
Abstract
Background
Breast cancer is one of the main malignant tumors that threaten the lives of women, which has received more and more clinical attention worldwide. There are increasing evidences showing that the immune micro-environment of breast cancer (BC) seriously affects the clinical outcome. This study aims to explore the role of tumor immune genes in the prognosis of BC patients and construct an immune-related genes prognostic index.
Methods
The list of 2498 immune genes was obtained from ImmPort database. In addition, gene expression data and clinical characteristics data of BC patients were also obtained from the TCGA database. The prognostic correlation of the differential genes was analyzed through Survival package. Cox regression analysis was performed to analyze the prognostic effect of immune genes. According to the regression coefficients of prognostic immune genes in regression analysis, an immune risk scores model was established. Gene set enrichment analysis (GSEA) was performed to probe the biological correlation of immune gene scores. P < 0.05 was considered to be statistically significant.
Results
In total, 556 immune genes were differentially expressed between normal tissues and BC tissues (p < 0. 05). According to the univariate cox regression analysis, a total of 66 immune genes were statistically significant for survival risk, of which 30 were associated with overall survival (P < 0.05). Finally, a 15 immune genes risk scores model was established. All patients were divided into high- and low-groups. KM survival analysis revealed that high immune risk scores represented worse survival (p < 0.001). ROC curve indicated that the immune genes risk scores model had a good reliability in predicting prognosis (5-year OS, AUC = 0.752). The established risk model showed splendid AUC value in the validation dataset (3-year over survival (OS) AUC = 0.685, 5-year OS AUC = 0.717, P = 0.00048). Moreover, the immune risk signature was proved to be an independent prognostic factor for BC patients. Finally, it was found that 15 immune genes and risk scores had significant clinical correlations, and were involved in a variety of carcinogenic pathways.
Conclusion
In conclusion, our study provides a new perspective for the expression of immune genes in BC. The constructed model has potential value for the prognostic prediction of BC patients and may provide some references for the clinical precision immunotherapy of patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Breast cancer is one of the main malignant tumors that threaten the lives of women, which has received more and more clinical attention worldwide. It is regarded as the second common malignant tumors in the world. It occupied 25% of all malignant tumors [1]. Triple-negative breast cancer (TNBC) was supposed to be the foremost malignant sub-type, accounting for approximately 20% [2, 3]. It is manifested as a large tumor, a high level of differentiation, a high risk of metastasis, and lymph node invasion [4,5,6]. TNBC is characterized by negative human epidermal growth factor receptor 2 (HER-2), progesterone receptor (PR) and estrogen receptor (ER), thus resistant to endocrine therapy and trastuzumab [7]. Due to the lack of targeted treatment strategies, chemotherapy remained the unique treatment option [8]. Therefore, it is significant and urgent to conduct a comprehensive bioinformatics study on gene expression of breast cancer to identify potential genes that can be used as therapeutic targets in BC.
Previous evidence has shown that the immune system has a contradictory influence on the occurrence and development of cancer, contributing to both cancer progression and inhibition [9,10,11,12]. The immune system has a practical impact on the progress of BC. What’s more, the response of BC patients to immunotherapy and traditional treatment is interfered by immune system [13, 14]. However, immune evasion remains a tough problem in the immunotherapy of BC, which brings a big challenge for the treatment for BC and improvement of prognosis of patients. Since there are significant diversities in the expression profiles of immune genes between BC and other cancer types [15, 16], further research is needed to determine which immune genes can play a role as therapeutic targets.
In this study, we explored the lineage and expression profiles of immune genes in BC and its impacts on the prognosis of BC patients. Besides, the functional features as well as mutated features of these immune-related genes were described. Furthermore, 15 immune-related genes closely associated with overall survival were selected and an independent risk score model was constructed for the prognosis of BC. Moreover, a nomogram was also constructed to further explore the model’s ability to predict.
Methods
Acquisition of data
First of all, a list of 2498 immune genes were downloaded from ImmPort database. Additionally, the gene expression profiles of BC patients was obtained from the TCGA database (https://portal.gdc.cancer.gov/), including 112 normal cases and 857 tumor cases. Meanwhile, corresponding clinical data were also obtained (Table 1). |LogFC | > 1 and P < 0.05 were used as the criteria for screening differential genes. Because TCGA is an open and publicly available database, ethical approval is not required.
Gene function enrichment analysis
Gene Ontology(GO) enrichment analysis is conducted to comprehend the biological process and molecular function of the differential genes, while Kyoto Encyclopedia of Genes and Genomes(KEGG) enrichment analysis is applied to identify potential related biological pathways. Gene enrichment analysis (GSEA) is performed between normal tissues and BC tissues in order to probe the biological pathways associated with immune genes risk scores.
Construction and validation of the immune genes risk scores
Cox regression tool was used for survival analysis. On the basis of differential expression, single factor cox significant and survival-related prognostic immune genes were screened out. Further, the least absolute shrinkage and selection operator (LASSO) regression analysis is execute to reduce the dimensionality, so as to screen out the optimal variables. Based on the variables obtained by LASSO and the corresponding regression coefficients, the risk scores were calculated. The median value was utilized to divide patients into a high- and a low-risk scores group. The prognostic correlation of immune gene risk score was obtained by Kaplan-Meier curve. The credibility and predictive value of the risk scoring model was evaluated through time-related ROC curve.
Analysis of copy number variation data and gene mutation analysis
Based on TCGA breast cancer data, the copy number variation (CNV) was analyzed using R-Circos package and R-ggplot2 package. Furthermore, the online tool website-cbioportal was used to analyze the genetic variation of hub genes. The threshold used was P < 0.05.
Statistical analysis
R3.6.1 was used for statistical analysis. The independent t test was used for continuous variables with normal distribution, and the Mann-Whitney U test was used for continuous variables with skewed distribution. A two-sided test was used, and a P value of < 0.05 was considered statistically significant.
Results
Differentially expressed immune genes (DEIGs) screening of BC
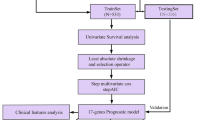
We designed a protocol for the analysis and construction of the prognostic model (Fig. 1). The analysis process was carried out in strict accordance with the protocol. A list of 2498 immune genes was obtained from ImmPort database. What’s more, the mRNA expression data of 857 cases of breast cancer and 122 cases of normal tissues were also obtained from the TCGA database for further bioinformatics analysis. The clinical features of samples were provided in Table 1. A total of 556 immune genes were determined as DEIGs between BC tissues and normal tissues, including 402 up-regulated and 154 down regulated (p < 0.05, Fig. 2a, Table 2). The heatmap spread out the top 10 up-regulated and top 10 down-regulated DEIGs (Fig. 2b).

Flow chart of research design

Identification of DEIGs. a volcano plots of 556 DEIGs in breast cancer and normal tissues from TCGA database. b Heatmap plots of top 10 up-regulated and top 10 down-regulated DEIGs. The colors in the heatmaps from green to red represent expression level from low to high. The red dots in the volcano plots represent up-regulation, the green dots represent down-regulation and black dots represent genes without differential expression
Functional annotation of these 556DEIGs
To study the potential mechanisms and molecular functions of the identified 556 DEIGs, we conducted the GO and KEGG analysis. The top three enriched GO terms for up-regulated DEIGs and down-regulated DEIGs were: T cell activation, lymphocyte differentiation and response to virus; cell chemotaxis, positive regulation of response to external stimulus and leukocyte migration, respectively (Fig. S1A). KEGG analysis revealed the top three enriched pathway for up-regulated DEIGs and down-regulated DEIGs were: Cytokine−cytokine receptor interaction, JAK − STAT signaling pathway and Chemokine signaling pathway; Cytokine−cytokine receptor interaction, JAK − STAT signaling pathway and EGFR tyrosine kinase inhibitor resistance pathway, respectively (Fig. S1B).
Establishment of immune prognosis model
Among the identified 556 DEIGs, 66 prognostic DEIGs were identified by utilizing univariate cox regression analyses (Fig. 3a). KM survival analysis showed that 30 of them were significantly correlated with OS. TCGA BC samples were randomly separated into two sets (training set: validation set, 2:1). Then, lasso regression analysis was applied to increase the robustness and select the optimal variables based on training set. Finally, 15DEIGs were got for the construction of immune prognostic index based on the optimal value (Fig. 3b, c, Table 3). After the establishment of the immune prognostic model, BC patients were stratified into high-risk and low-risk subgroups based on the cut-off risk score (Fig. 3d, e). Heatmap was utilized to visualize the difference of gene expression profile in low- and high- risk patients in BC training set (Fig. 3f). The results from KM analysis revealed that high risk patients possessed lower overall survival in both training group and validation group (P < 0.001) (Fig. 4a, b, c). R software was utilized to draw the time-dependent ROC curvesand the AUC was calculated at different time points to estimate the predictive performance of our prognostic model in training cohort, testing cohort and the entire TCGA cohort. The ROC curve prompted that the risk scores model had dominant credibility and predictive value (AUC = 0.752, AUC = 0.704 for 5 years overall survival in training and validation group, respectively) (Fig. 4d, e, f).

a Univariate survival analysis by Cox proportional hazards models to select prognostic key immune genes. b-c LASSO Cox regression model for19 prognostic immune genes used to construct immune genes risk score model. d Distribution of immune risk scores in breast cancer patients. e Distribution of survival status in breast cancer patients. f Distribution of specific risk factors in the high- and low-risk groups (divided by median value). (*P < 0.05)

a Kaplan-Meier curve analysis of high-risk and low-risk patients in the training cohort. b Kaplan-Meier curve analysis of high-risk and low-risk patients in the testing cohort. c Kaplan-Meier curve analysis of high-risk and low-risk patients in the entire TCGA cohort. d Time-dependent ROC curve analysis of the training cohort. e Time-dependent ROC curve analysis of the testing cohort. f Time-dependent ROC curve analysis of the entire TCGA cohort
Validation in external cohort and TCGA independent cohort
To evaluate the operability and accuracy of the prognostic model in clinical practice, we further conducted the external validation analysis. As was shown in Fig. 5a and b, the AUC value was 0.624 for the 5-year OS in GSE7390 validation set and 0.635, 0.606, 0.622 for the 1-, 3-, 5-year OS, respectively, in GSE21653 validation set. What’s more, according to the KM curves, high risk scores were significantly associated with poor prognoses both in GSE7390 and GSE21653 validation set. (P = 0.002 and 0.012, respectively) (Fig. 5c, d) The results were consistent with those of the training set. In further univariate cox analysis, age, pathological stage, pathological T, N, M stage and high risk scores were associated with poor survival (Fig. 6a). In multivariate Cox model, only age and risk score worked as independent predicted factors (P < 0.001) (Fig. 6b). To establish a quantitative visualization model of breast cancer prognosis, multiple clinical factors were combined to establish a nomogram (Fig. 6c). The calibration of nomogram suggested that there was strong coherence between the predicted and actual 3- and 5-year overall survival (Fig. 6d, e).

External validation set of the prognostic model. a ROC curve and AUC of the 15-gene signature in GSE7390 testing cohort. b ROC curve and AUC of the 15-gene signature in GSE21653 testing cohort. c KM survival analysis of the 15-gene signature in GSE7390 testing cohort. d KM survival analysis of the 15-gene signature in GSE21653 testing cohort

Cox’s proportional hazard model of correlative factors in breast cancer patients. a Univariate COX regression analysis for seven clinicopathological parameters affecting the overall survival. b Multivariate COX regression analysis for seven clinicopathological parameters affecting the overall survival. c An established nomogram to predict breast cancer survival based on cox model. d-e Plots displaying the calibration of each model comparing predicted and actual 3- and 5-year overall survival
Recognition of gene sets for genome variation
Based on TCGA breast cancer data, we analyzed the copy number variation (CNV) of 15 model genes and showed the frequency of copy number variation through R-Circos package and R-ggplot2 package (Fig. S2A, Fig. S4). The results showed that the top three genes with the highest CNV frequency were RAC2, ULBP1, and SERPINA3. (Fig. S2B) Furthermore, we analyzed the single nucleotide polymorphism composition (SNPs) of 15 model genes (Fig. S2C). The results showed that NR3C2 had the most SNPs, including missense mutation and silent. Finally, the online tool website-cbioportal was utilized to analyze the genetic variation of 15 immune genes (Fig. S2D).
Clinical and prognostic correlation of 15 model genes and the risk score
The proportion of 15 model genes in different clinical and pathological stages was investigated. Correlation analysis between tumor, node, metastasis stage, pathologic stage and 15 model genes expression in breast cancer cases were explored (Fig. S3A-D). Based on the results, it seemed that IL17B, NFKBIE and SERPINA3 mainly prompted the development of breast cancer. In addition, survival analysis showed that all model genes were significantly associated with survival (Fig. S4). Meanwhile, we found that the expression of RAC2, CD79A and IFNG were significantly associated with the infiltration of Macrophage M0 and Macrophage M2 (Fig. S5). Regard to the immune genes risk score, a strong correlation with age, sex, pathological stage and clinical T stage was identified (Fig. 7).

Correlation between immune genes risk scores and various clinical factors. a Age. b Sex. c Stage. d T stage. e N stage. f M stage
Gene set enrichment analysis of risk scores
To explore the biological correlation of risk scores involved in progression of breast cancer, a GSEA analysis of risk scores was performed based on the TCGA breast cancer cohort. GSEA analysis indicated high risk scores were associated with E2F_TARGETS, G2M_CHECKPOINT, GLYCOLYSIS, MTORC1_SIGNALING and PROTEIN_SECRETION pathway (Fig. 8a). In addition, low risk scores were associated with APOPTOSIS, COMPLEMENT, IL2_STAT5_SIGNALING, INFLAMMATORY_RESPONSE and P53 pathway (Fig. 8b).

Gene set enrichment analysis of immune genes risk scores. a high risk scores. b low risk scores
Discussion
BC is regarded as the most common malignant tumor in women. Although great efforts have been made to improve diagnosis and treatment strategies, it still poses a fatal threat to patients. Accumulation of evidence have shown that Cancer immunotherapy, especially the treatment of immune checkpoint inhibitors, has become an important part of the treatment of certain types of cancer, and has provided a continuous therapeutic effect for specific groups of patients [17]. Immune genes, such as cytokines, not only act locally, but rapidly spread within the tumor and affect the activation and dissemination of tumor immune cells [18, 19]. Obviously, different types of cancer have different immune gene subgroups. Therefore, the examination of immune gene subgroups is essential for judging the risk of tumors and exploring immunotherapy.
In our research, we performed a detailed and comprehensive evaluation of immune genes in BC. All gene expression data and patients clinical characteristics information were downloaded from TCGA dataset. Two thousand four hundred ninety-eight immune genes from ImmPort database were analyzed between breast cancer and normal tissues. Eventually, 556 DEIGs were verified. Moreover, we identified and constructed a 15 immune genes risk scores model for breast cancer through univariate and lasso regression analysis, including TSLP, IL17B, NR3C2, RAC2, SERPINA3, HSPA2, CD79A, UNC93B1, NFKBIE, SDC1, IFNG, IRF7, GALP, TNFRSF18 and ULBP1. Furthermore, to study the clinical and biological relevance of risk scores, the KM, ROC and GSEA analysis were conducted. Indeed, the high risk group received a lower survival, and possessed a higher histological grade.
Several DEIGs in the immune genes risk scores model have been investigated in human cancers. Thymic interstitial lymphopoietin (TSLP), a key inflammatory cytokine that induces type 2 inflammation, predicts a poor prognosis in oropharyngeal squamous cell carcinoma (OPSCC) [20]. With regard to breast cancer, Emma et al. has demonstrated that TSLP, which induced by IL-1 produced by breast tumors, act as a critical survival factor for the tumor [21]. This may indicate that TSLP can be a potential therapeutic intervention for breast cancer. Interleukin-17 (IL-17), a member of the interleukin family, is a cytokine that plays a role in inflammation and cancer, and can enhance lung cancer invasion/migration ability [22]. Seth et al. has found that the mammary tumor can induce IL17-producing γδ T cells, which can drive systemic expansion and polarization of neutrophils towards a CD8+ T cell-suppressive phenotype and subsequent metastasis formation in distant organs [23]. It seems that IL-17 plays a negative role in the prognosis of BC patients. In addition, studies have found that knocking down RAC2 can inhibit the progression of osteosarcoma by inhibiting the wnt signaling pathway [24]. Besides, the up regulation of hnRNP-K transcriptional activity mediated by SERPINA3 promotes the survival and proliferation of HCC cells, which may be an indicator of poor prognosis in HCC patients [25]. So far, overexpression of SERPINA3 has been observed in several cancer types including breast cancer and the high expression level has been demonstrated to positively correlate with poor prognosis in patients with breast cancer, which means SERPINA3 can be associated with a shorter OS [26]. AS a putative oncoprotein, Heat shock protein family a member 2 (HSPA2) is often up-regulated in human malignancies and promotes aggressive phenotype of tumors [27]. It seemed that overexpression of HSPA2 may be associated with worse clinical outcome. A recent study indicated that HSPA2 might play an important role in breast cancer development and progression by promoting cell growth, migration and invasion in xenografted mice [28]. However, it remains a controversy whether HSPA2 is a positive or negative regulator of carcinogenesis. NFKBIE aberrations are common genetic events in trans-b-cell malignancies, and NFKBIE deletion is a new marker of poor prognosis in primary mediastinal B-cell lymphoma (PMBL) [29]. The remaining genes have also been confirmed to be interrelated to malignant origin, aggressive behavior of tumors.
Similarly, Lai et al. [30] established a panel of 4 autophagy-related genes (ARG) signatures consisting of SERPINA1, ATG4A, NRG1 and IFNG to predict the prognosis of breast cancer, which can help clinicians make judgments and decisions on determining effective treatment strategies. Wang et al. [31] identified a six differentially-expressed genes (DEGs) model consisting of IGHA2, SERPINA1, GFALS, SPDYC, PAX7, and ADRB1 by using Cox regression survival modeling for breast cancer. In another study [32], the authors constructed a prognostic risk scoring system containing 6 genes (SCUBE3, RDH16, SPC24, SPC25, CCDC69 and DGAT2), suggesting that these mRNAs may serve a driving role in the progression of Her2-positive BC. The construction of this risk scoring system is conducive to identifying high-risk HER2-positive BC patients, and it subserve to help achieve personalized targeted therapy. Different from previous studies, our study provides novel insights into the role of Immune-related genes in the genesis and progression of BC. We first focused on DEIGs, and established and verified a novel DEIGs risk scores prediction model. And our prognostic model showed good predictive performance with regard to survival, which may contribute to the development of new prognostic indicators for BC. Besides, the Immune-related gene marker showed strongly association with immunoinfiltrating cells, which demonstrates that these Immune-related genes could be used in clinical adjuvant treatments.
Nevertheless, there still remain some weak points in our research. Firstly, our results are based on bulk RNA sequencing of single omics. The heterogeneity and diversity between cells in the tumor microenvironment is ignored. Secondly, only gene expression and gene mutation levels are concerned, while tumor burden, methylation levels and other equally important events in tumor progression are ignored.
Conclusion
In conclusion, our study reveals the biological effects of immune genes in the origin and development of BC. The immune gene risk score model has advantages in predicting the prognosis of BC, which is an independent factor affecting the prognosis of BC. In addition, our findings may be of great guiding value in make a thorough inquiry of novel strategies for cancer immunological diagnosis and treatment. With the rapid development of high-throughput sequencing technology, it is reasonable to believe that this scoring system can provide recommendations for patients’ immune status as well as clinical risk assessment and treatment strategies.
Availability of data and materials
The data was available in the ImmPort database (https://www.immport.org/) and The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/).
Abbreviations
- OS:
-
Over survival
- TNBC:
-
Triple negative breast cancer
- ER:
-
Estrogen receptor
- PR:
-
Progesterone receptor
- HER-2:
-
Human epidermal growth factor receptor 2
- TIICs:
-
Tumor infiltrating immune cells
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GO:
-
Gene ontology
- DAVID:
-
Database for Annotation, Visualization, and Integrated Discovery
- GSEA:
-
Gene enrichment analysis
- CNV:
-
Copy number variation
- LASSO:
-
Least absolute shrinkage and selection operator
- DEIGs:
-
Differentially expressed immune genes
- SNPs:
-
Single nucleotide polymorphism
- TME:
-
Tumor micro-environment
- TSLP:
-
Thymic stromal lymphopoietin
- OPSCC:
-
Oropharyngeal squamous cell carcinoma
- HSPA2:
-
Heat shock protein family a member 2
- PMBL:
-
Primary mediastinal B-cell lymphoma
- ARGs:
-
Autophagy-related genes
- DEGs:
-
Differentially expressed genes
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. https://doi.org/10.1002/ijc.29210.
Reis-Filho JS, Tutt AN. Triple negative tumours: a critical review. Histopathology. 2008;52(1):108–18. https://doi.org/10.1111/j.1365-2559.2007.02889.x.
Schneider BP, Winer EP, Foulkes WD, Garber J, Perou CM, Richardson A, Sledge GW, Carey LA. Triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res. 2008;14(24):8010–8. https://doi.org/10.1158/1078-0432.CCR-08-1208.
van Roozendaal LM, Smit LHM, Duijsens G, et al. Risk of regional recurrence in triple-negative breast cancer patients: a Dutch cohort study. Breast Cancer Res Treat. 2016;156(3):465–72. https://doi.org/10.1007/s10549-016-3757-4.
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. https://doi.org/10.1172/JCI45014.
Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SAW, Savage MI, Osborne CK, Hilsenbeck SG, Chang JC, Mills GB, Lau CC, Brown PH. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21(7):1688–98. https://doi.org/10.1158/1078-0432.CCR-14-0432.
Liedtke C, Rody A. New treatment strategies for patients with triple-negative breast cancer. Curr Opin Obstet Gynecol. 2015;27(1):77–84. https://doi.org/10.1097/gco.0000000000000137.
Mayer IA, Abramson VG, Lehmann BD, Pietenpol JA. New strategies for triple-negative breast cancer--deciphering the heterogeneity. Clin Cancer Res. 2014;20(4):782–90. https://doi.org/10.1158/1078-0432.CCR-13-0583.
Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. 2015;33(1):445–74. https://doi.org/10.1146/annurev-immunol-032414-112043.
Biswas SK. Metabolic reprogramming of immune cells in Cancer progression. Immunity. 2015;43(3):435–49. https://doi.org/10.1016/j.immuni.2015.09.001.
Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–22. https://doi.org/10.1038/ni.2703.
Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, Jiang P, Shen H, Aster JC, Rodig S, Signoretti S, Liu JS, Liu XS. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17(1):174. https://doi.org/10.1186/s13059-016-1028-7.
Varn FS, Mullins DW, Arias-Pulido H, Fiering S, Cheng C. Adaptive immunity programmes in breast cancer. Immunology. 2017;150(1):25–34. https://doi.org/10.1111/imm.12664.
Vonderheide RH, Domchek SM, Clark AS. Immunotherapy for breast Cancer: what are we missing? Clin Cancer Res. 2017;23(11):2640–6. https://doi.org/10.1158/1078-0432.CCR-16-2569.
Bates JP, Derakhshandeh R, Jones L, Webb TJ. Mechanisms of immune evasion in breast cancer. BMC Cancer. 2018;18(1):556. https://doi.org/10.1186/s12885-018-4441-3.
Emens LA. Breast Cancer immunotherapy: facts and hopes. Clin Cancer Res. 2018;24(3):511–20. https://doi.org/10.1158/1078-0432.CCR-16-3001.
Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–72. https://doi.org/10.1038/nri.2017.49.
Bremnes RM, Al-Shibli K, Donnem T, et al. The role of tumor-infiltrating immune cells and chronic inflammation at the tumor site on cancer development, progression, and prognosis: emphasis on non-small cell lung cancer. J Thorac Oncol. 2011;6(4):824–33. https://doi.org/10.1097/JTO.0b013e3182037b76.
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, Li B, Liu XS. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77(21):e108–e10. https://doi.org/10.1158/0008-5472.CAN-17-0307.
Lin CM, Lin LW, Chen YW, Ye YL. The expression and prognostic impact of proinflammatory cytokines and their associations with carcinogens in oropharyngeal squamous cell carcinoma. Cancer Immunol Immunother. 2020;69(4):549–58. https://doi.org/10.1007/s00262-020-02488-w.
Kuan EL, Ziegler SF. A tumor-myeloid cell axis, mediated via the cytokines IL-1alpha and TSLP, promotes the progression of breast cancer. Nat Immunol. 2018;19(4):366–74. https://doi.org/10.1038/s41590-018-0066-6.
Yang YF, Lee YC, Lo S, Chung YN, Hsieh YC, Chiu WC, Yuan SSF. A positive feedback loop of IL-17B-IL-17RB activates ERK/beta-catenin to promote lung cancer metastasis. Cancer Lett. 2018;422:44–55. https://doi.org/10.1016/j.canlet.2018.02.037.
Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJM, Ciampricotti M, Hawinkels LJAC, Jonkers J, de Visser KE. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–8. https://doi.org/10.1038/nature14282.
Xia P, Gao X, Shao L, Chen Q, Li F, Wu C, Zhang W, Sun Y. Down-regulation of RAC2 by small interfering RNA restrains the progression of osteosarcoma by suppressing the Wnt signaling pathway. Int J Biol Macromol. 2019;137:1221–31. https://doi.org/10.1016/j.ijbiomac.2019.07.016.
Ko E, Kim JS, Bae JW, Kim J, Park SG, Jung G. SERPINA3 is a key modulator of HNRNP-K transcriptional activity against oxidative stress in HCC. Redox Biol. 2019;24:101217. https://doi.org/10.1016/j.redox.2019.101217.
Koivuluoma S, Tervasmaki A, Kauppila S, et al. Exome sequencing identifies a recurrent variant in SERPINA3 associating with hereditary susceptibility to breast cancer. Eur J Cancer. 2021;143:46–51. https://doi.org/10.1016/j.ejca.2020.10.033.
Yang YL, Zhang Y, Li DD, Zhang FL, Liu HY, Liao XH, Xie HY, Lu Q, Zhang L, Hong Q, Dong WJ, Li DQ, Shao ZM. RNF144A functions as a tumor suppressor in breast cancer through ubiquitin ligase activity-dependent regulation of stability and oncogenic functions of HSPA2. Cell Death Differ. 2020;27(3):1105–18. https://doi.org/10.1038/s41418-019-0400-z.
Jagadish N, Agarwal S, Gupta N, Fatima R, Devi S, Kumar V, Suri V, Kumar R, Suri V, Sadasukhi TC, Gupta A, Ansari AS, Lohiya NK, Suri A. Heat shock protein 70-2 (HSP70-2) overexpression in breast cancer. J Exp Clin Cancer Res. 2016;35(1):150. https://doi.org/10.1186/s13046-016-0425-9.
Mansouri L, Noerenberg D, Young E, Mylonas E, Abdulla M, Frick M, Asmar F, Ljungström V, Schneider M, Yoshida K, Skaftason A, Pandzic T, Gonzalez B, Tasidou A, Waldhueter N, Rivas-Delgado A, Angelopoulou M, Ziepert M, Arends CM, Couronné L, Lenze D, Baldus CD, Bastard C, Okosun J, Fitzgibbon J, Dörken B, Drexler HG, Roos-Weil D, Schmitt CA, Munch-Petersen HD, Zenz T, Hansmann ML, Strefford JC, Enblad G, Bernard OA, Ralfkiaer E, Erlanson M, Korkolopoulou P, Hultdin M, Papadaki T, Grønbæk K, Lopez-Guillermo A, Ogawa S, Küppers R, Stamatopoulos K, Stavroyianni N, Kanellis G, Rosenwald A, Campo E, Amini RM, Ott G, Vassilakopoulos TP, Hummel M, Rosenquist R, Damm F. Frequent NFKBIE deletions are associated with poor outcome in primary mediastinal B-cell lymphoma. Blood. 2016;128(23):2666–70. https://doi.org/10.1182/blood-2016-03-704528.
Lai J, Chen B, Mok H, Zhang G, Ren C, Liao N. Comprehensive analysis of autophagy-related prognostic genes in breast cancer. J Cell Mol Med. 2020;24(16):9145–53. https://doi.org/10.1111/jcmm.15551.
Wang F, Tang C, Gao X, et al. Identification of a six-gene signature associated with tumor mutation burden for predicting prognosis in patients with invasive breast carcinoma. Ann Transl Med. 2020;8(7):453. https://doi.org/10.21037/atm.2020.04.02.
Gao C, Zhuang J, Li H, Liu C, Zhou C, Liu L, Feng F, Sun C, Wu J. Development of a risk scoring system for evaluating the prognosis of patients with Her2-positive breast cancer. Cancer Cell Int. 2020;20(1):121. https://doi.org/10.1186/s12935-020-01175-1.
Acknowledgments
Not applicable.
Funding
None.
Author information
Authors and Affiliations
Contributions
DR, YY and CL designed this work. CL and DY wrote the manuscript. DY and PY performed the bioinformatics analysis. ZY, LP, CC, WJ and LJ performed the data review. LP and PY polished the manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
GO (A) and KEGG(B) enrichment analysis of DEIGs.
Additional file 2: Figure S2.
Analysis of copy number variation and single nucleotide polymorphism of 15 model immune genes.
Additional file 3: Figure S3.
Correlation analysis between TNM&Stage and 15 model genes in breast cancer cases. (A) Correlation analysis between tumor stage and 15 model genes expression in breast cancer cases. (B) Correlation analysis between node stage and 15 model genes expression in breast cancer cases. (C) Correlation analysis between metastasis stage and 15 model genes in breast cancer cases. (D) Correlation analysis between pathologic stage and 15 model genes expression in breast cancer cases.
Additional file 4: Figure S4.
Survival analysis of 15 model immune genes.
Additional file 5: Figure S5.
Correlation between 15 model immune genes and immune cell infiltration.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, L., Dong, Y., Pan, Y. et al. Identification and development of an independent immune-related genes prognostic model for breast cancer. BMC Cancer 21, 329 (2021). https://doi.org/10.1186/s12885-021-08041-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08041-x