
Abstract
Background
Genomic selection (GS) allows estimation of the breeding value of individuals, even for non-phenotyped animals. The aim of the study was to examine the potential of identity-by-descent genomic selection (IBD-GS) in genomic selection for a binary, sib-evaluated trait, using different strategies of selective genotyping. This low-cost GS approach is based on linkage analysis of sparse genome-wide marker loci.
Findings
Lowly to highly heritable (h2 = 0.15, 0.30 or 0.60) binary traits with varying incidences (10 to 90%) were simulated for an aquaculture-like population. Genotyping was restricted to the 30% best families according to phenotype, using three genotyping strategies for training sibs. IBD-GS increased genetic gain compared to classical pedigree-based selection; the differences were largest at incidences of 10 to 50% of the desired category (i.e. a relative increase in genetic gain greater than 20%). Furthermore, the relative advantage of IBD-GS increased as the heritability of the trait increased. Differences were small between genotyping strategies, and most of the improvement was achieved by restricting genotyping to sibs with the least common binary phenotype. Genetic gains of IBD-GS relative to pedigree-based models were highest at low to moderate (10 to 50%) incidences of the category selected for, but decreased substantially at higher incidences (80 to 90%).
Conclusions
The IBD-GS approach, combined with sparse and selective genotyping, is well suited for genetic evaluation of binary traits. Genetic gain increased considerably compared with classical pedigree-based selection. Most of the improvement was achieved by selective genotyping of the sibs with the least common (minor) binary category phenotype. Furthermore, IBD-GS had greater advantage over classical pedigree-based models at low to moderate incidences of the category selected for.
Similar content being viewed by others

Findings
Background
An earlier study by Ødegård and Meuwissen [1] described how identity-by-descent (IBD) genomic selection (IBD-GS) for a Gaussian trait can use sparse marker data combined with selective genotyping of the phenotypically best families and the sibs with the most extreme (high/low) phenotypes within these families. Binary traits take only two possible values, and thus, truly extreme phenotypes cannot be identified at moderate frequencies (since both categories are common), while at high/low frequencies the least common category may be defined as phenotypically extreme.
The aim of the study was to quantify to what extent different types of selective genotyping schemes, using sparse markers, combined with IBD-GS could increase genetic gain for a sib-evaluated binary trait, compared with classical pedigree-based selection schemes, as applied in aquaculture breeding. The IBD-GS method uses linkage analysis to trace genomic IBD relationships in the population [2].
Methods
Data was simulated using the QMSim software [3]. The simulated datasets were essentially identical to the datasets reported in Ødegård and Meuwissen [1], except that the simulated (underlying) Gaussian phenotype z (standard normal) was converted to a binary phenotype y, with an incidence P by assuming: \( y=\left\{\begin{array}{c}\hfill 0\ if\ z\le k\hfill \\ {}\hfill 1\ if\ z>k\hfill \end{array}\right. \), where k = probit(1 − P). Here, 1 is defined as the desired category, and the incidence P was set to values 0.1, 0.2, 0.5, 0.8 and 0.9.
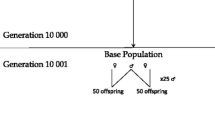
A total of 50 replicates were generated, assuming an underlying heritability of 0.15, 0.30 or 0.60. For the final generations, 100 full-sib families were produced, each consisting of 120 sibs, of which 100 were used for training and 20 were selection candidates (non-phenotyped).
Marker density was low (~40 markers/M) and genotypes were stored only for the phenotypically best 30 families (high incidence) in the last generation (and parents and grandparents). Genotypes were available on selection candidates (20 per family) and for varying fractions of their phenotyped training sibs (100 per family). Three strategies were tested:
Full genotyping (FG): Genotyping all training sibs.
Top-bottom genotyping (TBG): Genotyping 40 training sibs per family, aiming at 20 bad (y = 0) + 20 good (y = 1) sibs. For families with less than 20 sibs of one category, additional sibs of the other category were genotyped.
Minor category genotyping (MCG): Selective genotyping of sibs with the least common (minor) binary category phenotype.
All strategies involved genotyping 600 selection candidates, but with different numbers of the training animals (FG: 3000, TBG: 1200, MCG: 3000*f, where f is the incidence of the minor category phenotype within the pre-selected families).
A general probit threshold model was used for analysis:
where λ i is the underlying liability of animal i, μ is the overall mean of the underlying liabilities, a is a vector of additive genetic breeding values of all animals included in the pedigree, and Z i is the i th row from the incidence matrix Z. Two sub-models (PED and IBD-GS) were defined that differed in their distributional assumptions for the additive breeding values:
where A is the numerator relationship matrix and G IBD is an IBD-based genomic relationship matrix estimated through linkage analysis with the available markers (for animals being genotyped), using the linkage disequilibrium multi-locus iterative peeling (LDMIP) method [4]. The DMU software package [5] was used in all statistical analyses, assuming known (true) underlying variance components.
Evaluation of genotyping strategies and models was carried out as follows. Selection candidates of the last generation were ranked based on their estimated breeding values (EBV) obtained from the PED and IBD-GS models, respectively (high EBV are favorable), which were used to select 200 parents for the next generation. Genetic gain was calculated as the average true breeding value of the chosen parents. To ensure realistic and similar levels of inbreeding for both models, restrictions on the number of selected families were imposed for PED, requiring parents to be selected from 20 different full-sib families, while no restrictions were imposed for IBD-GS, since lower inbreeding is generally expected based on a shift from pure family selection (PED) to individual selection (IBD-GS).
Results and discussion
Descriptive statistics of the simulation study are in Table 1. Restrictions on inbreeding (20 families used in the breeding program) were only practiced for the PED model, since higher inbreeding is generally expected for this model. Despite no restrictions on the number of families to be used, the IBD-GS model selected parents from nearly all the 30 pre-selected families (Table 2), and inbreeding levels were similar for the two models (not shown).
As expected from the large family sizes, the average genetic gains through classical family-based selection were robust to both heritability and incidence, with genetic gain over one generation of classical selection differing only slightly (1.5 to 1.7 genetic standard deviations) between scenarios. The lowest gain was obtained at the lowest heritability and highest incidence of the desired category, while the highest gain was obtained for the highest heritability and at 50% or lower incidences of the desired category. Relative to PED, in all cases, the IBD-GS model gave substantial and significant (P < 1.0e-10) increases in genetic gain over one generation (Figures 1, 2 and 3). The relative increase in genetic gain of IBD-GS compared to pedigree-based models was largest (15 to 36%) at low to moderate (0.1 to 0.5) incidences of the category selected for, but smaller (3 to 21%) at higher incidences. Furthermore, the relative advantage of IBD-GS increased as the heritability of the underlying trait increased. If the trait has a high underlying heritability and at low to moderate incidence, the relative improvement was comparable to results obtained for a Gaussian trait of moderate heritability (0.30) [1]. For binary traits, expected heritability on the observed scale is always lower than the underlying heritability, and decreases symmetrically when the incidence departs from 50% [6]. However, the relevance of this statistic is limited when considering the efficiency of individual (within-family) selection. For IBD-GS, the realized genetic gain was far from symmetric around the incidence of 50%. Genetic gain was higher when the desired category phenotype was rare and lower when the desired category phenotype was common. For binary traits, the pre-selected (best) families will necessarily have above-mean incidences of the desired category phenotype, which is statistically favorable at low overall incidences, but unfavorable at high incidences. Furthermore, when incidence approaches extreme values, animals with the most common binary phenotype will be less informative, since their expected (average) underlying liability approaches the family means, while the opposite is true for animals that have the least common binary phenotype.

Increase in genetic gain achieved when using genomic selection (IBD-GS) relative to classical pedigree-based selection (PED) for a binary trait with an underlying heritability of 0.15. Presented by incidence of the desired category (10 to 90%) and genotyping strategy (FG = full genotyping, TBG = top-bottom genotyping, MCG = minor-category genotyping).

Increase in genetic gain achieved when using genomic selection (IBD-GS) relative to classical pedigree-based selection (PED) for a binary trait with an underlying heritability of 0.30. Presented by incidence of the desired category (10 to 90%) and genotyping strategy (FG = full genotyping, TBG = top-bottom genotyping, MCG = minor-category genotyping).

Increase in genetic gain achieved when using genomic selection (IBD-GS) relative to classical pedigree-based selection (PED) for a binary trait with an underlying heritability of 0.60. Presented by incidence of the desired category (10 to 90%) and genotyping strategy (FG = full genotyping, TBG = top-bottom genotyping, MCG = minor-category genotyping).
The differences between the three genotyping strategies were small in terms of genetic gain, but strategy FG was, as expected, slightly superior (0 to 6%) to TBG and MCG (Figures 1, 2 and 3). Albeit small, differences in genetic gains between FG and the other genotyping methods were highly significant (P < 1.0e-10) at moderate (20 to 50%) incidences, but not necessarily so at higher (5.0e-3 ≤ P ≤ 0.43) or lower (2.8e-11 ≤ P ≤ 0.11) incidences. Both TBG and MCG focus genotyping towards animals with the minor category phenotype within each family, and these animals are likely the most informative in prediction of Mendelian deviations from the family mean.
For normally distributed traits, Ødegård & Meuwissen [1] found that genetic predictions (regression of true on predicted breeding values) were slightly biased with selective genotyping. In this work, we also detected some bias, but its magnitude was similar in the pedigree-based and IBD-GS models with different genotyping strategies. Average regression coefficients of true breeding values on EBV were 0.82, 1.07 and 1.21 for underlying heritabilities of 0.1, 0.3 and 0.6, respectively. Hence, predicted EBV appear inflated at low heritabilities and deflated at high heritabilities.
References
Ødegård J, Meuwissen THE. Identity-by-descent genomic selection using selective and sparse genotyping. Genet Sel Evol. 2014;46:3.
Luan T, Woolliams JA, Ødegård J, Dolezal M, Roman-Ponce SI, Bagnato A, et al. The importance of identity-by-state information for the accuracy of genomic selection. Genet Sel Evol. 2012;44:28.
Sargolzaei M, Schenkel FS. QMSim: a large-scale genome simulator for livestock. Bioinformatics. 2009;25:680–1.
Meuwissen THE, Goddard ME. The use of family relationships and linkage disequilibrium to impute phase and missing genotypes in up to whole-genome sequence density genotypic data. Genetics. 2010;185:1441–9.
Madsen P, Jensen J. DMU: a user's guide. A package for analysing multivariate mixed models. 6–5.2 edition. Tjele, Denmark: Center for Quantitative Genetics and Genomics, Dept. of Molecular Biology and Genetics, University of Aarhus; 2013.
Dempster ER, Lerner IM. Heritability of threshold characters. Genetics. 1950;35:212–36.
Acknowledgements
This study was partly financed by the Research Council of Norway through project no. 225181. Nina Santi, AquaGen is acknowledged for contribution of valuable ideas regarding methods of selective genotyping.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JØ had the original idea, performed the simulations and statistical analyses and was responsible for drafting the manuscript. THEM developed the linkage analysis methodology (LDMIP software). Both authors contributed in writing the final manuscript. Both authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Ødegård, J., Meuwissen, T.H. Identity-by-descent genomic selection using selective and sparse genotyping for binary traits. Genet Sel Evol 47, 8 (2015). https://doi.org/10.1186/s12711-015-0090-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12711-015-0090-z