
Abstract
Background
Hypoxic pulmonary vasoconstriction (HPV) optimizes the match between ventilation and perfusion in the lung by reducing blood flow to poorly ventilated regions. Sepsis and endotoxemia impair HPV. We previously showed that nitric oxide synthase 2 (NOS2) is required, but not sufficient, for the effect of endotoxin on HPV. The aim of the current study was to identify additional factors that might contribute to the impairment of HPV during endotoxemia.
Methods
Gene expression profiling was determined using pulmonary tissues from NOS2-deficient (NOS2−/−) and wild-type mice subjected to endotoxin or saline challenge (control). HPV was accessed as the percentage increase in left pulmonary vascular resistance (LPVR) in response to left main bronchus occlusion (LMBO) in wild-type mice.
Results
Among the 22,690 genes analyzed, endotoxin induced a greater than three-fold increase in 59 and 154 genes in the lungs of wild-type and NOS2−/− mice, respectively. Of all the genes induced by endotoxin in wild-type mice, arginase 1 (Arg1) showed the greatest increase (16.3-fold compared to saline treated wild-type mice). In contrast, endotoxin did not increase expression of Arg1 in NOS2−/− mice. There was no difference in the endotoxin-induced expression of Arg2 between wild-type and NOS2-deficient mice. We investigated the role of arginase in HPV by treating the mice with normal saline or the arginase inhibitor Nω-hydroxy-nor-L-arginine (norNOHA). In control mice (in the absence of endotoxin) treated with normal saline, HPV was intact as determined by profound LMBO-induced increase in LPVR (121 ± 22% from baseline). During endotoxemia and treatment with normal saline, HPV was impaired compared to normal saline treated control mice (33 ± 9% vs. 121 ± 22%, P < 0.05). HPV was restored in endotoxin-exposed mice after treatment with the arginase inhibitor norNOHA as shown by the comparison to endotoxemic mice treated with normal saline (113 ± 29% vs, 33 ± 9%, P < 0.05) and to control mice treated with normal saline (113 ± 29% vs, 121 ± 22%, P = 0.97).
Conclusions
The results of this study suggest that endotoxemia induces Arg1 and that arginase contributes to the endotoxin-induced impairment of HPV in mice.
Similar content being viewed by others


Background
Hypoxic pulmonary vasoconstriction (HPV) diverts blood flow away from poorly- to better-ventilated regions of the lung, thereby optimizing gas exchange. The sensor and effector mechanisms responsible for HPV are intrinsic to pulmonary arterial smooth muscle cells [1]. Although it has been shown that HPV is modulated by a number of vasoactive mediators including nitric oxide (NO) and arachidonic acid metabolites, the precise mechanisms that mediate HPV remain incompletely understood [1,2,3,4,5].
HPV is impaired in patients with sepsis and in animal models of endotoxemia [2,3,4,5,6,7]. Impaired HPV (i.e., reduced pulmonary vasoconstrictor response in hypoxic lung regions) contributes to systemic hypoxemia during sepsis or endotoxemia. NO is produced by NO synthases (NOS1, NOS2, and NOS3). While NOS3 is constitutively expressed in vascular endothelial cells and importantly regulates pulmonary vascular tone, NOS2 is induced by inflammatory stimuli such as endotoxin. In a previous study, we showed that endotoxin impairs HPV in wild-type mice but not in mice with congenital NOS2 deficiency (NOS2−/−) leading to lower arterial oxygen tensions in wild-type mice than in NOS2−/− mice after endotoxin challenge. In saline-treated wild-type and NOS2−/− mice, the inhalation of 40 ppm NO for 22 h did not affect HPV, suggesting that a high level of NO, by itself, is not sufficient to impair HPV. When endotoxemic wild-type and NOS2−/− mice inhaled 40 ppm NO for 22 h, HPV was impaired to the same degree in both genotypes [2]. The results of these studies show that NO and at least one additional endotoxin-induced factor are required for endotoxin to impair HPV.
In the current study, we hypothesized that one or more gene(s) are differentially expressed in the lungs of wild-type and NOS2−/− mice after endotoxin challenge. We used an unbiased transcriptional array approach to identify genes that may have increased or decreased expression after endotoxin exposure in wild-type mice, but not NOS2−/− mice. Here, we report that expression of arginase 1 (Arg1) was markedly increased in the lungs of wild-type mice after endotoxin challenge. Endotoxin marginally decreased the expression of Arg1 in NOS2−/− mice. The arginase inhibitor Nω-hydroxy-nor-L-arginine restored HPV in wild-type mice. These observations identify a critical role for arginase in endotoxin-induced impairment of HPV.
Materials and methods
Animals and experimental setting
Male NOS2−/− mice and their wild-type controls (C57BL/6 J) were purchased from The Jackson Laboratory (Bar Harbor, Maine). The body weight ranged between 21 and 27 g. Mice were challenged with intravenous administration of either normal saline (0.1 ml/10 g body weight) or lipopolysaccharide (LPS, Escherichia coli O111:B4, Sigma, St Louis, MO; 20 mg/kg, dissolved in normal saline 0.1 ml/10 g body weight).
Microarray analysis
For microarray analysis, lungs were harvested from wild-type mice 22 h after challenge with either saline (N = 4) or LPS (N = 3) or from NOS2−/− mice 22 h after saline (N = 1) or LPS (N = 3) challenge. RNA, cDNA, and labeled cRNA were generated as previously described [8]. Fragmented cRNA was hybridized to Affymetrix mouse genome MOE430A Array containing 22,690 gene entries with the GeneChip Fluidics Station 450 and scanned with the GeneChip® Scanner 3000. Affymetrix GeneChip 5.0 software was used for data analysis according to Affymetrix protocols. Microarray data processing was performed in the R environment. Bioconductor package “affy” was used for data input, background correction, normalization and calculation of expression values. Data sets were annotated with official gene symbols originating from package “moe430a.db” using “annotate”. Resulting gene sets were annotated using in-house scripts and technical information from Affymetrix.
Measurement of gene expression
To confirm the increase in pulmonary Arg1 expression found in the microarray analysis of endotoxemic wild-type mice, but not NOS2−/− mice, additional wild-type and NOS2−/− mice received either normal saline or LPS (N = 6 for each of the four groups). Twenty-two hours later, total RNA was isolated from homogenized lungs using Trizol (Invitrogen Life Technologies, Carlsbad, CA, USA). cDNA was generated with MMLV reverse transcriptase (Promega, Madison, WI, USA) and random primers (Promega, Madison, WI, USA). Real-time qPCR was performed using the ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) using FAM MGB primers for Arg1 and arginase 2 (Arg2, Applied Biosystems, Foster City, CA, USA). 18S ribosomal RNA was detected using 18S VIC MGB primers (Applied Biosystems, Foster City, CA, USA) and Taqman Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Changes in Arg1 and Arg2 mRNA expression were determined using the ∆∆Ct method with normalization to 18S ribosomal RNA.
Measurement of Arg1 protein expression
Immunoblots were performed to assess levels of Arg1 and GAPDH. Lungs from wild-type and NOS2−/− mice with and without 22 h of endotoxemia were harvested, homogenized at 4 °C in PBS with 5 mM EGTA and protease inhibitor mix (Roche Diagnostics GmbH, Germany), and centrifuged at 10,000 g at 4 °C for 10 min. Protein in the supernatant protein was subjected to electrophoresis, transferred to a PVDF membrane, and probed with anti-Arg1 antiserum (1500, Lifespan Biosciences, Seattle, WA, USA) and anti-GAPDH antiserum (1:10,000, MerckMillipore, Darmstadt, Germany). Proteins were visualized using a LICOR infrared imager (Odyssey CLx) and quantitative densitometric analysis was performed (Odyssey Image Studio v3.1). The amount of arginase relative to GAPDH was determined.
Immunoenzyme staining
To localize Arg1 expression, lungs of wild-type mice were fixed in paraformaldehyde 22 h after challenge with saline (N = 5) or endotoxin (N = 5). Immunoenzyme stainings were performed on 2 μm paraffin-embedded sections using standard avidin-biotin anti-alkaline phosphatase technique (Vector Laboratories, Burlingame, CA) according to the manufacturer’s instructions. Tris-buffered saline supplemented with 0.2% bovine serum albumin (Biotrend, Cologne, Germany) was used as buffer. Primary antibody dilutions of rabbit polyclonal antibody, 1/50 (H-52, Santa Cruz, Dallas, Texas) and an isotype- and concentration-matched rabbit control Ig (Dianova, Hamburg, Germany) were prepared in this buffer and incubated for 1 h at room temperature. A biotinylated donkey anti-rabbit IgG Ab, 1/100 (Jackson ImmunoResearch, Newmarket, UK), was used as a secondary reagent (30 min at room temperature). Naphthol AS-biphosphate (Sigma) with New-fuchsin (Merck, Darmstadt, Germany) was used as the substrate for alkaline phosphatase. Slides were counterstained with hematoxylin (Sigma).
Measurement of hypoxic pulmonary vasoconstriction in mice
To investigate the effect of arginase on HPV, mice were initially challenged with either LPS or normal saline (control). Twenty-one hours later, mice were treated with an intravenous bolus of the arginase inhibitor Nω-hydroxy-nor-L-arginine (norNOHA, Cayman Chemical, Ann Arbor, Michigan; 20 mg/kg or 40 mg/kg) or normal saline. One hour later, HPV was measured as described previously [3,4,5, 7]. Mice were anesthetized and mechanically ventilated with a respiratory frequency of 100–110 breaths per minute, a tidal volume of 10 ml/kg body weight, the peak inspiratory pressure of approximately 10 cm H2O, positive end-expiratory pressure 2 cm H2O, and at an inspired oxygen fraction of 1.0. An arterial line was placed in the left carotid artery, and a left-sided thoracotomy was performed. A custom-made polyethylene catheter was positioned in the main pulmonary artery, and a flow probe was placed around the left pulmonary artery. Heart rate, systemic arterial pressure (SAP), pulmonary arterial pressure (PAP), and left pulmonary arterial blood flow (QLPA) were continuously measured and recorded (DI 720; Dataq Instruments, Akron, OH). Left lung alveolar hypoxia and collapse was induced by left mainstem bronchus occlusion (LMBO). A 50% transient occlusion of the inferior vena cava was repeated three times before and during LMBO to estimate left pulmonary vascular resistance (LPVR). LPVR was calculated from the slope of the PAP/QPLA relationship. The LMBO-induced increase in LPVR was expressed as the percentage increase of LPVR during LMBO from the baseline.
Measurement of arterial blood gases
After hemodynamic measurements were obtained, blood was sampled from the left carotid artery, anticoagulated with heparin, and blood gas analyses were performed using a Rapid Lab 840 (Chiron Diagnostics, Medfield, MA).
Statistical analysis
Data are expressed as mean ± SD. P values less than 0.05 were considered statistically significant. Statistical analyses were performed using Sigma Stat 3.0 (Systat Software Inc., Richmond, CA). Normality distribution of data was checked using the Kolmogorov-Smirnov test with Lillefors correction. For the comparison between the groups, data were analyzed using ANOVA with a post-hoc Tukey test for normally distributed data or using Kruskal Wallis ANOVA on ranks with a post-hoc Turkey test for not normally distributed data. Hemodynamic changes before (baseline) and during LMBO were compared with a paired t-test.
For the microarray data, the differential expression gene set annotation was performed as follows. The data analysis from wild-type and NOS2−/− mice after challenge with either saline or LPS was conducted with “limma” (Linear Models for Microarray and RNA-Seq, https://bioconductor.org) [9]. Empirical Bayes statistics for differential expression was calculated for both genotypes and conditions. To allow comparison of data sets moderated F-statistics were applied. Differentially up-regulated genes were selected that showed linear expression values of at least 3-fold after 22 h after LPS challenge in comparison to saline challenge. Gene candidates were considered as down-regulated if the respective expression values were decreased to 0.3-fold or less. Probability values were adjusted for multiple testing using the Benjamini–Hochberg procedure. Genes that were either up-regulated (≥ 3.0) or down-regulated (≤ 0.3) after treatment with LPS compared to saline were identified in NOS2−/− and wild-type mice. Then genes with endotoxin-induced increased expression (≥ 3.0) or decreased expression (≤ 0.3) were compared in wild-type and NOS2−/− mice. The Supplemental files summarize the results as follows: Additional file 1. Genes with increased expression in endotoxemic wild-type, but not in endotoxemic NOS2−/− mice; Additional file 2. Genes with decreased expression in endotoxemic wild-type, but not endotoxemic NOS2−/− mice; Additional file 3. Genes with increased expression in endotoxemic NOS2−/−, but not endotoxemic wild-type mice; Additional file 4. Genes with a decreased expression endotoxemic NOS2−/−, but not endotoxemic wild-type mice.
Results
Endotoxin challenge increases Arg1 levels in the lungs; dependence on NOS2
Endotoxin impairs HPV in wild-type mice, but not in NOS2−/− mice [2]. We hypothesized that differences in endotoxin-induced gene expression between wild-type mice and NOS2−/− mice might explain the different response to endotoxin in the two genotypes. Therefore, we compared the transcriptional profile of lungs from wild-type and NOS2−/− mice 22 h after endotoxin challenge using RNA microarrays and analyzed the data for up- and down-regulated gene expression. The microarrays assessed the expression of 22,690 genes. Compared to the lungs of control wild-type mice treated with saline, endotoxin challenge induced a greater than three-fold increase in 59 genes and a three-fold decrease in 72 genes in the lungs of wild-type mice (Additional file 1 and Additional file 2). Compared to the lungs of NOS2−/− mice challenged with saline, LPS caused an up-regulation of 154 genes and a down-regulation of 93 genes in the lungs of NOS2−/− mice (Additional file 3 and Additional file 4). Of the 800 genes that had three-fold altered expression by endotoxin, 346 were up-regulated or down-regulated in the lungs of both genotypes (Fig. 1). However, the results of the microarray analysis indicated that the effect of endotoxin on Arg1 expression was discordant in the two genotypes: Expression of Arg1 was up-regulated 16.3-fold in the lungs of LPS-treated wild-type mice; LPS induced a 10% decrease in the expression of Arg1 in LPS-treated NOS2−/− mice. In wild-type mice, LPS challenge induced a 4.8-fold increase in Arg2 gene expression compared to saline-treated wild-type mice. Exposure of NOS2−/− mice to LPS caused a similar, 6.1-fold increase in Arg2 gene expression compared to saline challenged NOS2−/− mice. These results suggest that endotoxin up-regulates Arg1 expression in the lungs of wild-type, but not NOS2−/− mice, while endotoxemia induces Arg2 expression to a similar extent in both genotypes.

Up-regulated genes in the lungs of endotoxemic wild-type, but not NOS2-deficient mice. Whole lung tissues from wild-type mice treated with (N = 3), and without (N = 4), endotoxin and from NOS2−/− mice treated with (N = 3), and without (N = 1), LPS were studied. The grey dots show the log2 fold change in gene expression upon LPS exposure; the x-axis represents changes in wild-type mice and the y-axis changes in NOS2−/− mice. The transcriptional profiles revealed a contrasting gene expression of pulmonary Arg1 in wild-type and NOS2−/− mice after LPS challenge: Arg1 expression increased in endotoxemic wild-type mice compared to saline challenged wild-type mice (positive scaling on the x-axis). In contrast, Arg1 expression did not increase in endotoxemic NOS2−/− mice compared to the saline-treated NOS2−/− mice (negative scaling on the y-axis). Arg2 expression increased in both strains after LPS challenge (positive scaling on the x- and y-axis).
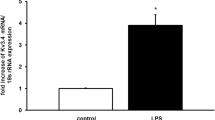
To verify the microarray results, pulmonary mRNA levels of Arg1 and Arg2 were measured using qPCR. LPS challenge increased Arg1 mRNA levels in the lungs of wild-type mice (17.9-fold increase compared to saline-challenged wild-type mice, P < 0.05) but not in NOS2−/− mice (0.67-fold decrease compared to saline challenged wild-type mice, P = 0.07) (Fig. 2a). Endotoxin increased Arg2 mRNA levels similarly in the lungs of wild-type mice and NOS2−/− mice (8.8-fold increase vs 9.1-fold increase, P = 0.90, Fig. 2b). Immunoblots were used to measure the protein levels of Arg1 in the lungs of wild-type and NOS2−/− mice after challenge with saline or LPS. LPS challenge increased the protein level of Arg1 in wild-type, but not in NOS2−/−, mice (Fig. 3). Taken together, the results confirm that endotoxin challenge induces a marked increase in Arg1 expression in the lungs. In contrast, induction of Arg2 expression is similar in the two genotypes.

Pulmonary mRNA expression of Arg1 and Arg2 in wild-type mice and NOS2−/− mice. Endotoxin increased the pulmonary Arg1 mRNA expression in wild-type mice but not in NOS2−/− mice (Panel a). The pulmonary Arg2 mRNA expression was similar in wild-type mice and NOS2−/− mice after LPS challenge (Panel b). Tissue samples were taken 22 h after challenge with either saline (control) or LPS. N = 6 per group; data are presented as mean ± SD; * P < 0.05 vs. saline-challenged wild-type mice, # P < 0.05 vs. saline-challenged NOS2−/− mice, and § P < 0.05 vs. endotoxin-challenged NOS2−/− mice

Pulmonary expression of Arg1 in wild-type and NOS2−/− mice. The level of Arg1 protein was low in the lungs of saline-treated wild-type mice (N = 6) and was markedly increased in the lungs of wild-type mice treated with LPS (N = 6). Arg1 protein level were low in NOS2−/− mice challenged with saline (control, N = 4) or endotoxin (N = 5). Lungs were harvested 22 h after challenge with either saline (control) or lipopolysaccharide (LPS). Data are presented as mean ± SD; * P < 0.05 vs. saline-challenged wild-type mice, # P < 0.05 vs. saline-challenged NOS2−/− mice, and § P < 0.05 vs. endotoxin-challenged NOS2−/− mice
Endotoxin increased Arg1 immunoreactive protein expression in small pulmonary arteries, bronchial epithelium and connective tissue
To evaluate the location of Arg1 protein expression, lung sections of wild-type mice were stained with Arg1 specific antibodies 22 h after challenge with saline (A) or endotoxin (B) (Fig. 4). Arg1 expression was found in walls of small pulmonary arteries, bronchial epithelium, and in connective tissue.

Arg1 immunostaining in lung sections of wild-type mice. Arg1 immunoreactivity in lungs of mice after saline (a) or endotoxin challenge (b). Immunoenzyme stainings were performed on paraffin-embedded sections using polyclonal rabbit anti-Arg1 and counterstained with hematoxylin. Representative image shows Arg1 immunoreactive protein (purple-brown) in bronchial epithelium (BR), connective tissue (CT) and small pulmonary arteries (PA)
An arginase inhibitor, norNOHA, restores HPV after LPS challenge
To determine the physiological impact of arginase on HPV, we examined the effects of the arginase inhibitor norNOHA on HPV in anesthetized wild-type mice. At baseline, the hemodynamic parameters did not differ between the mice in five experimental groups: 1) saline-challenged control mice treated with normal saline 21 h after the initial challenge; 2) control mice treated with norNOHA 21 h after the saline challenge; 3) LPS-challenged mice treated with normal saline 21 h after the initial challenge; 4) LPS-challenged mice treated with 20 mg/kg norNOHA 21 h after the initial challenge, and 5) LPS-challenged mice treated with 40 mg/kg norNOHA 21 h after the initial challenge (Table 1).
During LMBO, the heart rates were comparable between the groups. The SAP remained stable before and during LMBO in all groups except control mice that were treated with norNOHA. In this group, the SAP decreased compared to baseline (97 ± 11 mmHg vs. 77 ± 7 mmHg, P < 0.05), suggesting that the arginase inhibitor caused systemic vasodilation. Endotoxemic mice that received normal saline (instead of norNOHA) had a small but statistically significant decrease in SAP during LMBO compared to baseline (96 ± 14 mmHg vs. 93 ± 16 mmHg, P < 0.05). The PAP increased compared to baseline during LMBO in each group (Table 1).
To investigate the effect of arginase inhibition on HPV, we measured the percentage increase in LPVR before and during LMBO. In control mice treated with normal saline, LMBO caused a ~ 121% increase in LPVR from baseline (Fig. 5a). In control mice treated with norNOHA, the LPVR response to LMBO was attenuated compared to control mice treated with saline (80 ± 27% vs 121 ± 22%, P < 0.05). Mice challenged with LPS and treated with normal saline had an impaired increase in LPVR in response to LMBO compared to control mice treated with normal saline (33 ± 9% vs. 121 ± 22%, P < 0.05). The increase in LPVR in response to LMBO was also smaller in the LPS-challenged mice treated with norNOHA at 20 mg/kg compared to the control mice treated with normal saline (46 ± 28% vs. 121 ± 22%, P < 0.05). In contrast, treatment with norNOHA at 40 mg/kg restored the increase in LPVR in response to LMBO in LPS-challenged mice to a level that was comparable to that of control mice treated with normal saline (113 ± 29% vs 121 ± 22%, P = 0.97, Fig. 5a). These results show that the arginase inhibitor norNOHA, at a dosage of 40 mg/kg but not 20 mg/kg, restores HPV when administered to mice 22 h after endotoxin challenge.

Hypoxic pulmonary vasoconstriction and arterial oxygenation. Panel A: Mice were challenged with saline (control) or endotoxin (LPS). After 21 h, the animals were treated with normal saline (NS) or the arginase inhibitor norNOHA (20 mg/kg or 40 mg/kg). HPV was assessed as the left pulmonary vascular resistance (LPVR) in response to left mainstem bronchus occlusion (LMBO). Treatment with the arginase inhibitor norNOHA (40 mg/kg) restored HPV in endotoxemic mice. Panel B: Arterial blood gas tensions were measured during LMBO at the end of the experiments. The restoration of HPV was associated with improved arterial oxygenation (PaO2) in endotoxemic mice treated with 40 mg/kg norNOHA compared to saline treatment. N = 7 per group; data presented as mean ± SD; * P < 0.05 vs. control group treated with NS, § P < 0.05 vs. LPS group treated with normal saline, † LPS group treated with 20 mg/kg norNOHA
Restoration of HPV in endotoxemic mice improves systemic arterial oxygen tensions during LMBO
To estimate the impact of HPV on intrapulmonary shunt and systemic oxygenation, arterial blood gas tensions were measured during LMBO at the end of the HPV measurements (Fig. 5a, Table 2). The arterial pH of endotoxemic mice treated with normal saline, 20 or 40 mg/kg of norHOHA was lower than the arterial pH of control mice treated with normal saline (P < 0.05 for all three comparisons) or control mice treated with 40 mg/kg norNOHA (P < 0.05 for all three comparisons). Accordingly, the arterial base excess of endotoxemic mice after treatment with normal saline, 20 or 40 mg/kg norNOHA was smaller than the arterial base excess of control mice that received normal saline (P < 0.05 for all three comparisons) or control mice treated with 40 mg/kg norNOHA (P < 0.05 for all three comparisons). There was no difference in the arterial carbon dioxide tension between all experimental groups, suggesting that endotoxin induced a metabolic acidosis. The hemoglobin concentrations were similar in all mice. The arterial oxygen tension (PaO2) was highest in control mice treated with normal saline during LMBO. There was a small but significant decrease in PaO2 in control mice treated with norNOHA compared to normal saline (200 ± 38 mmHg vs. 267 ± 38 mmHg, P < 0.05). During LMBO, the PaO2 was lower in LPS-challenged mice treated with normal saline or 20 mg/kg norNOHA compared to control mice treated with normal saline (P < 0.05 for each comparison). In contrast, treatment of endotoxemic mice with 40 mg/kg norNOHA restored the PaO2 to levels that were comparable to those in control mice (not challenged with LPS) treated with saline (not treated with norNOHA) (234 ± 25 mmHg vs. 267 ± 38 mmHg, P = 0.35). These results suggest that treatment with 40 mg/kg norNOHA restores HPV and decreases intrapulmonary shunt in endotoxemic mice with an improvement in systemic arterial oxygenation.
Discussion
The current study used expression microarrays to identify one or more genes that might contribute, together with NO, to the endotoxin-induced impairment of HPV. Transcriptional profiling revealed that Arg1 was the most up-regulated gene in the lungs of wild-type mice after endotoxin challenge and that Arg1 was marginally decreased in endotoxemic NOS2−/− mice. The endotoxin-induced increase in Arg1 mRNA in wild-type, but not in NOS2−/− mice was confirmed using qPCR. Immunoblots showed that the differences in Arg1 mRNA levels resulted in corresponding differences in Arg1 protein levels. Immunohistochemical analysis revealed that endotoxin induced Arg1 expression in the walls of small pulmonary artery, bronchial epithelium, and connective tissues in wild-type mice. The functional importance of arginase in the endotoxin-induced impairment of HPV was supported by the restoration of HPV by the arginase inhibitor norNOHA. Taken together, these results reveal a critical role for arginase in the impairment of HPV during endotoxemia in mice.
The production of NO by NOS depends on the availability of L-arginine because no other amino acid or guanidino-containing compound can substitute as a substrate for NOS. L-arginine is metabolized by NOS to NO and L-citrulline and by arginase to urea and L-ornithine (Fig. 6a) [10]. Because L-arginine is the substrate for both NOS and arginase, one might expect that inhibition of arginase would increase the availability of L-arginine for NOS to produce more NO and thereby impair HPV. In this study, the pulmonary level of Arg1 and Arg2, were low at baseline (Figs. 2 and 3), suggesting that inhibition of arginase would have little effect on healthy mice. However, we observed that administration of norNOHA to healthy control mice modestly attenuated HPV. It is possible that, even in the presence of low baseline levels of arginase, norNOHA was able to increase the level of L-arginine available to NOS (primarily NOS3), resulting in more NO production, which exerted pulmonary vasodilating effects that oppose HPV (Fig. 6b).

Hypothetical roles of arginase and arginase inhibition on HPV. a Arginase and nitric oxide synthase (NOS) compete for the common substrate L-arginine. b In healthy control mice, the arginase inhibitor norNOHA may shift L-arginine to NOS thereby increasing the level of NO that in turn attenuates HPV. c Endotoxemia increases expression of NOS2 and Arg1. NOS2-dependent S-nitrosylation may promote Arg1 expression and activity. Increased Arg1 may decrease L-arginine concentration locally. Low local arginine levels might promote NOS2 uncoupling and reactive oxygen species (ROS) production. Increased NO levels and redox imbalance impairs HPV. Upregulated Arg1 also potentially increases downstream products including L-ornithine and polyamines, which may also impair HPV. d Inhibition of arginase by norNOHA restores HPV by preventing NOS2 uncoupling and ROS production by shifting more L-arginine to NOS2 and decreasing polyamines
To the best of our knowledge, this is the first study that showed that NOS2 is required for the ability of endotoxin to induce Arg1 in the lung. Although detailed signaling mechanism of NOS2-dependent Arg1 induction was not explored in the current study, protein S-nitrosylation induced by NOS2-derived NO has been shown to regulate transcription. For example, NOS2-dependent S-nitrosylation inhibits deacetylase sirtuin1 (SIRT1) during inflammation [11]. Inhibition of SIRT1 in turn activates signal transducer and activator of transcription 3 (STAT3) by increasing acetylation and phosphorylation of STAT3 [12]. Therefore, NOS2-dependent S-nitrosylation may activate STAT3-dependent transcription. Because STAT6/STAT3 are known promoters of Arg1 expression, it is conceivable that endotoxin-induced NOS2 upregulated Arg1 expression via STAT3 activation [13].
In this study, endotoxin challenge increased Arg1 mRNA and protein expression in homogenized lung tissue. The immunohistochemistry revealed a marked increase of Arg1 expression in the small pulmonary arteries, bronchial epithelium, and connective tissues. HPV is intrinsic to the pulmonary arterial smooth muscle cells. However, HPV is also modified by extrinsic factors derived from neighboring cell types such as vascular endothelial cells and macrophages [1]. Arg1 is typically expressed in liver, red blood cells, and macrophages, but can be expressed in vascular endothelial cells and smooth muscle cells [14, 15]. It is conceivable that Arg1 induction in multiple cell types modulates HPV during endotoxemia.
By reducing blood flow to poorly ventilated regions, HPV optimizes the match between ventilation and perfusion in the hypoxic regions of the lung and improves arterial oxygenation [1]. In this study, we observed that endotoxin challenge impaired HPV and decreased arterial oxygenation. In contrast, administration of arginase inhibitor during endotoxemia restored HPV and improved arterial oxygenation to the levels comparable to healthy control mice treated with normal saline. These results indicate Arg1 as a potential new therapeutic target for endotoxin-induced ventilation perfusion mismatch associated with systemic hypoxemia.
Twenty-two hours after endotoxin challenge, HPV was impaired in mice treated with normal saline, whereas administration of 40 mg/kg norNOHA restored HPV. Because inhibition of arginase can theoretically make more L-arginine available for NOS to produce NO, the observed protective effect of norNOHA on HPV is counterintuitive. However, the L-arginine levels, and therefore the substrate availability for NOS to produce NO, differs under healthy and pro-inflammatory conditions. Sepsis and endotoxemia reduce L-arginine levels [16, 17]. It has also been shown that NOS2-dependent S-nitrosylation activates Arg1 by stabilizing Arg1 trimer thus shifting more L-arginine to Arg1-dependent signaling [18]. In any case, decreased L-arginine availability can result in uncoupling of NOS (predominantly NOS2 under pro-inflammatory condition), with decreased production of NO and increased production of reactive oxygen species (ROS) (Fig. 6c) [19]. Increased oxidative stress and decreased anti-oxidant capacity have been reported in LPS-induced lung injury [19, 20]. We previously showed that treatment with ROS-scavengers attenuates endotoxin-induced impairment of HPV [7]. It has been reported that arginase inhibition preserves NOS coupling thereby reduces ROS production and vascular endothelial dysfunction in aging rats [21]. It is conceivable that norNOHA attenuated NOS-uncoupling, ROS production, and redox imbalance to preserve HPV in endotoxemic mice (Fig. 6d).
An alternative explanation for the observed restoration of HPV in endotoxemic mice treated with norNOHA is that the inhibitor may decrease the level of arginase downstream products, such as ornithine and polyamines. The polyamines putrescine, spermidine, and spermine are small polycationic molecules that rectify ion channel conductance [22]. Altered ion conductance in pulmonary artery smooth muscle cells modulates HPV [1]. Polyamine catabolism has also been shown to increase oxidative stress which may promote redox imbalance [23]. The potential role of arginine metabolites in certain cell populations (such as vascular smooth muscle cells) in the regulation of HPV remains to be defined.
It is possible that other factors, in addition to NOS2 and Arg1, contribute to the impairment of HPV. For example, endothelial protein C receptor (also known as activated protein C receptor) was the second most up-regulated gene in the lungs of endotoxemic wild-type mice after Arg1 (Additional file 1). Richard and colleagues reported that administration of activated protein C worsened oxygenation, presumably due to increased ventilation-perfusion mismatching, in a porcine model of acute lung injury [24]. The potential role of the endothelial protein C receptor in regulating HPV during endotoxemia warrants further studies.
A potential limitation of this investigation is the small number of lungs that were used in the microarray analyses. However, the transcriptional profiling was designed as a pilot study to identify candidate genes. Additional qPCRs and immunoblots were performed to confirm that endotoxin induced Arg1 gene expression in wild-type, but not NOS2−/− mice. A second limitation concerns the immunohistochemistry. After endotoxin challenge, Arg1 positive immunostaining was found in bronchial epithelium, small pulmonary arteries, and the connective tissue. However, no double staining was performed leaving the specific cell type uncertain. Further studies are needed to identify which cell types in lung tissue are responsible for increased arginase activity.
Conclusions
In summary, transcriptional profiling identified Arg1 as a factor that might be required for endotoxin to inhibit HPV in wild-type mice. The arginase inhibitor norNOHA, at a dose of 40 mg/kg, acutely restored HPV in endotoxemic mice. These results identify arginase as a novel molecular target to improve pulmonary ventilation-perfusion matching during endotoxemia.
Abbreviations
- Arg1:
-
Arginase 1
- Arg2:
-
Arginase 2
- BE:
-
Base excess
- Hb:
-
Hemoglobin
- HPV:
-
Hypoxic pulmonary vasoconstriction
- HR:
-
Heart rate
- LMBO:
-
Left main bronchus occlusion
- LPS:
-
Lipopolysaccharide
- LPVR:
-
Left pulmonary vascular resistance
- NO:
-
Nitric oxide
- norNOHA:
-
Nω-hydroxy-nor-L-arginine
- NOS2:
-
Nitric oxide synthase 2
- NOS2−/− :
-
NOS2-deficiency
- NOS3:
-
Nitric oxide synthase 3
- NS:
-
Normal saline
- ODC:
-
L-ornithine decarboxylase
- PaCO2 :
-
Arterial carbon dioxide tension
- PaO2 :
-
Arterial oxygen tension
- PAP:
-
Pulmonary arterial pressure
- pHa :
-
Arterial pH
- Procr:
-
Endothelial protein C receptor (activated protein C receptor)
- QLPA:
-
Left pulmonary arterial blood flow
- ROS:
-
Reactive oxygen species
- SAP:
-
Systemic arterial pressure
- SIRT1:
-
Sirtuin1
- STAT3:
-
Signal transducer and activator of transcription 3
References
Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev. 2012;92:367–520.
Ullrich R, Bloch KD, Ichinose F, Steudel W, Zapol WM. Hypoxic pulmonary blood flow redistribution and arterial oxygenation in endotoxin-challenged NOS2-deficient mice. J Clin Invest. 1999;104:1421–9.
Ichinose F, Zapol WM, Sapirstein A, Ullrich R, Tager AM, Coggins K, Jones R, Bloch KD. Attenuation of hypoxic pulmonary vasoconstriction by endotoxemia requires 5-lipoxygenase in mice. Circ Res. 2001;88:832–8.
Ichinose F, Hataishi R, Wu JC, Kawai N, Rodrigues AC, Mallari C, Post JM, Parkinson JF, Picard MH, Bloch KD, Zapol WM. A selective inducible NOS dimerization inhibitor prevents systemic, cardiac, and pulmonary hemodynamic dysfunction in endotoxemic mice. Am J Physiol Heart Circ Physiol. 2003;285:H2524–30.
Petersen B, Austen KF, Bloch KD, Hotta Y, Ichinose F, Kanaoka Y, Zapol WM. Cysteinyl leukotrienes impair hypoxic pulmonary vasoconstriction in endotoxemic mice. Anesthesiology. 2011;115:804–11.
Marshall BE, Hanson CW, Frasch F, Marshall C. Role of hypoxic pulmonary vasoconstriction in pulmonary gas exchange and blood flow distribution. Pathophysiology Intensive Care Med. 1994;20:379–89.
Baboolal HA, Ichinose F, Ullrich R, Kawai N, Bloch KD, Zapol WM. Reactive oxygen species scavengers attenuate endotoxin-induced impairment of hypoxic pulmonary vasoconstriction in mice. Anesthesiology. 2002;97:1227–33.
Steinbicker AU, Liu H, Jiramongkolchai K, Malhotra R, Choe EY, Busch CJ, Graveline AR, Kao SM, Nagasaka Y, Ichinose F, Buys ES, Brouckaert P, Zapol WM, Bloch KD. Nitric oxide regulates pulmonary vascular smooth muscle cell expression of the inducible cAMP early repressor gene. Nitric Oxide. 2011;30:294–302.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Morris SM Jr. Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol. 2009;157:922–30.
Shinozaki S, Chang K, Sakai M, Shimizu N, Yamada M, Tanaka T, Nakazawa H, Ichinose F, Yamada Y, Ishigami A, Ito H, Ouchi Y, Starr ME, Saito H, Shimokado K, Stamler JS, Kaneki M. Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase SIRT1 to increase acetylation and activation of p53 and p65. Sci Signal. 2014;7:ra106.
Nie Y, Erion DM, Yuan Z, Dietrich M, Shulman GI, Horvath TL, Gao Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol. 2009;11:492–500.
KE S, Shandilya H, Kepka-Lenhart D, Poljakovic M, Ghosh A, Morris SM Jr. Shaping the murine macrophage phenotype: IL-4 and cyclic AMP synergistically activate the arginase I promoter. J Immunol. 2013;191:2290–8.
Caldwell RW, Rodriguez PC, Toque HA, Narayanan SP, Caldwell RB. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol Rev. 2018;98:641–65.
Hochstedler CM, Leidinger MR, Maher-Sturm MT, Gibson-Corley KN, Meyerholz DK. Immunohistochemical detection of arginase-I expression in formalin-fixed lung and other tissues. J Histotechnol. 2013;36:128–34.
Luiking YC, Hallemeesch MM, Vissers YL, Lamers WH, Deutz NE. In vivo whole body and organ arginine metabolism during endotoxemia (sepsis) is dependent on mouse strain and gender. J Nutr. 2004;134:2768S–74S discussion 2796S-2797S.
Luiking YC, Ten Have GA, Wolfe RR, Deutz NE. Arginine de novo and nitric oxide production in disease states. Am J Physiol Endocrinol Metab. 2012;303(10):E1177–89.
Santhanam L, Lim HK, Lim HK, Miriel V, Brown T, Patel M, Balanson S, Ryoo S, Anderson M, Irani K, Khanday F, Di Costanzo L, Nyhan D, Hare JM, Christianson DW, Rivers R, Shoukas A, Berkowitz DE. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ Res. 2007;101:692–702.
Gross CM, Rafikov R, Kumar S, Aggarwal S, Ham PB 3rd, Meadows ML, Cherian-Shaw M, Kangath A, Sridhar S, Lucas R, Black SM. Endothelial nitric oxide synthase deficient mice are protected from lipopolysaccharide induced acute lung injury. PLoS One. 2015;10:e0119918.
Zhang HX, Liu SJ, Tang XL, Duan GL, Ni X, Zhu XY, Liu YJ, Wang CN. H2S attenuates LPS-induced acute lung injury by reducing oxidative/Nitrative stress and inflammation. Cell Physiol Biochem. 2016;40:1603–12.
Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, Santhanam L, Webb A, Camara A, Sikka G, Nyhan D, Shoukas AA, Ilies M, Christianson DW, Champion HC, Berkowitz DE. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol. 2009;107:1249–57.
Weiger TM, Hermann A. Cell proliferation, potassium channels, polyamines and their interactions: a mini review. Amino Acids. 2014;46:681–8.
Stewart TM, Dunston TT, Woster PM, Casero RA. Polyamine catabolism and oxidative damage. J Biol Chem. 2018;293:18736–45.
Richard JC, Bregeon F, Leray V, Le Bars D, Costes N, Tourvieille C, Lavenne F, Devouassoux-Shisheboran M, Gimenez G, Guerin C. Effect of activated protein C on pulmonary blood flow and cytokine production in experimental acute lung injury. Intensive Care Med. 2007;33:2199–206.
Acknowledgements
We thank Dr. Zapol for valuable comments and support. Histology and immunohistology services were provided by the Tissue Bank for Inflammatory Diseases Heidelberg (GEZEH), which was funded by the DFG (SFB 938 TP Z2 to F.L.). We acknowledge financial support by Deutsche Forschungsgemeinschaft within the funding program Open Access Publishing, by the Baden-Württemberg Ministry of Science, Research and the Arts and by Ruprecht-Karls-Universität Heidelberg.
Funding
This study was supported by NIH grant GM79360 (FI).
Availability of data and materials
The microarray data including information requested by the Microarray Gene Expression Data society can be accessed in the microarray database at NCBI Gene Expression Omnibus (GEO) using the following link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130936. Data sets of microarray analysis (MOE430A Annotations, CSV format, Release 36, 4/13/16) are obtainable at http://www.affymetrix.com/support/technical/annotationfilesmain.affx).
Author information
Authors and Affiliations
Contributions
BP and CJB carried out the experiments, analyzed the data, and wrote the manuscript with support from DBB and FI. GS contributed to sample preparation. DS performed the microarray analysis. FL carried out experiments. KDB and FI devised the project. FI and DBB interpreted the data and revised the manuscript. FI approved the final version of the manuscript to be published. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
All animal experiments were approved by the Subcommittee in Research Animal Care of the Massachusetts General Hospital.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Up-regulated genes in the lungs of endotoxemic wild-type, but not NOS2-deficient mice. We previously showed that HPV is preserved in endotoxemic NOS2-deficient (NOS2−/−) mice. [2]. The objective of this study was to identify gene products that may contribute to endotoxin-induced impairment of HPV in wild-type mice. Transcription profiling was performed on homogenized lung tissue from wild-type mice 22 h after LPS administration (N = 3) and compared to the profile produced by the lungs of wild-type mice challenged with saline LPS (N = 4). Transcription profiling was also performed on homogenized lung tissue from NOS2−/− mice 22 h after LPS administration (N = 3) and was compared to the profile produced by the lungs of NOS2−/− mice challenged with saline (N = 1). The microarray assessed the expression of 22,690 genes. A subset of 59 genes (shown in this table) was increased greater than three-fold in wild-type mice challenged with endotoxin, but was not enhanced three-fold in the lungs of NOS2−/− mice challenged with endotoxin. The microarray results indicated that Arg1 was the most highly expressed gene in endotoxemic wild-type mice, and that Arg1 expression was not increased in the lungs of endotoxemic NOS2−/− mice. (DOCX 1424 kb)
Additional file 2:
Down-regulated genes in the lungs of endotoxemic wild-type mice, but not endotoxemic NOS2-deficient mice. Expression of 72 genes was decreased to 0.3-fold or less by endotoxin in wild-type mice, but was not decreased by endotoxin in NOS2−/− mice. (DOCX 2071 kb)
Additional file 3:
Up-regulated genes in the lungs of endotoxemic NOS2−/− mice, but not in endotoxemic wild-type mice. Expression of 155 genes was increased more than three-fold by endotoxin in NOS2−/− mice, but was not similarly increased by endotoxin in wild-type mice. (DOCX 3039 kb)
Additional file 4:
Down-regulated genes in the lungs of endotoxemic NOS2−/− mice, but not in endotoxemic wild-type mice. Ninety-three genes were decreased to 0.3-fold or less by endotoxin in NOS2−/− mice, but were not decreased by endotoxin in wild-type mice. (DOCX 2346 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Petersen, B., Busch, C.J., Schleifer, G. et al. Arginase impairs hypoxic pulmonary vasoconstriction in murine endotoxemia. Respir Res 20, 109 (2019). https://doi.org/10.1186/s12931-019-1062-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-019-1062-6