
Abstract
Background
Human protein kinases, the key players in phosphoryl signal transduction, have been actively investigated as drug targets for complex diseases such as cancer, immune disorders, and Alzheimer’s disease, with more than 60 successful drugs developed in the past 30 years. However, many of these single-kinase inhibitors show low efficacy and drug resistance has become an issue. Owing to the occurrence of highly conserved catalytic sites and shared signaling pathways within a kinase family, multi-target kinase inhibitors have attracted attention.
Results
To design and identify such pan-kinase family inhibitors (PKFIs), we proposed PKFI sets for eight families using 200,000 experimental bioactivity data points and applied a graph convolutional network (GCN) to build classification models. Furthermore, we identified and extracted family-sensitive (only present in a family) pre-moieties (parts of complete moieties) by utilizing a visualized explanation (i.e., where the model focuses on each input) method for deep learning, gradient-weighted class activation mapping (Grad-CAM).
Conclusions
This study is the first to propose the PKFI sets, and our results point out and validate the power of GCN models in understanding the pre-moieties of PKFIs within and across different kinase families. Moreover, we highlight the discoverability of family-sensitive pre-moieties in PKFI identification and drug design.
Similar content being viewed by others

Introduction
Over 300 protein kinases share a common biological function as ATP-dependent phosphorylation enzymes [1], with a significant role in signal transduction, particularly in the progression of complex diseases such as cancers [2], immune system misfunctions, and Alzheimer’s disease [3]. Accordingly, they fall under the category of intensively investigated drug targets [2, 4], with 61 US Food and Drug Administration (FDA)-approved kinase inhibitors to date [5]. Due to the highly conserved catalytic sites of protein kinases, investigation of kinase inhibitor selectivity in the kinome space has been a challenge [6]. On the contrary, protein kinases within a single kinase family regulate shared cancer-related pathways [7]; therefore, inhibition of a single target leads to drug adaptation and resistance [8,9,10,11,12].
To overcome these issues of drug resistance, various studies have suggested drug combinations or multi-targeting drugs to be an effective approach for complex diseases [8, 9, 13,14,15,16,17,18]. Moreover, several approved kinase inhibitors were originally designed as pan-kinase-family inhibitors (PKFIs) to target multiple proteins of the kinase families [16, 17, 19,20,21], such as the epidermal growth factor receptor (EGFR)/HER2 dual-targeting inhibitor lapatinib [19] and the pan-vascular endothelial growth factor (VEGF) inhibitor sorafenib [22, 23]. To discover potential inhibitors within the large chemical space, deep learning techniques have been applied to rapidly identify potential inhibitors against a single target within the kinome [21, 24, 25]; however, these studies have seldom addressed the multi-targeting issue or the lack of explainability for the trained model’s judgment.
Graph convolutional network (GCN)[26] is a recently developed deep learning architecture that is designed to extract the spectrum information on the topological data. Due to the no-distanced and no-ordered properties of the topological data, it is hard to be operated by previous machine learning and deep learning techniques until the GCN architecture is brought onto the stage. The power that the GCN model provides is on the capability of self-organizing the surrounding information of each atom in the compound, and extracting the chemical substructures with different sizes. Therefore, with the help of GCN architecture, now we have the chance to achieve our aim: self-organizing the pre-moieties within families without using pre-defined fingerprints. On the other hand, some explainability methods, like gradient-weighted CAM (Grad-CAM)[27], for GCNs were developed to help identify functional groups or substructures on small molecules for biological molecular properties.
In this study, we aimed to develop GCN models to identify PKFIs and to highlight the chemical pre-moieties. First, we collected PKFIs from the ChEMBL database [28, 29] and kinase profiling data [30], and a total of 60,122 compounds of 384 kinases from 103 families within 195,802 data points were obtained. We then selected two families for each of the four kinase groups, tyrosine kinase (TK), AGC, CMGC, and calmodulin-dependent protein kinase (CAMK), and built GCN models for each selected family. Then, we applied gradient-weighted class activation mapping (Grad-CAM) [27] method to explain each inhibitor’s prediction. Our results indicate that our GCN model can aid in judging the viability of identifying family-sensitive pre-moieties in PKFIs.
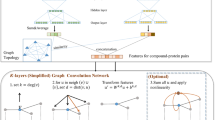
An overview of our method and models for identifying PKFIs is presented in Fig. 1. First, we collected 195,802 sets of kinase-compound activity data and defined the PKFI sets, followed by the featurization of each inhibitor into atomic features and Laplacian matrix-based topologies. The GCN model was constructed for each family to identify the PKFIs. Three indices, accuracy, the area under the receiver operating characteristic curve (AUROC), and Matthews correlation coefficient (MCC), were used in addition to applying the Grad-CAM sample-wise explanation to examine the predictability and reliability of our models and to further determine the family-sensitive pre-moieties.

Scheme for utilizing atom-wise featurization and topological information on compounds for the identification of pan-kinase family inhibitors (PKFIs) using graph convolutional network (GCN) models. A Schematic of the research framework. B 195,802 test datasets of 60,122 chemicals and 384 kinases were collected from the ChEMBL database and kinase profiling. C Eight families in four kinase groups were targeted in this study. D Each compound is transformed into atom features and a structure graph for GCN architectures to identify PKFIs. E A visualized explanation was made using the grad-CAM method
Results
PKFI sets and model performance
To assess the PKFI differences between each kinase family, we applied GCN models on eight families in four kinase groups (Table 1) individually. The configuration of our GCN architecture includes three graph convolutional layers with 64, 32, and 16 kernels respectively, and all. All convolutional feature maps were followed by a GAP layer, and then concatenate all GAP feature vectors were concatenated before applying a softmax classifier (Fig. 5). Each model was trained for 100 epochs, with an 80:20 training/testing distribution of each PKFI set utilizing the ADAM optimizer with a learning rate of 0.001, β_1 = “0.9”, β_2 = “0.999” based on cross-entropy as a loss function. All models were implemented in Keras [35] with a Tensorflow backend [36].
To evaluate the performance of each model, three metrics were applied: (1) accuracy (ACC) for general precision, (2) Matthew correlation coefficient (MCC) for measuring the quality of classification according to class-wise distributions, and (3) AUROC for measuring the composite index of sensitivity and specificity. The evaluation metrics of the test results of the eight PKFI sets are presented in Table 1. The average ACC (0.84) and AUROC (0.89) of all models were high enough to distinguish PKFIs. However, the MCC scores of the four GCN models were significantly lower in terms of their misbalanced distribution in the training sets: protein kinase D (PKD) family in the CAMK group (balance: 0.15 positive/negative), cGMP-dependent protein kinase (PKG) family in the AGC group (balance: 0.13), GSK family (balance: 0.37), and CLK family (balance: 0.42) in the CMGC group. Overall, the average ratio of balance of eight families was 0.62, where the balance values of the other four families were all above 0.5 and showed qualified MCC scores. This indicates that increasing the data size may aid in overcoming the imbalanced data distribution.
Discover common/specific pre-moieties across kinase families
Explanation and visualization were generated using the Grad-CAM method (see method, Eq. (6) and (7)). As shown in Fig. 2, we compared different explanations based on three families: the EGFR family [37, 38], the Janus kinase (JAK) family [39, 40] in the TK group, and the serine/threonine kinase PIM family [41] in the CAMK group. To demonstrate the rationality of the Grad-CAM explanation, lapatinib, a highly selective [42] EGFR/HER2 targeting dual inhibitor used in the treatment of breast cancer [43, 44], was introduced and explained by the EGFR model (Fig. 2). The preferences of the positive class are highlighted by green circles (Fig. 2A). The preference on the double nitrogen atoms in ‘middle naphthalenyl structure’ (structure of aromatic double ring) indicates that the hydrogen bond formable environments on aromatic rings were preserved in pan-EGFR inhibitors and could be captured by the EGFR model. We refer to these conserved chemical environments as family-sensitive pre-moieties, as they only retain parts from fixed moieties; the chemical characteristics are already indicated. This observation has also been validated in X-ray-crystalized complexes wherein the naphthalenyl nitrogen actually interacts with the main chain atoms of the hinge region allowing hydrogen bonding, thereby enabling ATP-competitive interaction to block the kinase activity, which is one of the key modes for designing kinase inhibitors [2, 45] (see Fig. 2C, deep blue).

Explanation of the GCN model’s prediction of lapatinib and other inhibitors in EGFR, JAK, and PIM models. (A) Grad-CAM preferences of lapatinib from the latest graph convolutional layer for both positive and negative classes. Circles are centered at each atom, with green ones for the positive class and orange for the negative class. The larger the circle, the more the atom contributes to the prediction of the model at a specific class. (B) Preferences for different inhibitors within and across families. Within the same family, conserved attention on similar environments is visualized, and family-sensitive pre-moieties can be seen by comparing cross-family inhibitors. (C) Crystallized complexes of the pan-EGFR inhibitor lapatinib (deep blue, PDB ID: 1XKK), pan-JAK inhibitor tofacitinib (light blue, PDB ID: 3EYG), and pan-PIM inhibitor LGH-447 (purple, PDB ID: 5DWR) demonstrated three different modes of kinase inhibition
After confirming the rationality of the explanations of Grad-CAM, we further compared the rationality of the explanations of inhibitors from different families (see Fig. 2B). The EGFR model pays intense attention to the hinge-interacting region, which is also represented in JAK family inhibitors. Explanations of baricitinib [46] and tofacitinib [47] indicate that this region has a minor effect on the formation of pan-family selectivity of JAKs, but is critical for selectivity of the EGFR family. In contrast, the major highlight of JAK inhibitors appears as triple-bonded nitrogen, which interacts with the p-loop structure (see Fig. 2C, light blue). This structure is essential for the transfer of phosphate onto substrates and disrupting it will interfere with phosphorylation. Indeed, within the same kinase group, inhibitors of EGFR and JAK families share partially similar structures but use different modes to interrupt kinase activity, as indicated by the explanation of our models as well as crystallized 3D structures.
In addition to the intra-TK-grouped comparison, we examined the inhibitors of the PIM family in the CAMK group to further assess the inter-grouped differentiation on the explanation of PKFIs. Aligned by the position of the hinge region as the center, LGH-447 and AZD-1208[21] of the PIM family showed no preference for the hinge region, but instead showed a preference for the cyclo-nitrogen regions of the tail (see Fig. 2B). These highlighted pre-moieties in PIM inhibitors (Fig. 2C, purple compound) actually undergo the third mode of interaction with the DFG motif of a kinase to destabilize the kinase structure and further interfere with its function [48].
Through the comparison of inhibitors across three families, family-sensitive pre-moieties and environments were demonstrated, along with the actual kinase-binding inhibitor structures.
Correlation of model explanation and statistics through current moiety-based fingerprint mapping
While revealing the family-sensitive pre-moieties (defined in Results, section B) by focusing along the decision process of the model, statistical significance must also be considered. Owing to the unfixed structure of pre-moieties, we utilized moiety-based fingerprint checkmol [49] to establish the statistical significance using odds ratio, as shown in Fig. 3. Most discovered pre-moieties are distinguishable (outline fingerprint in Fig. 3A) by checkmol descriptors, such as the triple-bonded nitrogen on baricitinib mapped to the #90 fingerprint and the cyclo-nitrogen on LGH-447 to #49 fingerprint, and also are significantly possessed by positive inhibitors within each PKFI set (i.e., #49 fingerprint concentrative possessed by pan-PIM inhibitors and #90 by pan-JAK inhibitors) (see Fig. 3A). However, there still remain several unmappable pre-moieties. For the naphthalenyl-nitrogen region of lapatinib, the only matched checkmol descriptor is #201 (any aromatic atoms) and #202 (any hetero-ring structures), and both are undistinguishable not only within the EGFR set but also across different PKFI sets (Fig. 3B).

Correlation between preferences and the mapping of current checkmol fingerprints. (A) The mapped region of checkmol fingerprint and their odds-ratio ranking (epsilon = 0.5, is added to prevent dividing by zero) of 204 checkmol descriptors within each family of the PKFIs set. Most pre-moieties associated with different modes of kinase inhibition (described previously) are mappable and correlated with the feature distribution in the training sets. However, several pre-moieties are still not precisely defined in the current fingerprint and thus are undistinguishable (i.e., naphthalenyl-nitrogen regions on lapatinib). (B) Overall odds-ratio distribution of checkmol descriptors on EGFR, JAK, and PIM datasets with mapped moieties is indicated. It should be noted that several super peaks are observed with relatively few compounds and thus are not currently discussed
The inconsistency between Grad-CAM-based family-sensitive pre-moieties and moiety-based checkmol fingerprint mapping might indicate that the information and rules of kinase-inhibitor interactions are hidden within the compound structures and compositions to a greater extent than we assumed. The GCN architecture is not only able to discover the specified local environments within inhibitors from each family but also shows the limitations of the current moiety-based describing methods. Furthermore, in this study, the GCN architecture has the potential to broaden the recognizable chemical moiety spaces and thus facilitate the rapid identification of potential inhibitors from large chemical spaces.
Conclusion and discussion
In this study for exploring kinase multi-targeting, we formulated pan-kinase-family inhibitor sets for the first time and used GCN architecture to identify the hidden information in the PKFI sets of EGFR, JAK, and PIM families. We further applied Grad-CAM to visualize the effects of chemical environments on inhibitors that were considered by the models to make decisions. Validated by the kinase-inhibitor complexes, we discovered that the family-sensitive pre-moieties contain information on kinase-inhibitor interactions and are associated with different modes of inhibition of kinase activity. By comparing our discovered pre-moieties and the checkmol moiety-based fingerprints, we demonstrated the insufficiency of current moiety-based descriptors, which can be overcome by the GCN architecture for recognizing family-specific chemical environments. In summary, the GCN technique has the power to identify PKFIs and learn the undefined pre-moieties in the field of potential drug design and optimization.
Methods
Datasets
To access the information on kinase-inhibitor reactivity, we collected 195,802 sets of kinase-chemical activity data from ChEMBL and kinase profiling, which contain about 60,122 compounds belonging to 384 kinases grouped into 103 kinase families.
The ChEMBL data was obtained from the ChEMBL version 25 (March 2019), which contains 15 million data points of 1.8 million chemicals and 12.5 thousand targets. We filtered our kinase-inhibitor set according to the following criteria: (1) selection of targets of 518 kinases [1] by UniProt ID with IC50 bioactivity, (2) exclusion of relation of “ ~ ” and “ > > ” for a certain activity, and (3) use of “Binding” assay type, “SINGLE PROTEIN” target type and confidence score 9 for retaining the direct experimental kinase-inhibitor interaction data. The resulting set contained 95,462 data points for 58,846 compounds and 382 kinases with IC50 < 500 nM as the activity cutoff.
To understand the complete test results between compounds and kinases, we collected kinase profiling data (containing 172 kinases and 3,858 compounds) published by Metz [30] and further applied the criteria below to obtain a reliable kinase-inhibitor set: (1) removal of ID-lacking, InChIKey-lacking, and InChIKey-duplicated chemicals, and (2) exclusion of pairs of blank activity results. The filtered dataset contained 1,421 compounds and 172 kinases, with a total of 100,786 test points and pKi > 6 as the activity cutoff.
After collecting these two sources of data, we merged them with “80% voting” for duplicates, which meant that the final active/inactive labels for the duplicated data points were in agreement with 80%-consistent answers among its duplicates, and those with maximum consistency below 80% were excluded. The final kinase bioactivity dataset contained 195,802 test points, with 60,122 compounds and 384 kinases.
4.2 Definition of pan-kinase-family inhibitors (PKFIs)
To establish the PKFI sets from kinase-inhibitor data, we further investigated the criteria for any compound \(C_{i}\) tested in the kinase family \(kF_{i}\) to be a PKFI.
where the total test points within compound i and family j must be no less than half of the total membership of family j with testing data on at least two kinase members. The final label of each PKFI is given as an answer when all the test points are consistently active or inactive.
Featurization of input compounds
To facilitate the classification of PKFIs with GCN frameworks, an attributed graph \({\mathcal{G}}_{i} = \left( {AF_{i} ,\tilde{L}_{i} } \right)\) is presented for each input compound Ci where \(AF_{i} \in {\mathbb{R}}^{{N \times d_{feat} }}\) is the node descriptions of atomic environments in the compounds, and atom types, chemo-properties, and charges, are described [26, 31] (Table 2). Following the previous work of Kipf and Welling [32], we used a modified normalized Laplacian matrix \(\tilde{L}_{i} \in {\mathbb{R}}^{N \times N}\) that encodes the topological structure of connections and bond order cross atoms (Fig. 4):
where \(\tilde{A} = A + I_{N}\) is the adjacency matrix of input compounds added by self-interaction, \(I_{N} \in {\mathbb{R}}^{N \times N}\) is the identity matrix, and \(\tilde{D}_{ii} = \mathop \sum \nolimits_{j} \tilde{A}_{ij}\) is the diagonal degree matrix based on \(\tilde{A}\). Given that the task of our GCN models is to identify PKFIs that potentially contain a different number of atoms, we enabled both the atomic environment AF and modified Laplacian matrix \(\tilde{L}\) to contain 50 heavy atoms (hydrogen excluded) maximally by padding up the blank region with zeros (Fig. 4).

Feature encoding of the input compounds. Each compound is encoded by atomic features and a structure graph. Atomic features contain 28 descriptors belonging to six types, including atom types, hybridization, charges, and chemo-properties. For a structure graph, a modified normalized Laplacian matrix (Eq. (4)) was applied as a representation of the compound’s topology information. Padding to 50 atoms with zeros was applied to contain variable numbers of atoms in the input compounds
Graph convolution network architecture
Following the presentation of \({\mathcal{G}} = \left( {AF,\tilde{L}} \right)\) for each input compound, the function of the graph convolution layer is defined as follows:
where \(F^{l}\) denotes the graph convolutional function at layer \(l\) and \(F^{0} = AF\) is the compound atomic environment, and, \(W^{l} \in {\mathbb{R}}^{{d_{l - 1} \times d_{l} }}\) is the trainable kernel set of the lth layer that responds to the spectral pattern recognition of compound local information. Figure 5 describes the operation of graph convolution upon the presentation of \({\mathcal{G}} = \left( {AF,\tilde{L}} \right)\) and the schematic of our GCN architecture. To understand the chemical environments of compounds that cannot be provided by atomic features, we utilized the atomic connection information provided by the modified Laplacian matrix \(\tilde{L}\) to gather the surrounding information of each atom and form local environments, which is the purpose of equation \(\tilde{L} \cdot F^{{\left( {l - 1} \right)}} \left( {AF,\widetilde{ L}} \right)\) (Eq. (5) & Fig. 5). To learn the composition of local environments, trainable kernel weights \(W^{l}\) were applied.

The operation of the graph convolution and GCN model architecture of binary classification is described in this paper. To include the surrounding environments of each atom (local environments), the multiplication of topology graph \(\tilde{L}\) and the compound features (either the input atomic features AF or the feature map F(l−1) from the last layer) is required, which should be further multiplied by Wl to learn the information provided by the local environment.
As the graph convolution layer is stacked along with the graph, the local environment centered on each atom expands with bond orders. Therefore, to facilitate the sensing of diverse spectral patterns from different sizes of local environments, our GCN model considered all outputs from the three graph convolution layers. The global average pooling (GAP) was then applied after each convolution output to eliminate overfitting upon the pseudo-atomic order stipulated just for utilizing the GCN framework. Then, the final softmax binary classifier was applied to obtain a concatenation of three pooled feature maps to predict PKFIs (Fig. 5).
Gradient-weighted class activation mapping as model explainability
Grad-CAM was originally designed and applied for convolutional neural networks (CNNs) [33], is an additional explanation for deep learning models that visualize the heat map of the attention region (which contributes the most to compounds for the prediction of the model) from the feature maps of each layer. Owing to the commonality of convolution and graph convolution, Pope and Kolouri further extended it to GCN frameworks [34]. The Grad-CAM method consists of two major steps. We first defined the class-specific weights \(\alpha\) for the \(k^{th}\) feature of class \(c\) at layer \(l\):
where \(y^{c} \leftarrow GCN\left( {AF_{i} ,{ }\tilde{L}_{i} } \right)\) is the predicted probability on class c of compound i, and the weight vector \({\upalpha }_{k}^{l,c}\) of k features is calculated by the summation of the back-propagated gradients along the atom order dimension.
By defining the weights for each feature k of compound i, we computed the Grad-CAM feature map of layer \(l\) through \({\upalpha }_{k}^{l,c}\):
with the help of Grad-CAM, we can evaluate how much attention a model pays to each atom during prediction and further visualize the pre-moieties regions of each compound.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available in the ChEMBL database [28, 29], https://www.ebi.ac.uk/chembl/.
Abbreviations
- PKFI:
-
Pan-kinase family inhibitor
- GCN:
-
Graph convolutional network
- Grad-CAM:
-
Gradient-weighted class activation mapping
- FDA:
-
Food and Drug Administration
- AUROC:
-
Area under the receiver operating characteristic curve
- MCC:
-
Matthews correlation coefficient
- GAP:
-
Global average pooling
- ACC:
-
Accuracy
- CNN:
-
Convolutional neural network
References
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–34.
Cohen P. Protein kinases–the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1(4):309–15.
Corbett A, et al. Drug repositioning for Alzheimer’s disease. Nat Rev Drug Discov. 2012;11(11):833–46.
Cohen P, Alessi DR. Kinase drug discovery–what’s next in the field? ACS Chem Biol. 2013;8(1):96–104.
U.S. Food & Drug Administration. (2020). New Drugs at FDA: CDER’s New Molecular Entities and New Therapeutic Biological Products. Available: https://www.fda.gov/drugs/development-approval-process-drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products
Smyth LA, Collins I. Measuring and interpreting the selectivity of protein kinase inhibitors. J Chem Biol. 2009;2(3):131–51.
Lee JY, et al. Identification of the PCA29 gene signature as a predictor in prostate cancer. J Bioinform Comput Biol. 2019;17(3):1940006.
Shen X, et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001;107(7):893–903.
Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25(11):1265–75.
Fabbro D, Cowan-Jacob SW, Moebitz H. Ten things you should know about protein kinases: IUPHAR Review 14. Br J Pharmacol. 2015;172(11):2675–700.
Kleczko EK, Kwak JW, Schenk EL, Nemenoff RA. Targeting the complement pathway as a therapeutic strategy in lung cancer. Front Immunol. 2019;10:954.
Abu-Humaidan AHA, Ekblad L, Wennerberg J, Sorensen OE. EGFR modulates complement activation in head and neck squamous cell carcinoma. BMC Cancer. 2020;20(1):121.
Rusnak DW, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1(2):85–94.
Mehta A, Tripathy D. Co-targeting estrogen receptor and HER2 pathways in breast cancer. Breast. 2014;23(1):2–9.
Islam S, et al. Co-targeting aurora kinase with PD-L1 and PI3K abrogates immune checkpoint mediated proliferation in peripheral T-cell lymphoma: a novel therapeutic strategy. Oncotarget. 2017;8(59):100326–38.
Battistello E, et al. Pan-SRC kinase inhibition blocks B-cell receptor oncogenic signaling in non-Hodgkin lymphoma. Blood. 2018;131(21):2345–56.
Luszczak S, et al. PIM kinase inhibition: co-targeted therapeutic approaches in prostate cancer. Signal Transduct Target Ther. 2020;5(1):7.
Reddy TP, et al. Simultaneous targeting of HER family pro-survival signaling with Pan-HER antibody mixture is highly effective in TNBC: a preclinical trial with PDXs. Breast Cancer Res. 2020;22(1):48.
Lackey KE. Lessons from the drug discovery of lapatinib, a dual ErbB1/2 tyrosine kinase inhibitor. Curr Top Med Chem. 2006;6(5):435–60.
Payton M, et al. Preclinical evaluation of AMG 900, a novel potent and highly selective pan-aurora kinase inhibitor with activity in taxane-resistant tumor cell lines. Cancer Res. 2010;70(23):9846–54.
Chen JQ, Chen HY, Dai WJ, Lv QJ, Chen CYC. Artificial intelligence approach to find lead compounds for treating tumors. J Phys Chem Lett. 2019;10(15):4382–400.
Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43–9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006;407:597–612.
Ishihara S, et al. Sorafenib inhibits vascular endothelial cell proliferation stimulated by anaplastic thyroid cancer cells regardless of BRAF mutation status. Int J Oncol. 2019;55(5):1069–76.
Popova M, Isayev O, Tropsha A. Deep reinforcement learning for de novo drug design. Sci Adv. 2018;4(7):11047.
Zhavoronkov A, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol. 2019;37(9):1038.
Kearnes S, McCloskey K, Berndl M, Pande V, Riley P. Molecular graph convolutions: moving beyond fingerprints. J Comput Aided Mol Des. 2016;30(8):595–608.
Selvaraju RR, Cogswell M, Das A, Vedantam R, Parikh D, Batra D. Grad-CAM: visual explanations from deep networks via gradient-based localization. Int J Comput Vision. 2020;128(2):336–59.
Gaulton A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:1100.
Mendez D, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47(D1):D930–40.
Metz JT, Johnson EF, Soni NB, Merta PJ, Kifle L, Hajduk PJ. Navigating the kinome. Nat Chem Biol. 2011;7(4):200–2.
Altae-Tran H, Ramsundar B, Pappu AS, Pande V. Low data drug discovery with one-shot learning. ACS Cent Sci. 2017;3(4):283–93.
Thomas MW, Kipf N. Semi-Supervised Classification with Graph Convolutional Networks. In: Presented at the Advances in neural information processing systems. 2017.
Zhou AKB, Agata L, Aude O, Antonio T. Learning Deep Features for Discriminative Localization. In: Presented at the CVPR. 2016.
Pope PE, Kolouri S, Rostami M, Martin CE, Hoffmann H. Explainability Methods for Graph Convolutional Neural Networks. In: 2019 IEEE/Cvf Conference on Computer Vision and Pattern Recognition (Cvpr 2019), pp. 10764–10773. 2019 (in English).
Chollet, F. keras. 2018. https://github.com/fchollet/
Abadi M et al. TensorFlow: a system for large-scale machine learning. In: Proceedings of Osdi'16: 12th Usenix Symposium on Operating Systems Design and Implementation, pp. 265–283. 2016 (in English).
Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65(10):1566–84.
Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb Perspect Biol. 2014;6(4):a020768.
Verma A, Kambhampati S, Parmar S, Platanias LC. Jak family of kinases in cancer. Cancer Metastasis Rev. 2003;22(4):423–34.
Yamaoka K, Saharinen P, Pesu M, Holt VE 3rd, Silvennoinen O, O’Shea JJ. The Janus kinases (Jaks). Genome Biol. 2004;5(12):253.
Narlik-Grassow M, Blanco-Aparicio C, Carnero A. The PIM family of serine/threonine kinases in cancer. Med Res Rev. 2014;34(1):136–59.
Davis MI, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11):1046–51.
Geyer CE, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–43.
de Azambuja E, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): survival outcomes of a randomised, open-label, multicentre, phase 3 trial and their association with pathological complete response. Lancet Oncol. 2014;15(10):1137–46.
Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23(21):5036–41.
Keystone EC, et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann Rheum Dis. 2015;74(2):333–40.
Sandborn WJ, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376(18):1723–36.
Treiber DK, Shah NP. Ins and outs of kinase DFG motifs. Chem Biol. 2013;20(6):745–6.
Haider N. Functionality pattern matching as an efficient complementary structure/reaction search tool: an open-source approach. Molecules. 2010;15(8):5079–92 (in English).
Acknowledgements
Not applicable.
Funding
The research work and the publication were funded by the Center for Intelligent Drug Systems and Smart Bio-devices (IDS2B) from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. And the Ministry of Science and Technology (MOST) of Taiwan: MOST110-2634-F-A49-005-, All Vista Healthcare Project (MOST110-2634-F-002-016-), Research Center for Epidemic Prevention Science (MOST111-2321-B-016-002, MOST111-2321-B-A49-007-), Joint Research Center for AI Technology (MOST110-2634-F-009-015-) and Artificial Intelligence to Precision Health: Integrating Dynamic Physiological Signals and EMR to build a Medical Digital Twins Platform sponsored (MOST110-2321-B-A49-003); National Yang Ming Chiao Tung University and National Health Research Institutes (NHRI-EX111-11017BI). The funding bodies had no role in the design of the study and collection, analysis, interpretation of data, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
XYL designed the model and the computational framework and analyzed the data and wrote the article. XYL, YWH, and YWF collected data. JMY supervised the project. All authors discussed the results and commented on the manuscript. All authors read and approved the final manuscript.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 23 Supplement 4, 2022: The 20th International Conference on Bioinformatics (InCoB 2021). The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-23-supplement-4.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, XY., Huang, YW., Fan, YW. et al. Identification of pan-kinase-family inhibitors using graph convolutional networks to reveal family-sensitive pre-moieties. BMC Bioinformatics 23 (Suppl 4), 247 (2022). https://doi.org/10.1186/s12859-022-04773-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-022-04773-0