
Abstract
Background
The host response to influenza A infections is strongly influenced by host genetic factors. Animal models of genetically diverse mouse strains are well suited to identify host genes involved in severe pathology, viral replication and immune responses. Here, we have utilized a dual RNAseq approach that allowed us to investigate both viral and host gene expression in the same individual mouse after H1N1 infection.
Results
We performed a detailed expression analysis to identify (i) correlations between changes in expression of host and virus genes, (ii) host genes involved in viral replication, and (iii) genes showing differential expression between two mouse strains that strongly differ in resistance to influenza infections. These genes may be key players involved in regulating the differences in pathogenesis and host defense mechanisms after influenza A infections. Expression levels of influenza segments correlated well with the viral load and may thus be used as surrogates for conventional viral load measurements. Furthermore, we investigated the functional role of two genes, Reg3g and Irf7, in knock-out mice and found that deletion of the Irf7 gene renders the host highly susceptible to H1N1 infection.
Conclusions
Using RNAseq analysis we identified novel genes important for viral replication or the host defense. This study adds further important knowledge to host-pathogen-interactions and suggests additional candidates that are crucial for host susceptibility or survival during influenza A infections.
Similar content being viewed by others


Background
Influenza A viruses have an adverse impact on human and animal health worldwide through seasonal epidemics, newly emerging pandemics, and reoccurring outbreaks in livestock. The most severe human pandemic in 1918 resulted in about 30 million fatal casualties [1]. In addition, seasonal influenza infections represent a major health hazard causing deaths and enormous losses of work force every year [2].
We and others have shown in animal models that the genetic background of the host strongly influences mortality and morbidity after influenza infections. In particular, major differences in susceptibility and resistance were observed between different mouse inbred strains [3–13]. Detailed analysis of the mouse strains C57BL/6J and DBA/2J revealed that C57BL/6J mice survived infections with a low pathogenic A/Puerto Rico/8/1934 H1N1 virus (PR8M) whereas DBA/2J mice rapidly lost weight and all infected mice died [3, 14]. Infected DBA/2J had higher viral loads in their lungs and also exhibited a stronger inflammatory response compared to C57BL/6J mice [3, 14, 15]. Therefore, the comparison of these two mouse strains represents a very suitable model system to identify genes that are associated with severe infection outcomes in humans [16].
During an acute influenza virus infection, highly dynamic and inter-related responses are triggered in the host which eventually results in clearance of the pathogen and establishment of a long-lasting immunity. We recently demonstrated that these host responses can be studied comprehensively by measuring changes in the gene expression levels after infection [17, 18].
Here, we expanded those earlier studies by utilizing a dual RNAseq approach that enabled us to investigate both virus as well as host gene expression in the same individual. We found several new host genes that are strongly correlated with virus gene expression. Host genes potentially involved in viral replication were identified by comparisons with candidates from previous siRNA studies. In addition, we identified host genes that exhibit differential expression between the C57BL/6J and DBA/2J mouse strains after infection. These genes may be crucial to direct the host response to influenza A infections and be causal for differences in susceptibility and resistance of genetically diverse hosts to influenza or other viral infections. We studied the role of two candidate genes and found that deletion Irf7 renders the host highly susceptible to H1N1 infection.
Results
Global expression profiles are distinct in C57BL/6J and DBA/2J mice
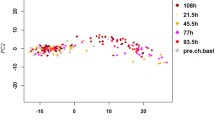
RNA was extracted from the lungs of C57BL/6J and DBA/2J mice infected with PR8M (a variant of A/Puerto Rico/8/1934 H1N1) as described in [14], and gene expression was quantified using RNA sequencing (RNAseq) technology. Principal component analysis (PCA) of normalized counts for host genes confirmed separate groupings of non-infected (controls) and infected lungs (Fig. 1). The transcriptome profiles of C57BL/6J mice and DBA/2J mice were distinct as shown by the second principle component, whereas the host response to the infection is mostly represented by the first principle component which explains 59 % of the expression variation. PC2 reveals distinct expression profiles for the two strains due to their different genetic backgrounds explaining 18 % of the expression variation. C57BL/6J mice exhibited a change in transcriptome profiles that was distinct for days 3, 5, 8, and 14 after infection. However, infected DBA/2J mice showed an early and stronger change in transcriptome profiles at day 3 post infection (p.i.) compared to C57BL/6J. Their expression profile did not show any major changes until day 5 when DBA/2J mice were moribund.

PCA analysis of normalized host gene counts for all samples. Principle component analysis reveals separate grouping of non-infected mice and infected mice for both mouse strains. Replicates for a given day p.i. grouped together well. For C57BL/6J mice, groups from different days p.i. were well separated. For DBA/2J a much stronger infection response was observed compared to C57BL/6J mice and individual mice at days 3 and 5 p.i. were not well separated. Note that for day 14 p.i., two of three samples were not separated and only two spots are visible. B6md1, D2md1 and B6md3, D2md3: C57BL/6J and DBA/2J mice mock-treated and analyzed at days 1 or 3 post treatment, respectively. Sample labels: C57BL/6J at days 1, 3, 5, 8 and 14 p.i.: B6d1, B6d3, B6d5, B6d8, B6d14, respectively; and DBA/2J mice at days 1, 3, 5 p.i.: D2d1, D2d3, D2d5, respectively
Expression levels of influenza gene segments correlate with viral load
In addition to expression profiling of host genes, RNAseq also allowed us to investigate transcripts of the eight viral segments. Expression levels of all influenza segments (calculated as RPKM: reads which map per kilobase of exon model per million mapped reads) changed in all infected mice over time and were highest at days 3 and 5 p.i. in C57BL/6J mice and at day 3 p.i. in DBA/2J mice (Fig. 2). In infected C57BL/6J mice, expression signals from influenza genes strongly decreased on day 8 p.i. and were at baseline levels of mock-treated controls on day 14 p.i. Expression levels of influenza transcripts were higher in DBA/2J mice compared to C57BL/6J mice at days 1 and 3 p.i. Most influenza RNA segments revealed a similar relative increase in expression, except for the segment encoding the neuraminidase (‘NA’) showing a lower increase compared to all other segments.

Expression levels of influenza genes. Normalized expression levels for influenza segments (PA, HA, M, NA, NS, PB1, PB2, NP) were calculated as mean expression values (log2 RPKM + 1), relative to respective mock treated animals (mock day 1 for day1 infected mice; mock day 3 for all other days p.i.). Lines represent expression levels from lungs of C57BL/6J at day 1, 3, 5, 8 and 14 p.i. (B6d1, B6d3, B6d5, B6d8, B6d14, respectively) and at day 1, 3, 5 p.i. for DBA/2J mice (D2d1, D2d3, D2d5, respectively). B6mock, D2mock: mock-treated C57BL/6J and DBA/2J control mice, respectively
Dynamics of the influenza gene expression levels determined by RNASeq correlated well to infectious viral particles [15] in C57BL/6J and DBA/2J mice (Fig. 3).

Correlation of expression levels and infectious particles. The part below the diagonal represents Spearman pairwise correlation factors in percent as pie charts; the part above the diagonal shows scatter plots for pairwise comparisons of RPKM and FFU in C57BL/6J and DBA/2J mice. For C57BL/6J mice, RPKM values from mock day3 and from days 1 to 14 p.i., and for DBA/2J, RPKM values from mock day 3 and days 1 to 5 p.i. were used for the analysis. Data were ordered by day, +1 added and then log2 transformed. FFU were taken from [15], offset by 1 and log2 transformed. Pearson pairwise correlations between RPKM and FFU for C57BL/6J were: corr = 0.8999419, p-value = 3.676e-07; and for DBA/2J: cor = 0.8832289, p-value = 0.0001401
Differentially expressed genes overlap with genes previously identified to be required for viral replication
Differentially expressed genes (DEG) between infected and mock-treated animals (log-fold change > |0.5|, FDR < 5 %) were determined for C57BL/6J infected mice at days 3, 5 and 8 p.i and for DBA/2J infected mice at days 3 and 5 p.i. (Table 1). We then compared these DEGs with genes that were identified previously in siRNA screens to be important for viral replication [19, 20]. The comparison with a gene list (34 genes) described by Stertz et al. [20] showed little overlap to our DEG gene lists (two to seven genes, data not shown). However, another study [19] used a combination of siRNA experiments and gene expression studies and identified 52 genes. Here, we found an overlap of 18 genes with DEGs from C57BL/6J and 25 with DEGs from DBA/2J (Table 2). Eighteen genes were common to both C57BL/6J and DBA/2J (Table 2, Fig. 4).

Expression changes of DEG genes overlapping with previously identified genes required for viral replication. The heatmap illustrates DEG genes from infected C57BL/6J and DBA/2J that overlap with previously identified genes [19]. Mean expression differences of DEG genes from infected C57BL/6J and DBA/2J at days 3 and 5 p.i. to mock-treated samples were calculated and values were scaled by rows. Colors display z-scores from −1.5 (dark green) to 1.5 (red) for normalized gene expression values. Rows: name of genes, columns: difB6d1: difference in expression levels of C57BL/6J infected mice at day 1 compared to mock day 1 treated animals; difB6d3 to difB6d14: difference in expression levels of C57BL/6J infected mice at days 3, 5, 8, 15 p.i. compared to mock-treated day 3 animals; difD2d1: difference in expression levels of DBA/2J infected mice at day 1 compared to mock-treated day 1 animals; difD2d3 to difD2d5: difference in expression levels of DBA/2J mice infected at days 3, 5 p.i. compared to mock-treated day 3 animals
From these 25 genes that overlapped with DEGs from DBA/2J, we selected Irf7 (Interferon regulatory factor 7) for further studies. We generated an Irf7 knock-out line on a C57BL/6J background by backcrossing to test the importance of Irf7 for the host response to influenza infection. After infection with 2x105 Focus Forming Unit (FFU) PR8M virus, Irf7 −/− mice lost significantly more body weight and exhibited increased mortality compared to wild type controls (Fig. 5a, b). These observations demonstrate that Irf7 plays an important role for the host defense to influenza A infection.

Body weight loss of Irf7 and Reg3g knock-out mice after influenza A infection. Female mice were infected with 2x105 FFU PR8M by intranasal application. Mice with a weight loss of more than 30 % of the starting weight had to be euthanized and were recorded as dead. a Homozygous Irf7 −/− mice showed higher body weight loss (on days 6 to day 8 p.i., p < 0.05, Mann Whitney U test) and (b) significantly increased mortality (Log-rank test, p < 0.01) compared to C57BL/6J control mice. c Mutant Reg3g −/− mice exhibited significant differences in body weight loss at days 4, 5, 6, 9, and 10 p.i. (Mann Whitney U test, p < 0.05) but no significant increase in mortality (Log-rank test). Please note that in (a) after day 6 p.i. only the surviving mice are shown and are thus not representative for the entire group
Host genes involved in virus defense and innate immune responses strongly correlate with changes in influenza gene expression
We selected significantly up- or down-regulated genes (FDR < 0.05 and minimal expression level of log2 = 1; FDR: false discovery rate) from C57BL/6J infected mice at days 1, 3, 5, 8, and 14 p.i. to identify host genes correlating with the expression of the viral genome. This analysis was restricted to C57BL/6J because we aimed to cover the period of increase in viral load until day 5 p.i. as well as the clearance phase after day 5 p.i. We found 182 host genes with a highly correlated expression (169 positively and 13 negatively) (Spearman correlation coefficient of larger than |0.8|, and FDR < 0.05) (Table 3, Additional file 1: Table S1). Gene Ontology (GO) enrichment analyses of positively correlated genes revealed enrichment for terms including ‘host immune response’, ‘regulation of virus genome replication’, ‘chemokine and cytokine production’ and ‘responses to virus’. Reactome pathway analysis of these 169 positively correlated genes revealed enrichment for terms including ‘interferon signaling’, ‘immune system’, and ‘cytokine signaling in immune system’.
Several genes are up-regulated in C57BL/6J mice but not in DBA/2J mice after infection
C57BL/6J mice exhibit a much lower viral load in their lungs after infection with H1N1 influenza A virus (PR8M) compared to DBA/2J mice [14, 15]. Furthermore, the host response in DBA/2J is characterized by a stronger inflammatory response [14]. Therefore, we searched for genes that were exclusively up-regulated in C57BL/6J but not in DBA/2J mice after infection with PR8M. We hypothesized that these genes may be responsible for the more efficient control of virus replication in C57BL/6J mice. In a first step, we performed an analysis of variance (ANOVA) for all genes in all groups to identify genes that were significantly up-regulated (FDR < 10 %). From this set, genes up-regulated only in C57BL/6J were selected. This filtering identified five DEGs that were significantly regulated at day 3 and 5 p.i. in C57BL/6J mice: Lhx2, 2210415F13Rik, Trim15, Reg3g, and Cd72. The very low expression levels of Lhx2, 2210415F13Rik and Trim15 make it unlikely that these are crucial candidates mediating the difference in susceptibility between C57BL/6J and DBA/2J. We therefore investigated Reg3g (regenerating islet-derived 3 gamma) in more detail.
Knock-out mice carrying a mutation in the Reg3g gene on a C57BL/6 N background were infected with influenza PR8M. Differences in body weight loss were observed in mutant compared to wild type mice at day 4 to 6 p.i (Fig. 5c). However, no significant difference in survival was observed between Reg3g knock-out and wild type C57BL/6 N mice after influenza A virus infection. Thus, Reg3g seems to play a minor role in the host defense to influenza virus H1N1 infection.
Discussion
Here, we performed RNAseq based analysis of gene expression changes in a murine influenza A infection model by comparing a resistant mouse strain, C57BL/6J, that survives PR8M (H1N1) infection, with a highly susceptible strain, DBA/2J, for which infection with PR8M is lethal. Our studies confirm differences in gene expression profiles between the two mouse strains that were described in a previous analysis using microarrays [18]. At day 3 p.i., 670 differentially expressed probesets in infected C57BL/6J and 1046 in infected DBA/2J mice, respectively, were identified previously by Alberts and colleagues [18] and also overlapped with DEGs found in this study.
Influenza virus transcripts carry a poly(A) tail similar to host mRNAs. Cellular mRNAs are polyadenylated through cleavage at the polyadenylation signal and subsequent addition of the poly(A) tail. In contrast, viral mRNAs obtain their polyadenylation through a stuttering mechanism in which the RNA-dependent RNA polymerase moves back and forth over a stretch of five to seven U residues shortly before the 5´end [21, 22]. Since we selected poly(A) RNAs for RNAseq, we were able to investigate expression of viral genes and, at the same time, to correlate changes in the host transcriptome with increase and decrease of virus gene expression. In this way, we could confirm that changes in expression levels of viral mRNA were correlated with viral load in the infected lungs. The kinetics of viral replication over time as well as the difference between C57BL/6J and DBA/2J was well reflected by changes in sequence counts determined by RNAseq. Thus, the relative changes in RNA expression may serve as a surrogate for virus replication and viral load in infected animals. Thus, RNAseq represents a big advantage compared to microarrays technology where a parallel detection of host gene expression and viral genome expression is not possible.
Zhou et al. [19] combined an siRNA screen with expression analysis in the human lung epithelial cell line A549 after infection with PR8 virus. They identified 300 genes as significantly up-regulated and subsequently performed a siRNA screen for those genes. That screen detected 52 genes as regulators of viral replication, including 40 genes that were not reported previously. We found 25 genes that overlapped with the 52 genes identified by [19] (Table 2).
From the 25 overlapping genes, six genes (Stat1, B2m, Lgals3bp, Dusp5, Nfkbia, Il15ra) were also identified as host factors involved in influenza virus replication by Shapira and colleagues [23]. They used human bronchial epithelial cells for transcriptional profiling and combined the data with results from a yeast two-hybrid approach where ten major viral proteins of PR8 were tested against 12,000 human proteins.
Furthermore, genes acting downstream of RIG-I binding to viral RNA like Irf7, Irf9, Stat1 and NF-kB were also found amongst the genes that overlapped with the list from Zhou et al. [19]. Amongst these factors, IRF7 has been described as an essential key mediator of interferon signaling activation and regulation and has been shown to be critical for innate immunity [24]. It is constitutively expressed in plasmacytoid dendritic cells which rapidly produce type I IFN in response to viral infection [25, 26]. This initial activation triggers a positive feedback loop regulation of Ifnα and Ifnβ genes by Irf7 in adjacent cells [27, 28]. The importance of Irf7 in influenza pathogenesis was also shown in several in vitro studies [29, 30]. Epithelial cells recognize influenza A virus via RIG-I/MAVS, leading to the activation of Irf7 and subsequent induction of type I and type III interferons in redundant amplification loops. In addition, a recently published study revealed an IRF7-dependent amplification of IFNs in an influenza patient carrying a mutation in that gene [31]. In contrast, no in vivo studies using Irf7 deficient mouse mutants have been published so far. Therefore, we selected Irf7 (Interferon regulatory factor 7) to generate knock-out mice on a C57BL/6J background by backcrossing and to investigate its role for host defense in vivo. After infection with PR8M, Irf7-deficient mice exhibited a more pronounced body weight loss and increased mortality compared to wild type mice after infection with H1N1 virus. These experiments demonstrate the in vivo relevance of Irf7 for the host response to influenza virus infection. Our studies confirm the potential role of Irf7 in influenza pathogenesis in an in vivo model system as suggested by previous in vitro studies [29, 30]. The potential functional roles of all other genes from the list in Table 2 are discussed in more detail in the supplements.
When comparing results from several RNAi screens [23, 32–35], Stertz and Shaw identified 34 genes with potential importance for viral replication that were found in at least two screens (reviewed in [20]). However, only seven genes overlapped with the 34 genes in DBA/2J at day 5 p.i. (Plk3, Rps10, Il17ra, Ptprn, Racgap1, Nhp2l1, Atp6v0c). One explanation for the small overlap may be that the RNAi screens were performed in cell culture whereas our studies identified differentially regulated genes in infected lungs. It should be noted that transcriptomes in lungs are much more complex due to the contribution from infiltrating immune cells. Thus, changes in expression of cultured cells may not reflect the entire spectrum of host responses well. More future studies will be necessary to further elucidate this aspect.
Since viral and host transcripts can be followed in the same individual, we were able to correlate changes in host gene expression with changes in the level of virus gene expression. We studied host gene expression in C57BL/6J lungs and viral transcripts including both the period of increasing viral load (day 1 to day 5 p.i.) as well as the period of decrease in viral load (day 8 to 14 p.i.). We found 182 host genes that were positively or negatively correlated with influenza gene expression in infected C57BL/6J mice (Table 3 shows the 20 positively correlated genes and all negatively correlated genes). Many of these genes exhibit well known functions in the host immune response which are discussed in more detail in the Additional file 2: Supplemental Material.
In contrast to C57BL/6J mice that survive, DBA/2J mice die on day six to seven after infection with PR8. A comparison of the DEGs in both mouse strains was performed to identify genes that are exclusively up-regulated in C57BL/6J. We hypothesize that these genes are candidates mediating the resistance of C57BL/6J against influenza infection. We identified two genes (Cd72 and Reg3g) that were significantly and strongly up-regulated in C57BL/6J mice compared to DBA/2J. The B cell co-receptor Cd72 is an important receptor regulating B cell activation [36], negatively regulating BCR signaling [37] and is additionally expressed on murine NK cells where it acts in an inhibitory manner through regulating cytokine production but not cytotoxicity [38]. In C57BL/6J we observed a two-fold higher up-regulation of Cd72 compared to DBA/2J. The resulting deficit in the inhibitory effect on NK cells and the following diminished regulation of cytokine amounts may be a good explanation for the exaggerated immune response observed in DBA/2J mice. More experiments will be needed to evaluate the possible role of Cd72 for the host response to influenza A virus.
For Reg3g an increase in expression levels was observed in IBD (inflammatory bowel disease), a murine bacterial reconstitution model [39] and after experimental intestinal infection with Listeria monocytogenes [40]. The role of Reg3g in lung infection was further elucidated by Choi et al. [41]. They were able to show that Reg3g expression is regulated by Stat3 and highly increased after MRSA (Methicillin-resistant Staphylococcus aureus) infection in the lung epithelium. Administration of recombinant Reg3g was able to restore mucosal immunity against MRSA in vivo, highlighting the therapeutic potential for Reg3g [41]. The fact that Reg3g was up-regulated in C57BL/6J, but not in DBA/2J may account for the differences in disease outcome. We therefore studied the possible role of Reg3g in knock-out mice. However, despite its strong up-regulation after influenza virus infection, deletion of this gene had no strong effect on the susceptibility of the host to infections with H1N1. It may, however, be possible that Reg3g deficient mice are susceptible to other influenza virus subtypes or other viral infections.
Conclusions
In conclusion, using RNAseq analysis we identified novel genes important for viral replication or host defense. This study adds further important knowledge to host-pathogen-interactions and suggests additional candidates that are crucial for host susceptibility or survival during influenza A infections.
Methods
Ethics statement
All experiments in mice were approved by an external committee according to the German national guidelines of the animal welfare law. The protocol used in these experiments has been reviewed and approved by an ethics committee as described in the regulations from the German Bundesministerium für Ernährung, Landwirtschaft und Verbraucherschutzand, and detailed in the “Tierschutzkommissions-Verordnung vom 23. Juni 1987 (BGBl. I S. 1557)” (http://www.gesetze-im-internet.de/bundesrecht/tierschkomv/gesamt.pdf). Subsequently, the protocol has been formally approved by the ‘Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany’ (Permit Number: 3392 42502-04-13/1234).
Virus and mice
The mouse-adapted virus strain influenza A/Puerto Rico/8/1934 H1N1 (PR8M) was produced as described previously [14, 42]. C57BL/6J and DBA/2J mice were obtained from Janvier, France. Mutant B6;129P2-Irf7 tm1Ttg were kindly provided by Tadatsugu Taniguchi [24]. B6;129P2-Irf7 tm1Ttg mice were backcrossed to C57BL/6J for 12 generations to generate B6.129P2-Irf7 tm1Ttg mice (Irf7 −/−). The background was confirmed by SNP-genotyping (Mouse Universal Genotyping Array (MUGA), Neogen Corporation, USA). The Reg3g knock-out mouse strain was created from ES cell clone EPD0309_D08, obtained from the KOMP Repository (www.komp.org) to generate B6-Reg3g tm1a(KOMP)Wtsi (Reg3g −/−) mice.
Mouse infections
Female, 10–12 weeks old mice were anesthetized by intra-peritoneal injection with Ketamine/Xylazine (85 % NaCl (0.9 %), 10 % Ketamine, 5 % Xylazine) with doses adjusted to the individual body weight. Mice were then intranasally infected with 20 μl virus solution (2x103 (RNASeq) or 2x105 (knock-out mice) FFU PR8M) or mock-infected with PBS.
RNA isolation
Mice were sacrificed and entire lungs were extracted from mice from both strains on days 1, 3 and 5 after infection. For mock-infected animals, mice were sacrificed at days 1 and 3 post treatment. In addition, lungs from C57BL/6J mice were also collected on days 8 and 14. For every treatment and day post infection (p.i.) 4–5 mice were prepared. The lungs were immediately transferred to RNAlater solution (Qiagen), kept at 4 °C for one day and subsequently stored at −20 °C. RNA was isolated using Qiagen Midi Kit as described previously [43]. RNA quality was controlled on a 2100 Bioanalyzer Instrument (Agilent). All RNA samples had a RNA Integrity Number (RIN) of ≥ 9.7. Three independent biological replicates were selected for each time point for subsequent RNA sequencing.
RNAseq library preparation, sequencing and analysis
Twenty μg of total RNA was enriched for poly A+ RNA using one cycle of the Poly A Purist Kit from Ambion according to the manufacturer’s standard protocol. The resulting enriched RNA samples were analyzed on an Agilent Bioanalyzer to determine the remaining amount of rRNA in the samples. If the amount was higher than 5 %, samples were subjected to another cycle of Poly A enrichment. One-hundred ng of the poly A+ enriched RNA was then used to prepare libraries for sequencing using the AB Library Builder™ Whole Transcriptome Core Kit for 5500 Genetic Analysis Systems on a Library Builder system. Libraries were amplified for 15 cycles before 5500 Wildfire primers were added using five cycles of fusion primer amplification as directed in the 5500 Wildfire manual. Before sequencing, small aliquots of libraries were pooled and sequenced on an Ion Torrent PGM 314 chip after additional amplification with PGM fusion primers. The library pools were quantified by Real-Time PCR and immobilized on flow cells for the SOLiD 5500 Wildfire instrument (Applied Biosystems) and sequenced (50 bp reads). The average number of reads per sample was 29.5. One sample had a high number of reads (230 million), and the others on average had 23 million reads. The mouse reference genome (GRCm38/mm10) was downloaded from ftp://hgdownload.cse.ucsc.edu/goldenPath/mm10/chromosomes/. Sequencing reads (XSQ format) from C57BL/6J samples were aligned to the C57BL/6J reference genome using the Whole Transcriptome mapping module of the LifeScope 2.5.1 software (http://www.lifetechnologies.com/lifescope). Similarly, sequencing reads from DBA/2J samples were aligned to the enhanced DBA/2J genome that was generated by substituting ~4.5 million DBA/2J SNPs in the reference genome. Filter reference containing polyA, polyC, polyG, polyT, rRNAs, tRNAs, as well as adaptor, barcode, and primer sequences was used to remove non-mRNAs reads prior to the mapping. We used the mouse RefSeq transcript annotation downloaded from UCSC genome browser (www.genome.ucsc.edu) to generate a junction reference library containing a list of exon-exon pairs. Reads were aligned against both the reference genome and the junction library. Reads that could not be mapped were realigned against the H1N1 viral contigs. Reads with minimum mapping quality of 10 were used to generate raw counts to be used for downstream differential and correlation analysis.
Bioinformatic analysis
Raw read counts were used for analysis with DESeq2 [44] statistical package after adding 1 to all values. The DESeq function rlogTransformation was used to normalize and log transform raw read counts and to calculate normalized expression counts. The normalized expression counts were then used for further analysis without applying any additional pre-processing filtering. Principal component analysis analysis and identification of differentially expressed genes were performed using DESeq2. DEGs were selected based on an adjusted p-value of 0.05 (FDR of 5 %) and exhibiting at least a 1.4-fold difference in expression levels (log2 = 0.5). Strip charts, scatter plots and heat maps were generated using the R software package [45]. Multi-group comparisons were performed with the LIMMA package [46] using BH correction for multiple testing [47]. Cell signature genes were identified based on the BioGPS database (GEO database ID GSE10246) and our previous analysis of gene expression patterns in a non-lethal infection [17]. Inflammatory genes that are expressed during influenza infections were selected based on our previous influenza transcriptome studies [17, 43]. For analysis of influenza transcripts, log2-transformed RPKM values were calculated from counts of sequences that aligned to influenza gene segments. Analysis of correlations between influenza (log2 RPKM counts of the sum of all genes) and host gene (normalized log2 counts) expression levels was performed with the R function cor using Spearman as method. Correlation graphs were generated using the R package ‘corrgram’ [48]. Adjusted p-values for correlated genes were calculated as FDR using cor.test. GO enrichment analysis and Reactome enrichment analysis (using the full gene list from normalized counts as reference) was performed with the R package clusterProfiler [49].
Availability of supporting data
The raw RNAseq data has been deposited at GEO (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE66040 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66040).
References
Johnson NP, Mueller J. Updating the accounts: global mortality of the 1918–1920 “Spanish” influenza pandemic. Bull Hist Med. 2002;76(1):105–15.
Fauci AS. Seasonal and pandemic influenza preparedness: science and countermeasures. J Infect Dis. 2006;194 Suppl 2:S73–6.
Srivastava B, Blazejewska P, Hessmann M, Bruder D, Geffers R, Mauel S, et al. Host genetic background strongly influences the response to influenza a virus infections. PLoS ONE. 2009;4(3), e4857.
Trammell RA, Toth LA. Genetic susceptibility and resistance to influenza infection and disease in humans and mice. Expert Rev Mol Diagn. 2008;8(4):515–29.
Ding M, Lu L, Toth LA. Gene expression in lung and basal forebrain during influenza infection in mice. Genes Brain Behav. 2008;7(2):173–83.
Boon AC, deBeauchamp J, Hollmann A, Luke J, Kotb M, Rowe S, et al. Host genetic variation affects resistance to infection with a highly pathogenic H5N1 influenza A virus in mice. J Virol. 2009;83(20):10417–26.
Boon AC, Debeauchamp J, Krauss S, Rubrum A, Webb AD, Webster RG, et al. Cross-reactive neutralizing antibodies directed against pandemic H1N1 2009 virus are protective in a highly sensitive DBA/2 influenza mouse model. J Virol. 2010;84(15):7662–7.
Otte A, Sauter M, Alleva L, Baumgarte S, Klingel K, Gabriel G. Differential host determinants contribute to the pathogenesis of 2009 pandemic H1N1 and human H5N1 influenza A viruses in experimental mouse models. Am J Pathol. 2011;179(1):230–9.
Boon AC, Finkelstein D, Zheng M, Liao G, Allard J, Klumpp K, et al. H5N1 influenza virus pathogenesis in genetically diverse mice is mediated at the level of viral load. MBio. 2011;2(5):e00171–11.
Trammell RA, Liberati TA, Toth LA. Host genetic background and the innate inflammatory response of lung to influenza virus. Microbes Infect. 2012;14:50–8.
Pica N, Iyer A, Ramos I, Bouvier NM, Fernandez-Sesma A, Garcia-Sastre A, et al. The DBA.2 mouse is susceptible to disease following infection with a broad, but limited, range of influenza A and B viruses. J Virol. 2011;85(23):12825–9.
Vanlaere I, Vanderrijst A, Guenet JL, De Filette M, Libert C. Mx1 causes resistance against influenza A viruses in the Mus spretus-derived inbred mouse strain SPRET/Ei. Cytokine. 2008;42(1):62–70.
Ferris MT, Aylor DL, Bottomly D, Whitmore AC, Aicher LD, Bell TA, et al. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog. 2013;9(2), e1003196.
Blazejewska P, Koscinski L, Viegas N, Anhlan D, Ludwig S, Schughart K. Pathogenicity of different PR8 influenza A virus variants in mice is determined by both viral and host factors. Virology. 2011;412(1):36–45.
Dengler L, Kuhn N, Shin DL, Hatesuer B, Schughart K, Wilk E. Cellular changes in blood indicate severe respiratory disease during influenza infections in mice. PLoS ONE. 2014;9(7), e103149.
Kollmus H, Wilk E, Schughart K. Systems biology and systems genetics-novel innovative approaches to study host-pathogen interactions during influenza infection. Curr Opin Virol. 2014;6C:47–54.
Pommerenke C, Wilk E, Srivastava B, Schulze A, Novoselova N, Geffers R, et al. Global transcriptome analysis in influenza-infected mouse lungs reveals the kinetics of innate and adaptive host immune responses. PLoS ONE. 2012;7(7), e41169.
Alberts R, Lu L, Williams RW, Schughart K. Genome-wide analysis of the mouse lung transcriptome reveals novel molecular gene interaction networks and cell-specific expression signatures. Respir Res. 2011;12:61.
Zhou Z, Cao M, Guo Y, Zhao L, Wang J, Jia X, et al. Fragile X mental retardation protein stimulates ribonucleoprotein assembly of influenza A virus. Nat Commun. 2014;5:3259.
Stertz S, Shaw ML. Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes Infect. 2011;13(5):516–25.
Samji T. Influenza A: understanding the viral life cycle. Yale J Biol Med. 2009;82:153–9.
Poon LL, Pritlove DC, Fodor E, Brownlee GG. Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J Virol. 1999;73(4):3473–6.
Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139(7):1255–67.
Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434(7034):772–7.
Izaguirre A, Barnes BJ, Amrute S, Yeow WS, Megjugorac N, Dai J, et al. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. J Leukoc Biol. 2003;74(6):1125–38.
Kerkmann M, Rothenfusser S, Hornung V, Towarowski A, Wagner M, Sarris A, et al. Activation with CpG-A and CpG-B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J Immunol. 2003;170(9):4465–74.
Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. Embo J. 1998;17(22):6660–9.
Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998;441(1):106–10.
Colina R, Costa-Mattioli M, Dowling RJ, Jaramillo M, Tai LH, Breitbach CJ, et al. Translational control of the innate immune response through IRF-7. Nature. 2008;452(7185):323–8.
Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog. 2013;9(11), e1003773.
Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. 2015;348(6233):448–53.
Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, et al. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454(7206):890–3.
Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463(7282):818–22.
Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139(7):1243–54.
Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, et al. Human host factors required for influenza virus replication. Nature. 2010;463(7282):813–7.
Parnes JR, Pan C. CD72, a negative regulator of B-cell responsiveness. Immunol Rev. 2000;176:75–85.
Li DH, Tung JW, Tarner IH, Snow AL, Yukinari T, Ngernmaneepothong R, et al. CD72 down-modulates BCR-induced signal transduction and diminishes survival in primary mature B lymphocytes. J Immunol. 2006;176(9):5321–8.
Alcon VL, Luther C, Balce D, Takei F. B-cell co-receptor CD72 is expressed on NK cells and inhibits IFN-gamma production but not cytotoxicity. Eur J Immunol. 2009;39(3):826–32.
Ogawa H, Fukushima K, Naito H, Funayama Y, Unno M, Takahashi K, et al. Increased expression of HIP/PAP and regenerating gene III in human inflammatory bowel disease and a murine bacterial reconstitution model. Inflamm Bowel Dis. 2003;9(3):162–70.
Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J Exp Med. 2007;204(8):1891–900.
Choi SM, McAleer JP, Zheng M, Pociask DA, Kaplan MH, Qin S, et al. Innate Stat3-mediated induction of the antimicrobial protein Reg3gamma is required for host defense against MRSA pneumonia. J Exp Med. 2013;210(3):551–61.
Wilk E, Schughart K. The mouse as model system to study host-pathogen interactions in influenza A infections. Curr Protoc Mouse Biol. 2012;2:177–205.
Alberts R, Srivastava B, Wu H, Viegas N, Geffers R, Klawonn F, et al. Gene expression changes in the host response between resistant and susceptible inbred mouse strains after influenza A infection. Microbes Infect. 2010;12(4):309–18.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
R_Core_Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2013. http://www.R-project.org/.
Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful apporach to multiple testing. J R Stat Soc. 1995;57:289–300.
Friendly M. Corrgrams: exploratory displays for correlation matrices. Am Stat. 2002;56:316–24.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Acknowledgements
This work was supported by intramural grants from the Helmholtz-Association (Program Infection and Immunity), a research grant FluResearchNet (No. 01KI07137) and a research grant ‘Infection challenge in the German Mouse Clinic’ from the German Ministry of Education and Research to KS. We would like to thank the animal caretakers at the Central Animal Facilities at the HZI for maintaining the mice for this study, Christin Fricke and Karin Lammert for excellent technical assistance. The original stock of influenza A virus was obtained from Stefan Ludwig (University of Münster). Part of this work has been performed as PhD thesis work (SL) at the University of Veterinary Medicine, Hannover. We thank Danny Arends for help with the analysis of expression data. We thank Tony Marion for final editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EW, SL and BH performed experiments, EW, SL, AP, MP, JW, CP, KS analyzed the data, KS and EW conceived experiments, EW, SL, AP and KS wrote the manuscript. All authors have read and approved the manuscript.
Esther Wilk, Ashutosh K. Pandey, Sarah R. Leist and Bastian Hatesuer contributed equally as first authors.
Additional files
Additional file 1: Table S1.
Genes expressed in infected C57BL/6J at days 1, 3, 5, 8, and 14 p.i. that strongly correlate with changes in expression of influenza segments. (PDF 76 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wilk, E., Pandey, A.K., Leist, S.R. et al. RNAseq expression analysis of resistant and susceptible mice after influenza A virus infection identifies novel genes associated with virus replication and important for host resistance to infection. BMC Genomics 16, 655 (2015). https://doi.org/10.1186/s12864-015-1867-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-1867-8