
Abstract
Background
Combined pulmonary fibrosis and emphysema (CPFE) is a heterogeneous clinico-radiological syndrome without a consensus definition. There are limited data on the relation between the amount of parenchymal fibrosis and prognosis. In this study, we assessed the prognostic implications of the extent of fibrosis assessed by an automated quantitative computed tomography (CT) technique and the radiological and functional change over time in patients with a broad spectrum of fibrotic interstitial lung diseases (ILDs) encountered in a real-world setting.
Methods
We conducted a single-centre, retrospective study of 228 consecutive patients with CPFE, encountered from 2007 to 2015 at Kameda Medical Center, Chiba, Japan. We investigated the prognostic value of automated CT fibrosis quantification and the subsequent course of CPFE.
Results
Among 228 patients with CPFE, 89 had fibrosis affecting < 5% of their lungs, 54 had 5 to < 10% fibrosis, and 85 had ≥ 10% fibrosis at the time of diagnosis. Lower volume of fibrosis correlated with lower rates of mortality and acute exacerbation (p < 0.001). In particular, among those with < 5% fibrosis, only 4.5% died and none experienced acute exacerbation during follow-up, whereas 57.6% and 29.4% of those with ≥ 10% fibrosis experienced death and acute exacerbation, respectively. Although, the ≥ 10% fibrosis group had the poorest overall survival as well as the highest incidence of acute exacerbation, the incidence of decline in pulmonary function tests, change per year in total lung volume, and progression of fibrosis on chest CT was highest in the 5 to < 10% fibrosis group. The Cox proportional hazard model for CPFE progression (defined by composite criteria of death, acute exacerbation, and decline in forced vital capacity or diffusing capacity) showed fibrosis proportion was a risk factor independent of age, sex, smoking pack-years, the Charlson Comorbidity Index, lung cancer, connective tissue disease, and idiopathic pulmonary fibrosis.
Conclusions
Less severe (< 5%) fibrosis at baseline was associated with disease stability and better prognosis compared to more severe fibrosis in CPFE occurring with fibrotic ILDs. Further studies including a validation cohort will be needed.
Trial Registration Retrospectively registered.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background
Combined pulmonary fibrosis and emphysema (CPFE) is a heterogeneous clinico-radiological syndrome, comprising emphysema in the upper lung and fibrosis in the lower lung. CPFE is strongly associated with cigarette smoking thus, considered a smoking-related interstitial lung disease (ILD) [1, 2]. Natural disease history differs between patients with CPFE and those with either pulmonary fibrosis or emphysema alone. The median survival time reported for patients with CPFE varies from 2.1 to 8.5 years, with an increased incidence of pulmonary hypertension and lung cancer [1]. A recent analysis revealed that patients with CPFE had a similar mortality rate with those with idiopathic pulmonary fibrosis (IPF) alone; however, it excluded patients with other fibrotic ILDs [3]. Although initial reports of CPFE cases were described in patients with IPF [4], recent studies included fibrotic ILDs with computed tomography (CT) findings inconsistent with a usual interstitial pneumonia (UIP) pattern [5]. In the absence of a consensus definition for CPFE, it seems important to analyse all patients with co-existent emphysema and fibrotic ILDs (rather than just IPF/UIP) seen on high resolution CT to clarify the prognostic implications of CPFE in the broader population [2].
Study populations in previous reports varied, and no studies have correlated the long-term evolution of CPFE to baseline CT findings and pulmonary function measures; therefore, the prognostic implications of these studies remain uncertain. This study aimed to assess the prognostic implications of the extent of fibrosis assessed by an automated quantitative CT technique and pulmonary function measures at the time of initial CPFE diagnosis.
Methods
Study design and participants
This was a single-centre cohort study designed to assess the prognostic value of fibrosis quantification in patients with CPFE adhered to the Strengthening the Reporting of Observational Studies in Epidemiology Statement [6]. The institutional review board of Kameda Medical Center approved the study and allowed use of the opt-out method, instead of requiring informed consent.
Study participants were retrospectively selected from electronic medical records using a computer-assisted search function of Kameda Medical Center, Chiba, Japan, from January 2007 to December 2015. Consecutive adult patients with bilateral fibrosis based on their chest CT radiology report were initially included in the study, instead of using the clinical diagnosis of CPFE, to avoid selection bias. From the chest CT reports, we considered fibrosis cases reported with relevant description, such as: fibrotic changes, ILD, and interstitial pneumonia in Japanese and English. We used the first chest CT scan and pulmonary function tests (PFTs) in the patient-selection period for initial evaluation and the date of diagnosis. In the absence of consensus criteria for CPFE, we avoided using the minimum fibrosis or emphysema criteria and analysed the entire patient cohort with any amount of bilateral lung fibrosis and emphysema to avoid selection bias. The exclusion criteria were: (1) no mention of emphysema in the chest CT report; (2) diagnosis of other specific types of ILD, following the prior publication by Cottin V, et al., such as drug-induced ILD, pneumoconiosis, hypersensitivity pneumonitis, sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, or eosinophilic pneumonias [5, 7]; (3) no history of cigarette smoking (to avoid other forms of cystic lung diseases) [2]; and (4) no follow-up chest CT or PFTs > 1 year after the initial evaluation, unless the patient had experienced acute exacerbation or had died.
The lung parenchyma of each eligible patient was automatically segmented from the CT dataset, and each pixel in the lung tissue was classified by the Computer-Aided Lung Informatics for Pathology Evaluation and Rating (CALIPER) [8, 9] software as normal parenchyma; hyperlucent area (moderate or severe low attenuation areas [LAAs]); or fibrosis (ground glass opacity [GGO], reticular densities, or honeycombing pattern). The whole-lung CALIPER results for each participant were classified into three groups based on previous studies [10, 11] as the proportion of fibrosis volume in CPFE and interstitial lung abnormality: (1) < 5%, (2) 5 to < 10%, and (3) > 10% to assess the relationship between the amount of fibrosis and disease progression. We examined the patients’ medical records from January 2007 to December 2018.
Data collection and CALIPER analysis
The following demographic, clinical, and laboratory data were collected at the time of the initial chest CT study: age, sex, data required for the Charlson Comorbidity Index (CCI) [12] and ILD-gender-age-physiology score [13], smoking history, clinical ILD diagnosis, anti-fibrotic agent use, pulmonary hypertension or lung cancer history, and PFT results. The initial and most recent PFT and chest CT results were collected.
Available chest CT results were evaluated by CALIPER (developed at Mayo Clinic, Minnesota, United States), a system based on histogram signature mapping, and was programmed using expert radiologist analysis of pathologically confirmed datasets, obtained through the Lung Tissue Research Consortium [9, 14], to quantify features of the lung parenchyma. CALIPER classified each lung parenchymal voxel into one of the following categories: normal lung, mild volume of LAAs, moderate LAA, severe LAA, reticular opacities, honeycombing, and GGO. CALIPER algorithm is specifically designed to detect low density pixels that could be within either emphysema or honeycombing [8, 14, 15]. A volumetric morphological analysis of the surrounding area is performed, to determine whether that pixel and its contiguously surrounding pixels should be classified as emphysema or honeycombing. When the algorithm identifies a rounded periphery of higher density surrounding an area of low density, it is classified as a honeycomb cyst; if not, it is classified as emphysema. Fibrosis extent was defined as the sum volume of honeycombing, ground glass, and reticular opacities [9, 15]. The amount of normal lung was defined as the sum volume of normal lung and mild LAAs [16]. Emphysema was defined as the sum volume of moderate and severe LAAs.
Outcome of interest
The primary outcome was CPFE progression defined by the following criteria: (1) death by any cause, (2) acute exacerbation, (3) decline in forced vital capacity (FVC) > 10% [17], and (4) decline in diffusing capacity of the lung for carbon monoxide (DLco) > 15% [17, 18]. We considered the disease had progressed when any of these criteria were met. The secondary outcome was overall survival calculated from the date of initial chest CT until death; progression-free survival was calculated from the date of the initial chest CT until a patient met any of the progression criteria. Per-year change in PFT and CALIPER quantification based on chest CT was calculated from the interval in the data, divided by the years between the baseline and the most recent test results.
Statistical analysis
Categorical variables are expressed as number and percentage and were compared using the chi-square and Fisher’s exact tests. Continuous variables are expressed as the median and interquartile range and were compared using the Mann–Whitney U-test. We adjusted for potential confounding covariates for the correlation between fibrosis extent and outcome, including age, sex, smoking amount (pack-years), and CCI before data collection to minimise selection bias [12]. Background lung-specific covariates that were adjusted for included lung cancer, connective tissue disease, and IPF. A Cox proportional hazard model for CPFE progression was adjusted by the following additional factors to determine fibrosis proportion: age, sex, pack-years, and CCI. Lung-specific adjustments were for lung cancer, connective tissue disease, and IPF. Overall survival and progression-free survival stratified by fibrosis extent on chest CT were analysed by Kaplan–Meier plots and log-rank tests. Statistical significance was indicated by p < 0.05. All analyses were performed using R ver. 3.5.2 (R Foundation for Statistical Computing, Vienna, Austria) [19] with the add-on package EZR ver. 1.36 (Saitama Medical Center, Jichi Medical University, Saitama, Japan) [20].
Results
Study participants
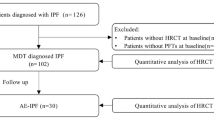
Figure 1 shows the patient’s selection process. The baseline characteristics of the final 228 patients included in the study are shown in Table 1, and more details are provided in the Additional file 1.

Patient selection flow chart
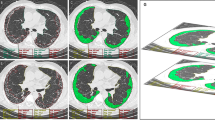
Representative images of the CALIPER evaluation are shown in Fig. 2. The ternary plot in Fig. 3 shows the distribution proportion of fibrosis, emphysema, and normal lung for all 228 participants at initial evaluation. Fibrosis was in the range of 30%; however, emphysema was distributed from approximately 0 to 80%. The distribution of the ternary plot from the lower left to the upper right suggests that the percentage of emphysema is negatively correlated to the percentage of fibrosis. Disease progression and death were associated with a higher percentage of fibrosis.

Representative high-resolution computed tomography images from three patients. Images of the upper lung (a1, b1, and c1) and lower lung (a2, b2, and c2) and 3D renderings analysed by computer-aided lung informatics for lung informatics pathology evaluation and rating shown on each right side (a3, 4; b3, 4; and c3, 4)

Ternary plot for the extent of fibrosis, emphysema, and normal lung; a death vs non-death, b progression vs non-progression
Outcomes
The study outcomes are shown in Table 2. The numbers of all deaths and acute exacerbations were significantly lower in patients with less severe fibrosis. The composite progression was highest in patients with 5 to < 10% fibrosis compared to other groups (p < 0.001). Regarding per-year change in PFTs and chest CT findings, the mean absolute and relative decline in FVC, decline in total lung volume, and progression of fibrosis were highest and lowest in patients with 5 to < 10% fibrosis and < 5% fibrosis, respectively (p < 0.001). Per-year change in DLco and emphysema severity was not significantly different across the three groups. The study outcomes in subgroups of emphysema extent are shown in Additional file 2 and none of them were significantly different.
Kaplan–Meier curves for overall survival and progression-free survival are shown in Fig. 4. Overall survival and progression-free survival significantly differed across the fibrosis groups. Overall survival and progression-free survival were 7.3 years (interquartile range [IQR]: 4.8 years–not applicable) and 3.7 years (IQR: 2.7–6.4 years), respectively, in the 5 to < 10% fibrosis group; and 5.7 years (IQR: 3.5–7.6 years) and 3.5 years (IQR: 2.4–4.2 years), respectively, in the ≥ 10% group. For the < 5% fibrosis group, overall survival could not be statistically calculated because the events were below 50%. In contrast, Kaplan–Meier curves for PFT progression showed the 5 to < 10% fibrosis group and the < 5% fibrosis group were most and least affected, respectively. Cox proportional hazard model for CPFE progression was used to assess fibrosis proportion (Table 3). In hazard models, fibrosis proportion was a risk factor independent of age, sex, smoking pack-years, CCI, lung cancer, connective tissue disease, and IPF.

Kaplan–Meier curve for a overall survival and b progression-free survival. Line: patients with < 5% fibrosis. Dash: patients with 5 to < 10% fibrosis. Dot: patients with ≥ 10% fibrosis
Comparison of patients with and without connective tissue disease
Since 20.6% of the study population had connective tissue diseases (CTDs), we performed subgroup analysis to compare characteristics and outcomes in patients with CTD versus those without CTD. The CTD group comprised significantly more female patients, of younger age, with fewer smoking pack-years, and lower lung cancer prevalence; CT findings and fibrosis quantity did not differ significantly (Additional file 3). Most patients with CTD (~ 60%) had rheumatoid arthritis. Among those without CTD, nearly 25% manifested positive autoantibody, consistent with the concept of interstitial pneumonia with autoimmune features formulated in 2015 [21]. Kaplan–Meier curves for overall survival showed no significant difference between the subgroups (Additional file 4). Multivariate analysis (Additional file 5) revealed mortality was not influenced by CTD or sex; instead, it was affected by the presence of IPF, lung cancer, and age.
Discussion
This study aimed to elucidate the relation between the baseline amount of fibrosis quantified by an automated CT technique and the subsequent course of patients with CPFE. Our results show progression in patients with baseline fibrosis < 5% was not as prominent as in the other CPFE groups and the ≥ 10% fibrosis group had the poorest overall survival as well as the highest incidence of acute exacerbation. Baseline amount of fibrosis was an independent and strong predictor of prognosis. In contrast, in terms of progression-free survival, 5 to < 10% fibrosis group showed a similar Kaplan–Meier curve compared to the ≥ 10% fibrosis group. The incidence of composite progression (especially decline in PFTs), change per year in total lung volume, and progression of fibrosis on chest CT was highest in the 5 to < 10% fibrosis group.
Although Choi SH et al. reported fibrosis extent to correlate with prognosis of CPFE, it was not identified to be an independent prognostic factor [22]. In the Cox regression analysis, we considered the following factors: age, sex, and amount of cigarette smoking, which were previously reported to be associated with emphysema, interstitial lung abnormality, and disease progression by a sub-analysis of the Framingham Heart Study [11, 23]. The CCI [12] was also included in the regression analysis. Finally, lung-specific variables were entered into the models such as lung cancer and IPF, both of which have been identified to be independent poor prognostic factors [24], and CTD-ILD, another common cause of CPFE [25, 26]. Our results revealed that age and male sex were associated with an increased amount of fibrosis. Moreover, patients with CPFE had a high prevalence of lung cancer (23.2%), CTD (20.6%), and IPF (14.9%). It appears that our modelling was appropriate when considering the results of patient characteristics in the current study. Given our Cox regression analysis, we concluded that the extent of fibrosis was an independent and strong prognostic factor for patients with CPFE.
Our subgroup analysis comparing patients with CTD and those without showed that the majority of CTD was rheumatoid arthritis and many others with CPFE had autoantibodies related to rheumatoid arthritis, reinforcing the potential role of cigarette smoking in both diseases [27, 28].
Most previous studies have investigated CPFE within the IPF population rather than investigate the broader CPFE population including those with other fibrotic ILDs, or analysed only patients above a certain threshold of fibrosis extent [5, 29]. To the best of our knowledge, our study included the largest cohort of patients with CPFE with the full spectrum of fibrosis extent encountered in the real-world setting, adding evidence regarding the independent prognostic value of fibrosis quantification.
Although Wiggins et al. [4] first reported combined cryptogenic fibrosing alveolitis and emphysema in eight cigarette smokers in the 1980s, with a unique presentation of severe breathlessness and low gas transfer without apparent airflow obstruction; CPFE remained poorly recognised until the 2000s [5]. In 2010, many studies conducted on CPFE in IPF recognised a poor prognosis due to the high prevalence of pulmonary hypertension and lung cancer [2, 5, 30]; the current study also showed that IPF is associated with poor outcome. Alsumrain et al. reported a similar proportion of IPF to that in our study in their CPFE cohort [10], and the remaining patients were either unclassifiable or diagnosed with secondary ILD using clinico-radiological characteristics. This suggests that careful assessment is needed to identify patients who may benefit from lung biopsy to diagnose UIP patterns or another potentially progressive ILD. We might consider such an intensive approach especially for patients with 5 to < 10% fibrosis who apparently showed radiological and functional progression, yet had less mortality and acute exacerbation than the > 10% fibrosis group. Conversely, patients with CPFE with < 5% showed stable disease behaviour for up to 10 years. We might say that the patients with < 5% do not require monitoring as closely as that required for patients with > 5% fibrosis.
This study has several limitations. First, its single-centre, retrospective design may limit external validity. However, an advantage of our general and tertiary hospital was that over 80% of board-certified pulmonary physicians were within a 50 km radius, making it possible to follow-up patients with multiple assessments over long periods, similar to that in a population-based study. Second, the inclusion and exclusion criteria are debatable. We included any proportion of emphysema and fibrosis. According to the American Thoracic Society/European Respiratory Society 2013 statement of idiopathic interstitial pneumonias (IIP) [29], CPFE was classified as a smoking-related IIP. Thus, there is no global consensus on the definition of CPFE. The number of excluded non-smokers, as the result of current analysis, were only 4. Thus, no impact on the results is expected. In addition, the exclusion criteria referred from the past literature [5] made easy to compare between studies, and it also made possible to exclude the other cystic diseases such as Langerhans cell histiocytosis possibly occurring as the other cystic diseases of smokers. Third, the definition of progression is a matter of debate. Cottinet et al. published a pooled analysis reporting that FVC measurements might be inappropriate for monitoring disease progression in patients with IPF and emphysema extent greater than or equal to 15% [31]. Certainly, our results for CPFE showed discrepancy in survival and progression in patients > 10% fibrosis. However, FVC remains the standard for evaluating disease progression. Further study is necessary to improve the prognostic predictability of CPFE. Finally, CT scans were obtained for clinical reasons and not at regular intervals as part of a prospective study; thus, there may have been more intensive follow-up in participants with more rapid disease progression. Automated quantification of CT data also might be made some difficulty on generalizability.
Conclusion
Patients with < 5% fibrosis showed relatively benign disease behaviour in terms of both overall survival and functional preservation. However, careful follow-up may be required for patients with > 5% fibrosis, and especially when the extent exceeds 10%. Further studies are required to validate the classification of CPFE.
Availability of data and materials
Not applicable.
Abbreviations
- CALIPER:
-
Computer-Aided Lung Informatics for Pathology Evaluation and Rating
- CCI:
-
Charlson Comorbidity Index
- CPFE:
-
Combined pulmonary fibrosis and emphysema
- CT:
-
Computed tomography
- CTD:
-
Connective tissue diseases
- DLco:
-
Diffusing capacity of the lung for carbon monoxide
- FVC:
-
Forced vital capacity
- GGO:
-
Ground glass opacity
- IIP:
-
Idiopathic interstitial pneumonias
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- LAAs:
-
Low attenuation areas
- PFT:
-
Pulmonary function tests
- UIP:
-
Usual interstitial pneumonia
References
Jankowich MD, Rounds SIS. Combined pulmonary fibrosis and emphysema syndrome: a review. Chest. 2012;141:222–31.
Kumar A, Cherian SV, Vassallo R, Yi ES, Ryu JH. Current concepts in pathogenesis, diagnosis, and management of smoking-related interstitial lung diseases. Chest. 2018;154:394–408.
Jiang CG, Fu Q, Zheng CM. Prognosis of combined pulmonary fibrosis and emphysema: comparison with idiopathic pulmonary fibrosis alone. Ther Adv Respir Dis. 2019;13:1753466619888119.
Wiggins J, Strickland B, Turner-Warwick M. Combined cryptogenic fibrosing alveolitis and emphysema: the value of high resolution computed tomography in assessment. Respir Med. 1990;84:365–9.
Cottin V, Nunes H, Brillet PY, Delaval P, Devouassoux G, Tillie-Leblond I, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26:586–93.
von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP, et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Ann Intern Med. 2007;147:573–7.
American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277–304.
Uppaluri R, Hoffman EA, Sonka M, Hunninghake GW, McLennan G. Interstitial lung disease: a quantitative study using the adaptive multiple feature method. Am J Respir Crit Care Med. 1999;159:519–25.
Bartholmai BJ, Raghunath S, Karwoski RA, Moua T, Rajagopalan S, Maldonado F, et al. Quantitative computed tomography imaging of interstitial lung diseases. J Thorac Imaging. 2013;28:298–307.
Alsumrain M, De Giacomi F, Nasim F, Koo CW, Bartholmai BJ, Levin DL, et al. Combined pulmonary fibrosis and emphysema as a clinicoradiologic entity: characterization of presenting lung fibrosis and implications for survival. Respir Med. 2019;146:106–12.
Araki T, Putman RK, Hatabu H, Gao W, Dupuis J, Latourelle JC, et al. Development and progression of interstitial lung abnormalities in the Framingham Heart Study. Am J Respir Crit Care Med. 2016;194:1514–22.
Quan H, Li B, Couris CM, Fushimi K, Graham P, Hider P, et al. Updating and validating the Charlson comorbidity index and score for risk adjustment in hospital discharge abstracts using data from 6 countries. Am J Epidemiol. 2011;173:676–82.
Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. 2014;145:723–8.
Maldonado F, Moua T, Rajagopalan S, Karwoski RA, Raghunath S, Decker PA, et al. Automated quantification of radiological patterns predicts survival in idiopathic pulmonary fibrosis. Eur Respir J. 2014;43:204–12.
Jacob J, Bartholmai BJ, Rajagopalan S, Kokosi M, Maher TM, Nair A, et al. Functional and prognostic effects when emphysema complicates idiopathic pulmonary fibrosis. Eur Respir J. 2017;50:1700379.
Occhipinti M, Paoletti M, Bartholmai BJ, Rajagopalan S, Karwoski RA, Nardi C, et al. Spirometric assessment of emphysema presence and severity as measured by quantitative CT and CT-based radiomics in COPD. Respir Res. 2019;20:101.
du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med. 2011;184:1382–9.
Latsi PI, du Bois RM, Nicholson AG, Colby TV, Bisirtzoglou D, Nikolakopoulou A, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med. 2003;168:531–7.
Hornik K. The Comprehensive R Archive Network. Wiley Interdiscip Rev Comput Stat. 2012;4:394–8.
Kanda Y. Investigation of the freely available easy-to-use software “EZR” for medical statistics. Bone Marrow Transplant. 2013;48:452–8.
Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46:976–87.
Choi SH, Lee HY, Lee KS, Chung MP, Kwon OJ, Han J, et al. The value of CT for disease detection and prognosis determination in combined pulmonary fibrosis and emphysema (CPFE). PLoS ONE. 2014;9(9):e107476.
Araki T, Nishino M, Zazueta OE, Gao W, Dupuis J, Okajima Y, et al. Paraseptal emphysema: prevalence and distribution on CT and association with interstitial lung abnormalities. Eur J Radiol. 2015;84:1413–8.
Kim DS, Collard HR, King TE Jr. Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3:285–92.
Cottin V, Nunes H, Mouthon L, Gamondes D, Lazor R, Hachulla E, et al. Combined pulmonary fibrosis and emphysema syndrome in connective tissue disease. Arthritis Rheum. 2011;63:295–304.
Cottin V. Combined pulmonary fibrosis and emphysema: bad and ugly all the same? Eur Respir J. 2017;50:1700846.
Wang D, Zhang J, Lau J, Wang S, Taneja V, Matteson EL, et al. Mechanisms of lung disease development in rheumatoid arthritis. Nat Rev Rheumatol. 2019;15:581–96.
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19.
Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–48.
Sugino K, Ishida F, Kikuchi N, Hirota N, Sano G, Sato K, et al. Comparison of clinical characteristics and prognostic factors of combined pulmonary fibrosis and emphysema versus idiopathic pulmonary fibrosis alone. Respirology. 2014;19:239–45.
Cottin V, Hansell DM, Sverzellati N, Weycker D, Antoniou KM, Atwood M, et al. Effect of emphysema extent on serial lung function in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196(9):1162–71.
Acknowledgements
We would like to thank Kenjin Nishioka and Hajime Inokuchi (Post Graduate Education Center, Kameda Medical Center, Kamogawa, Japan) for the data collection. We would also like to thank Editage (www.editage.jp) for English language editing.
Funding
This research did not receive any specific grants from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Y.N. participated in the concept and design of the study. B.B. participated in the concept and study design as well as in the manuscript writing. M.N. also participated in the concept and study design, provided study materials/patients, was responsible for the collection and assembly of data, data analysis and interpretation, and contributed to the manuscript writing. J.R. participated in the concept, design of the study, and manuscript writing and is responsible for the administrative support. T.N. and S.M. provided study materials or patients. HM participated in the data analysis and interpretation. KY participated in the data analysis and interpretation as well as in the manuscript writing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The institutional review board of Kameda Medical Center approved the study (ID18-102) and allowed use of the opt-out method, instead of requiring informed consent.
Consent for publication
Not applicable.
Competing interests
Dr. Bartholmai reports royalties from Imbio, LLC, during the conduct of the study. The CALIPER software is licensed to Imbio, LLC and sold as Imbio Lung Texture Analysis (LTA). Royalties are due to Mayo Clinic and Dr. Bartholmai and were paid during this study, but no fees were given to the authors or to Mayo Clinic directly related to the content of the manuscript. Dr. Bartholmai also received personal fees from Promedior, LLC, grants from NIH/NHLBI, personal fees from Imbio, LLC, and Mayo Clinic received fees from Boehringer Ingelheim, outside the submitted work in the last 3 years. In addition, Dr. Bartholmai has a patent systems and methods for analyzing in vivo tissue volumes using medical imaging pending.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Detailed baseline characteristics.
Additional file 2.
Study outcomes in patients with emphysema subgrouping.
Additional file 3.
Baseline characteristics in patients with or without connective tissue disease.
Additional file 4.
Kaplan–Meier curve for overall survival. Line: patients without connective tissue disease. Dash: patients with connective tissue disease.
Additional file 5.
Predictors of mortality by logistic regression analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nemoto, M., Nei, Y., Bartholmai, B. et al. Automated computed tomography quantification of fibrosis predicts prognosis in combined pulmonary fibrosis and emphysema in a real-world setting: a single-centre, retrospective study. Respir Res 21, 275 (2020). https://doi.org/10.1186/s12931-020-01545-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-020-01545-3