial microcirculatory damage and organ dysfunction if they occur systemically. RBC, red blood cell.
Monocytes and macrophages, together with platelets, have long been regarded as the leading actors in activation of the coagulation system; however, recent evidence has suggested that neutrophils also play important roles. For instance, the attenuation of thrombotic manifestations after neutrophil depletion demonstrates the significant contribution of these cells [4]. In the early 1970s Lerner and colleagues [5] suggested a critical role for neutrophils in the thrombotic process, but their theory has only recently been accepted. The cornerstone in this field was a report on neutrophil extracellular traps (NETs), which comprise the extracellular components of neutrophils, including DNA, histones, and neutrophil granule constituents [6]. The observation that NETs express pro-thrombotic and pro-coagulant functions has attracted much attention. In addition to causing microcirculatory disturbances, thrombin generation by neutrophils has been implicated in the induction of further inflammation through the activation of protease activated receptors (PARs) [7, 8]. PARs are G-protein-coupled membrane receptors expressed by a variety of cells, including vascular cells. Thrombin and other coagulation factors such as factor VIIa and factor Xa activate platelets and regulate the behavior of other cells through the activation of these receptors. For example, PAR1 is activated when thrombin binds to and cleaves its amino-terminal exodomain to unmask a new receptor amino terminus. This new amino terminus then serves as a tethered peptide ligand, binding intramolecularly to the body of the receptor to effect transmembrane signaling [9]. PARs are also activated by a variety of proteases other than the coagulation factors, such as trypsin and cathepsin G, leading to induction of proinflammatory responses in a variety of cells. Activation of PARs upregulates endothelial expression of tissue factor (TF), mobilization of Weibel Palade bodies and release of von Willebrand factor [10, 11]. In this regard, coagulation and inflammation are the two wheels of the host response to infection.
Consequently, knowledge of neutrophil functions is quite important, not only for understanding how the coagulation system is activated but also for elucidating the mechanisms involved in inflammation and host defense. Although anticoagulant therapy could be the treatment of choice for patients with sepsis [12], concurrent effects on neutrophils should be considered. We expect that such approaches may lead to the development of new strategies for controlling severe sepsis.
Tissue factor expression
TF is a trans-membrane protein that initiates the coagulation cascade and results in thrombin generation [13]. It binds to the coagulation factor VIIa to activate factor X, forming a transient ternary complex. During sepsis, TF is generally accepted to be the main initiator of coagulation. Physiologically, circulating blood is strictly isolated from TF to avoid unexpected clot formation [14]. In contrast, a wide variety of blood cells, as well as endothelial cells, express TF under certain conditions [15, 16]. The question arises, however, as to what are the major cellular sources of TF in sepsis? Several cell types that are hemostatically inactive under normal conditions can transform their membrane into a powerful procoagulant surface when exposed to agonists. In particular, endothelial cells and mononuclear phagocytes have been intensively studied and reported to induce synthesis of TF after exposure to stimulating agents during sepsis [17]. Egorina and colleagues [18] reported that lipopolysaccharide induced TF expression in human monocytes in vitro. TF expression has also been observed in monocytes from patients with bacteremia [19] or with viral infection [20]. As well as hematopoietic cells, vascular endothelial cells also express TF. One study reported co-localization of TF and von Willebrand factor in the baboon model of sepsis [21].
In contrast, the ability of neutrophils to express TF has been a matter of debate. Although Rapaport and Rao [22] reported in 1995 that TF expressed in neutrophils plays an important role in the activation of coagulation, the significance of neutrophils in thrombus formation had been considered minor. Twenty years have passed since then, and several studies have reported the in vivo and ex vivo production of TF by neutrophils [23, 24]. Maugeri and colleagues [23] and Ritis and colleagues [24] reported that neutrophils produce and express functional TF in response to inflammatory agents, such as C5a, bacterial peptide fMLP and P-selectin. Todoroki and colleagues [25] also reported that TF-positive neutrophils were observed in the circulation in a primate model of sepsis. We also recognized TF expression on the cellular surface of the neutrophils stimulated by lipopolysaccharide (unpublished data; Figure 2). In 2012, Darbousset and colleagues [26] reported that neutrophils are the main source of blood-borne TF using a thrombosis model. Moreover, neutrophils treated with inflammatory mediators reportedly express TF at the mRNA level. On the other hand, Osterud and colleagues [27] reported that Escherichia coli phagocytosis in the absence of inflammatory mediators was not sufficient for TF production, despite the observed upregulation of TF mRNA, and this finding is in accordance with the previously reported inability of lipopolysaccharide-treated granulocytes to produce TF [28].

Tissue factor expression by neutrophils. Representative micrographs of leukocytes (original magnification 100×). Leukocytes extracted from mouse were stimulated by lipopolysaccharide. Eight hours later, the neutrophils were fixed on the slide and stained with immunofluorescent antibodies to both neutrophil elastase (middle panel) and tissue factor (right panel). The neutrophils were stained by both agents, while lymphocytes (arrows) were negative for the staining.
Other studies have also reported that neutrophils do not synthesize TF, but can acquire TF by binding monocyte/platelet-derived microparticles [29]. Regardless of whether TF is produced de novo or is acquired from other sources, we think neutrophil-derived TF plays some roles in coagulopathy during sepsis since neutrophils are recognized as a core part of thrombus formation. Indeed, it seems reasonable to consider the activity of TF produced and/or acquired by neutrophils as part of the pathogenesis of thrombotic events during sepsis [30], since neutrophils are the most abundant leukocyte population and enormous numbers of neutrophils take part in the fight against invading microorganisms at the front line of the infection site. Further investigation is required to clarify the major cellular source of septic coagulopathy.
Neutrophil extracellular trap formation
NETs, which are composed of nuclear and granule constituents of neutrophils, were first described by Brinkmann and colleagues [6]. They primarily entrap and dispose of microbes, thereby contributing to host defense [31]. An alternative effect of NETs is initiation of the coagulation cascade [32, 33]. The polyanionic NET surface readily activates contact phase proteins such as factor XII. The contact system (intrinsic cascade) is initiated when blood comes into contact with an anionic surface, and relies on factors intrinsic to flowing blood. In contrast, the extrinsic pathway has received its name because it is initiated by TF, a protein that is not normally in contact with the bloodstream. The participation of the contact pathway is dispensable for physiological hemostasis, and its significance with respect to its role in the activation of coagulation during sepsis is still uncertain. Frick and colleagues [34] reported that the contact system works as a branch of innate immunity and is partly responsible for the activation of sepsis-associated coagulopathy, although many others think that its role is minor. Studies of experimental bacteremia or endotoxemia have shown that activation of coagulation under these circumstances is exclusively mediated by the TF-VIIa route [35]. Blocking contact activation by means of a monoclonal antibody to factor XIIa did not affect E. coli-induced disseminated intravascular coagulation in baboons [36]. Thus, further study is needed concerning this issue.
NETs stimulate platelets via histones H3 and H4 to promote the thrombotic reaction [37]. Furthermore, a part of the mechanism for activation of coagulation is explained by the extracellular delivery of TF by NETs. Kambas and colleagues [38] demonstrated the autophagy-mediated delivery of TF to NETs in sepsis patients. The role of NETs was further examined using experimental models of deep vein thrombosis [39]. These models have provided new information on the initiation and progression of clot formation and the specific impact of NETs; for example, citrullinated histone H3 [40], which is a signature feature of expelling NETs, was observed in thrombi, and the intravenous administration of histones accelerated clot formation [41]. In a mouse model of thrombosis induced by lipopolysaccharide, the earliest event observed in the vessel is the adhesion of leukocytes to endothelium, platelet aggregation and the subsequent endothelial damage (Figure 3). These phenomena indicate the important role of neutrophils in venous thrombus formation [42, 43]. The association of TF with NETs can also target thrombin generation and fibrin clot formation to the sites of infection [4]. Kambas and colleagues [38, 44] demonstrated that an autophagy-based mechanism is implicated in the extracellular localization of TF in NETs. The linkage between NETs and thrombosis suggests a critical role for neutrophils in the interaction between inflammatory and thrombotic pathways.

Neutrophil adhesion to blood vessels. Left panel: intra-vital microscopic view of the mouse mesenteric vein at 1 hour after lipopolysaccharide infusion. The leukocyte (arrow) adheres to the endothelium and the platelets aggregate in the surrounding area (arrow heads) (objective lens × 20). Right panel: fluorescence live imaging of the mesenteric vein in mouse showing leukocytes in the thrombus (arrows). Fluorescent microscopic examination was performed at 1 hour after infusion of lipopolysaccharide and 4′,6-diamidino-2-phenylindole (DAPI). Nuclei of the damaged endothelial cells were also stained in blue (arrow head) (objective lens × 40).
As well as TF expression on NETs, histones are also involved in initiation of the coagulation cascade. According to a report by Xu and colleagues [41], histones H3 and H4 have a highly damaging effect on endothelial cells; an intense accumulation of neutrophils is seen in the lungs when histones are injected intravenously into mice, and peri-bronchoalveolar bleeding and thrombus formation is observed, findings that were minimized when an antibody against H4 was administered. We also confirmed the remarkable lung edema, bleeding and thrombus formation in a mouse model after the intravenous administration of histone H3 [45] (Figure 4). Histones are DNA-binding proteins with a positive charge and their toxicity can be diminished by binding of heparins, which are highly sulfated and negatively charged. However, because high-dose heparin increases the risk of bleeding, Wildhagen and colleagues [46] have developed nonanticoagulant heparin, which was reported to improve survival in sepsis.Other circulating NET components, such as nucleosomes and DNA, also activate the coagulation system, and plasma samples obtained from severe sepsis patients demonstrated the positive correlation between the nucleosome and fibrin/fibrinogen degradation products (FDPs), and histone H3 and FDPs (unpublished data; Figure 5).

Histone H3-induced lung injury. Histological changes in mouse lung 3 hours after 50 mg/kg intravenous histone H3. Macroscopic findings of the lung demonstrate massive bleeding and edema compared to the control (lower left corner). Remarkable bleeding was recognized in the surrounding part of the trachea and the vessels microscopically. (Hematoxylin-eosin staining; ×20 objective lens).

Relationship between fibrin/fibrinogen degradation products and neutrophil extracellular traps. Circulating levels of nucleosome and histone H3 were measured in 10 patients (17 points) with severe sepsis. Positive correlation was observed both between nucleosome and fibrin/fibrinogen degradation products (FDP; R2 = 0.258), and between histone H3 and FDP (R2 = 0.459), suggesting that neutrophil extracellular traps play some role in the activation of coagulation.
Histone/DNA complexes are potent activators of human platelets via Toll-like receptor-2 and -4. Histones also interact with the A1 domain of von Willebrand factor, leading to further acceleration of platelet activation [47]. Furthermore, histones induce endothelial cell cytotoxicity and maximize the platelet-endothelial interaction [48]. Two serine protease components of NETs, neutrophil elastase and cathepsin G, also contribute to coagulation (discussed below).
As mentioned above, NET release is thought to play important roles in the pathophysiology of sepsis and activation of coagulation, although it remains controversial whether NETs contribute significantly to sepsis-associated coagulopathy and pathogen killing. To prove the significance of NET release during sepsis, more sophisticated imaging techniques will be needed with appropriate fluorescent dyes to delineate NETs.
As discussed, it is assumed that NETs play at least some role in the development of sepsis-associated coagulopathy; however, the contribution of neutrophils to the activation of coagulation is not limited to infectious diseases. NETs also contribute to coagulopathy in non-infectious diseases such as autoimmune disease [49] and malignancies [50]. Kambas and colleagues [49] demonstrated that the expression of TF in NETs and neutrophil-derived microparticles plays important roles in the induction of thrombosis in active antibody-associated vasculitis.
Microparticles
Microparticles are released from a variety of cells by budding of the plasma membrane, a process known as ‘ectocytosis’ [51]. These microparticles express the specific antigens of their parental cells [52], and their procoagulant activity depends on negatively charged phospholipids, such as phosphatidylserine, TF, and inclusion proteins [44, 53]. Circulating levels of microparticles increase under critical conditions, such as severe sepsis and disseminated intravascular coagulation [54, 55]. These reports indicate the substantial increase in interest in the role of microparticles as effectors in the tight tuning of adaptive responses such as inflammation, immunity, and hemostasis [56]. Among microparticles, TF-rich microparticles have been investigated most thoroughly [27, 57]. Monocytes/macrophages have been suggested to be the main source of TF-rich microparticles [58], although recent studies have demonstrated that neutrophil-derived microparticles also express TF [44]. In addition, neutrophil-derived microparticles expose phosphatidylserine on their membrane [59] and contain bioactive enzymes such as neutrophil elastase and myeloperoxidase, which all contribute to activation of coagulation [60]. The possibility of neutrophil-derived microparticle-driven coagulation is further supported by a report by Nieuwland and colleagues [61]. They reported elevated levels of neutrophil-derived microparticles in septic patients with disseminated intravascular coagulation and suggested that the coagulation is enhanced by phospholipid and surface TF on neutrophil-derived microparticles. Although the results are not always consistent [60, 62], neutrophil-derived microparticles are, at least to some degree, involved in the pathogenesis of coagulation disorder during sepsis.
Serine proteases
In addition to the aforementioned mechanism of neutrophil-transmitted activation of coagulation, protease-related systems also accelerate coagulation. Neutrophil elastase and cathepsin G are the two major proteases related to coagulation, with neutrophil elastase considered to be dominant. In mice lacking neutrophil elastase but expressing cathepsin G, a significant reduction in fibrin formation has been reported [63]. Neutrophil elastase associated with NETs cleaves tissue factor pathway inhibitor (TFPI) [63, 64], a primary inhibitor of TF that is synthesized mainly by endothelial cells and platelets and is presented in the blood as well as on cell surfaces. The increased procoagulant activity induced by the degradation of TFPI is crucial for preventing the spread of invading microorganisms. NET components cooperate efficiently to degrade TFPI, and polyanionic nucleosomes, expelled along with NET ejection, serve as the platform for the binding of neutrophil elastase [63]. In addition, neutrophil proteases have been implicated in the control of platelet responses to vascular injury by enhancing matrix thrombogenicity [65] and also by the cleavage of von Willebrand factor [66]. It is an intriguing scenario that two major bacteriocidal components - the nucleosome and neutrophil elastase - cooperate in clot formation to amplify the entrapment of microbes and facilitate pathogen killing.
Another important function of neutrophil elastase is the inactivation of anticoagulants. Plasma levels of physiological anticoagulants such as antithrombin and activated protein C are known to decrease markedly because of their increased consumption, suppressed synthesis, and degradation by neutrophil proteases [67]. Since the supplementation of these anticoagulants is practically possible and the development of other anticoagulants is currently underway [68], determining the indication, timing and dose for their use is quite important.
Conclusion
Immunothrombosis is a major element of the intravascular innate immune system. Neutrophils expressing TF are recruited into the circulatory system and the release of TF-bearing neutrophil-derived microparticles, NETs and their components intensifies activation of the coagulation cascade, leading to thrombus formation and the activation of other cell populations, such as platelets and endothelial cells. These cellular and molecular responses all participate in clot formation. In addition, neutrophil-derived proteases, such as neutrophil elastase and cathepsin G, further accelerate these sequential reactions. However, the magnitude of the contribution of neutrophils to the activation of coagulation still remains to be elucidated. The debate on the ability of neutrophils to produce functional TF is ongoing, and the intracellular localization of TF in neutrophils raises a concern regarding its ability to activate the coagulation system. These issues should be addressed in the future. Is the neutrophil a ‘prima donna’ or a ‘corps de ballet’ in the procoagulant process during sepsis? Further evidence is needed to answer this question.
Abbreviations
- FDP:
-
fibrin/fibrinogen degradation product
- NET:
-
neutrophil extracellular trap
- PAR:
-
protease activated receptor
- TF:
-
tissue factor
- TFPI:
-
tissue factor pathway inhibitor.
References
Luo D, Szaba FM, Kummer LW, Plow EF, Mackman N, Gailani D, Smiley ST: Protective roles for fibrin, tissue factor, plasminogen activator inhibitor-1, and thrombin activatable fibrinolysis inhibitor, but not factor XI, during defence against the Gram-negative bacterium Yersinia enterocolitica. J Immunol. 2011, 187: 1866-1876. 10.4049/jimmunol.1101094.
Engelmann B, Massberg S: Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013, 13: 34-45.
Opal SM, Esmon CT: Bench-to-bedside review: Functional relationships between coagulation and the innate immune response and their respective roles in the pathogenesis of sepsis. Crit Care. 2003, 7: 23-38.
von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Köllnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, et al: Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012, 209: 819-835. 10.1084/jem.20112322.
Lerner RG, Goldstein R, Cummings G, Lange K: Stimulation of human leukocyte thromboplastic activity by endotoxin. Proc Soc Exp Biol Med. 1971, 138: 145-148. 10.3181/00379727-138-35848.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A: Neutrophil extracellular traps kill bacteria. Science. 2004, 303: 1532-1535. 10.1126/science.1092385.
Pawlinski R, Pedersen B, Schabbauer G, Tencati M, Holscher T, Boisvert W, Andrade-Gordon P, Frank RD, Mackman N: Role of tissue factor and protease-activated receptors in a mouse model of endotoxemia. Blood. 2004, 103: 1342-1347.
Schouten M, Wiersinga WJ, Levi M, van der Poll T: Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. 2008, 83: 536-545.
Coughlin SR: How the protease thrombin talks to cells. Proc Natl Acad Sci U S A. 1999, 96: 11023-11027. 10.1073/pnas.96.20.11023.
Langer F, Morys-Wortmann C, Kusters B, Storck J: Endothelial protease-activated receptor-2 induces tissue factor expression and von Willebrand factor release. Br J Haematol. 1999, 105: 542-550. 10.1111/j.1365-2141.1999.01356.x.
Storck J, Kusters B, Zimmermann ER: The tethered ligand receptor is the responsible receptor for the thrombin induced release of von Willebrand factor from endothelial cells (HUVEC). Thromb Res. 1995, 77: 249-258. 10.1016/0049-3848(95)91612-O.
Iba T, Nagaoka I, Boulat M: The anticoagulant therapy for sepsis-associated disseminated intravascular coagulation. Thromb Res. 2013, 131: 383-389. 10.1016/j.thromres.2013.03.012.
Mitroulis I, Kambas K, Anyfanti P, Doumas M, Ritis K: The multivalent activity of the tissue factor-thrombin pathway in thrombotic and non-thrombotic disorders as a target for therapeutic intervention. Expert Opin Ther Targets. 2011, 15: 75-89. 10.1517/14728222.2011.532788.
Drake TA, Morrissey JH, Edgington TS: Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989, 134: 1087-1097.
Colucci M, Balconi G, Lorenzet R, Pietra A, Locati D, Donati MB, Semeraro N: Cultured human endothelial cells generate tissue factor in response to endotoxin. J Clin Invest. 1983, 71: 1893-1896. 10.1172/JCI110945.
Semeraro N, Biondi A, Lorenzet R, Locati D, Mantovani A, Donati MB: Direct induction of tissue factor synthesis by endotoxin in human macrophages from diverse anatomical sites. Immunology. 1983, 50: 529-535.
Brozna JP: Cellular regulation of tissue factor. Blood Coagul Fibrinolysis. 1990, 1: 415-426. 10.1097/00001721-199010000-00009.
Egorina EM, Sovershaev MA, Bjorkoy G, Gruber FX, Olsen JO, Parhami-Seren B, Mann KG, Osterud B: Intracellular and surface distribution of monocyte tissue factor: application to intersubject variability. Arterioscler Thromb Vasc Biol. 2005, 25: 1493-1498. 10.1161/01.ATV.0000168413.29874.d7.
Osterud B, Flaegstad T: Increased tissue thromboplastin activity in monocytes of patients with meningococcal infection: related to an unfavourable prognosis. Thromb Haemost. 1983, 49: 5-7.
Funderburg NT, Mayne E, Sieg SF, Asaad R, Jiang W, Kalinowska M, Luciano AA, Stevens W, Rodriguez B, Brenchley JM, Douek DC, Lederman MM: Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood. 2010, 115: 161-167. 10.1182/blood-2009-03-210179.
Drake TA, Cheng J, Chang A, Taylor FB: Expression of tissue factor, thrombomodulin, and Eselectin in baboons with lethal Escherichia coli sepsis. Am J Pathol. 1993, 142: 1458-1470.
Rapaport SI, Rao LV: The tissue factor pathway: how it has become a ‘prima ballerina’. Thromb Haemost. 1995, 74: 7-17.
Maugeri N, Brambilla M, Camera M, Carbone A, Tremoli E, Donati MB, de Gaetano G, Cerletti C: Human polymorphonuclear leukocytes produce and express functional tissue factor upon stimulation. J Thromb Haemost. 2006, 4: 1323-1330. 10.1111/j.1538-7836.2006.01968.x.
Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, Rafail S, Kartalis G, Sideras P, Lambris JD: A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006, 177: 4794-4802. 10.4049/jimmunol.177.7.4794.
Todoroki H, Nakamura S, Higure A, Okamoto K, Takeda S, Nagata N, Itoh H, Ohsato K: Neutrophils express tissue factor in a monkey model of sepsis. Surgery. 2000, 127: 209-216. 10.1067/msy.2000.103027.
Darbousset R, Thomas GM, Mezouar S, Frère C, Bonier R, Mackman N, Renné T, Dignat-George F, Dubois C, Panicot-Dubois L: Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood. 2012, 120: 2133-2143. 10.1182/blood-2012-06-437772.
Osterud B, Rao LV, Olsen JO: Induction of tissue factor expression in whole blood: lack of evidence for the presence of tissue factor expression in granulocytes. Thromb Haemost. 2000, 83: 861-867.
Østerud B: Tissue factor expression in blood cells. Thromb Res. 2010, 125 (Suppl 1): S31-S34.
Egorina EM, Sovershaev MA, Olsen JO, Østerud B: Granulocytes do not express but acquire monocyte-derived tissue factor in whole blood: evidence for a direct transfer. Blood. 2008, 111: 1208-1216.
Aras O, Shet A, Bach RR, Hysjulien JL, Slungaard A, Hebbel RP, Escolar G, Jilma B, Key NS: Induction of microparticle- and cell-associated intravascular tissue factor in human endotoxemia. Blood. 2004, 103: 4545-4553. 10.1182/blood-2003-03-0713.
Papayannopoulos V, Zychlinsky A: NETs: a new strategy for using old weapons. Trends Immunol. 2009, 30: 513-521. 10.1016/j.it.2009.07.011.
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A: Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007, 176: 231-241. 10.1083/jcb.200606027.
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH, Kubes P: Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007, 13: 463-469. 10.1038/nm1565.
Frick IM, Björck L, Herwald H: The dual role of the contact system in bacterial infectious disease. Thromb Haemost. 2007, 98: 497-502.
Levi M, ten Cate H: Disseminated intravascular coagulation. N Engl J Med. 1999, 341: 586-592. 10.1056/NEJM199908193410807.
Pixley RA, De La Cadena R, Page JD, Kaufman N, Wyshock EG, Chang A, Taylor FB, Colman RW: The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia: in vivo use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J Clin Invest. 1993, 91: 61-68. 10.1172/JCI116201.
Fuchs TA, Bhandari AA, Wagner DD: Histones induce rapid and profound thrombocytopenia in mice. Blood. 2011, 118: 3708-3714. 10.1182/blood-2011-01-332676.
Kambas K, Mitroulis I, Apostolidou E, Girod A, Chrysanthopoulou A, Pneumatikos I, Skendros P, Kourtzelis I, Koffa M, Kotsianidis I, Ritis K: Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS One. 2012, 7: e45427-10.1371/journal.pone.0045427.
Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, Bhandari AA, Wagner DD: Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012, 10: 136-144. 10.1111/j.1538-7836.2011.04544.x.
Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA: Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009, 184: 205-213. 10.1083/jcb.200806072.
Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT: Extracellular histones are major mediators of death in sepsis. Nat Med. 2009, 15: 1318-1321. 10.1038/nm.2053.
Iba T, Kidokoro A, Fukunaga M, Fuse S, Suda M, Kunitada S, Hara T: Factor Xa-inhibitor (DX-9065a) modulates the leukocyte-endothelial cell interaction in endotoxemic rat. Shock. 2002, 17: 159-162. 10.1097/00024382-200202000-00013.
Gardiner EE, Ward CM, Andrews RK: The NET effect of clot formation. J Thromb Haemost. 2012, 10: 133-135. 10.1111/j.1538-7836.2011.04551.x.
Kambas K, Mitroulis I, Ritis K: The emerging role of neutrophils in thrombosis - the journey of TF through NETs. Front Immunol. 2012, 3: 385-
Iba T, Murai M, Nagaoka I, Tabe Y: Neutrophil extracellular traps, damage-associated molecular patterns, and cell death during sepsis. Acute Med Surg. 2014, 1: 2-9. 10.1002/ams2.10.
Wildhagen KC, García de Frutos P, Reutelingsperger CP, Schrijver R, Aresté C, Ortega-Gómez A, Deckers NM, Hemker HC, Soehnlein O, Nicolaes GA: Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood. 2014, 123: 1098-1101. 10.1182/blood-2013-07-514984.
Ward CM, Tetaz TJ, Andrews RK, Berndt MC: Binding of the von Willebrand factor A1 domain to histone. Thromb Res. 1997, 86: 469-477. 10.1016/S0049-3848(97)00096-0.
Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT: Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012, 7: e32366-10.1371/journal.pone.0032366.
Kambas K, Chrysanthopoulou A, Vassilopoulos D, Apostolidou E, Skendros P, Girod A, Arelaki S, Froudarakis M, Nakopoulou L, Giatromanolaki A, Sidiropoulos P, Koffa M, Boumpas DT, Ritis K, Mitroulis I: Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann Rheum Dis. 2013, doi: 10.1136/annrheumdis-2013-203430.
Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, Scadden DT, Wagner DD: Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A. 2012, 109: 13076-13081. 10.1073/pnas.1200419109.
Stein JM, Luzio JP: Ectocytosis caused by sublytic autologous complement attack on human neutrophils: the sorting of endogenous plasma membrane proteins and lipids into shed vesicles. Biochem J. 1991, 274: 381-386.
Mause SF, Weber C: Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res. 2010, 107: 1047-1057. 10.1161/CIRCRESAHA.110.226456.
Leroyer AS, Tedgui A, Boulanger CM: Role of microparticles in atherothrombosis. J Intern Med. 2008, 263: 528-537. 10.1111/j.1365-2796.2008.01957.x.
Schouten M, Wiersinga WJ, Levi M, van der Poll T: Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. 2008, 83: 536-545.
Joop K, Berckmans RJ, Nieuwland R, Berkhout J, Romijn FP, Hack CE, Sturk A: Microparticles from patients with multiple organ dysfunction syndrome and sepsis support coagulation through multiple mechanisms. Thromb Haemost. 2001, 85: 810-820.
Morel O, Toti F, Hugel B, Bakouboula B, Camoin-Jau L, Dignat-George F, Freyssinet JM: Procoagulant microparticles: disrupting the vascular homeostasis equation?. Arterioscler Thromb Vasc Biol. 2006, 26: 2594-2604. 10.1161/01.ATV.0000246775.14471.26.
Wang JG, Manly D, Kirchhofer D, Pawlinski R, Mackman N: Levels of microparticle tissue factor activity correlate with coagulation activation in endotoxemic mice. J Thromb Haemost. 2009, 7: 1092-1098. 10.1111/j.1538-7836.2009.03448.x.
Aleman MM, Gardiner C, Harrison P, Wolberg AS: Differential contributions of monocyte- and platelet-derived microparticles towards thrombin generation and fibrin formation and stability. J Thromb Haemost. 2011, 9: 2251-2261. 10.1111/j.1538-7836.2011.04488.x.
Gasser O, Schifferli JA: Microparticles released by human neutrophils adhere to erythrocytes in the presence of complement. Exp Cell Res. 2005, 307: 381-387. 10.1016/j.yexcr.2005.03.011.
Delabranche X, Boisramé-Helms J, Asfar P, Berger A, Mootien Y, Lavigne T, Grunebaum L, Lanza F, Gachet C, Freyssinet JM, Toti F, Meziani F: Microparticles are new biomarkers of septic shock-induced disseminated intravascular coagulopathy. Intensive Care Med. 2013, 39: 1695-1703. 10.1007/s00134-013-2993-x.
Nieuwland R, Berckmans RJ, McGregor S, Böing AN, Romijn FP, Westendorp RG, Hack CE, Sturk A: Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood. 2000, 95: 930-935.
Guervilly C, Lacroix R, Forel JM, Roch A, Camoin-Jau L, Papazian L, Dignat-George F: High levels of circulating leukocyte microparticles are associated with better outcome in acute respiratory distress syndrome. Crit Care. 2011, 15: R31-10.1186/cc9978.
Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, Konrad I, Kennerknecht E, Reges K, Holdenrieder S, Braun S, Reinhardt C, Spannagl M, Preissner KT, Engelmann B: Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010, 16: 887-896. 10.1038/nm.2184.
Glaser CB, Morser J, Clarke JH, Blasko E, McLean K, Kuhn I, Chang RJ, Lin JH, Vilander L, Andrews WH: Oxidation of a specific methionine in thrombomodulin by activated neutrophil products blocks cofactor activity. A potential rapid mechanism for modulation of coagulation. J Clin Invest. 1992, 90: 2565-2573. 10.1172/JCI116151.
Wohner N, Keresztes Z, Sótonyi P, Szabó L, Komorowicz E, Machovich R, Kolev K: Neutrophil granulocyte-dependent proteolysis enhances platelet adhesion to the arterial wall under high-shear flow. J Thromb Haemost. 2010, 8: 1624-1631. 10.1111/j.1538-7836.2010.03890.x.
Raife TJ, Cao W, Atkinson BS, Bedell B, Montgomery RR, Lentz SR, Johnson GF, Zheng XL: Leukocyte proteases cleave von Willebrand factor at or near the ADAMTS13 cleavage site. Blood. 2009, 114: 1666-1674. 10.1182/blood-2009-01-195461.
Levi M: The coagulant response in sepsis. Clin Chest Med. 2008, 29: 627-642. 10.1016/j.ccm.2008.06.006.
Iba T: Harmonized guidance for DIC from the ISTH and the current status of anticoagulant therapy in Japan. J Thromb Haemost. 2013, 11: 2076-2078. 10.1111/jth.12344.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Iba, T., Miki, T., Hashiguchi, N. et al. Is the neutrophil a ‘prima donna’ in the procoagulant process during sepsis?. Crit Care 18, 230 (2014). https://doi.org/10.1186/cc13983
Published:
DOI: https://doi.org/10.1186/cc13983