
Abstract
Venetoclax has been approved by the United States Food and Drug Administration since 2016 as a monotherapy for treating patients with relapsed/refractory chronic lymphocytic leukemia having 17p deletion. It has led to a breakthrough in the treatment of hematologic malignancies in recent years. However, unfortunately, resistance to venetoclax is inevitable. Multiple studies confirmed that the upregulation of the anti-apoptotic proteins of the B-cell lymphoma 2 (BCL2) family mediated by various mechanisms, such as tumor microenvironment, and the activation of intracellular signaling pathways were the major factors leading to resistance to venetoclax. Therefore, only targeting BCL2 often fails to achieve the expected therapeutic effect. Based on the mechanism of resistance in specific hematologic malignancies, the combination of specific drugs with venetoclax was a clinically optional treatment strategy for overcoming resistance to venetoclax. This study aimed to summarize the possible resistance mechanisms of various hematologic tumors to venetoclax and the corresponding clinical strategies to overcome resistance to venetoclax in hematologic malignancies.
Similar content being viewed by others

Background
Apoptosis is a form of cell death, which is important in the development of the body, immune responses, and homeostasis. Apoptosis inhibition is also one of the characteristics of tumors, and the induction of apoptosis has become an important strategy for tumor therapy. The apoptotic pathway is regulated by the extrinsic and intrinsic pathways [1, 2]. The intrinsic apoptotic pathway is controlled by B-cell lymphoma-2 (BCL2) family proteins that can regulate the permeability of the outer mitochondrial membrane through protein interactions [3, 4]. The expression of BCL2 family proteins is usually dysregulated in hematologic malignancies [5]. A variety of tumor cells increased the expression of BCL2 family proteins through multiple mechanisms to ensure cell survival and proliferation, including chromosomal translocation, gene amplification, and downregulation/deletion of microRNAs that degrade BCL2 RNA [6, 7]. For example, BCL2 is usually overexpressed in multiple myeloma (MM) cells with t(11;14) [8]. The deletion of tumor suppressor genes microRNA-15 (miR-15) and microRNA-16 (miR-16) located in the 13q14 chromosome can also lead to increased expression of BCL2 [9, 10]. Therefore, targeting BCL2 can be used as one of the treatment strategies for tumors with high expression of BCL2.
Venetoclax, a BH3-mimetic, is a novel, oral, highly selective BCL2 inhibitor with high affinity for the BH3-binding groove of BCL2 [11]. It overcomes the apoptosis resistance and cell proliferation caused by the high expression of BCL2 in tumor cells. Previous clinical studies on venetoclax dose escalation showed 79% overall response rate (ORR) and 20% complete response rate (CR) in patients with chronic lymphocytic leukemia (CLL) having 17p deletion who were resistant to conventional chemotherapy. Within the dose range of 400–1200 mg, patients had similar treatment response rates and progression-free survival rate after 15 months [12]. In April 2016, venetoclax was approved by the United States Food and Drug Administration (FDA) as a monotherapy for treating patients with relapsed and refractory (RR) CLL having 17p deletion [12]. In addition, venetoclax has also shown significant activity in many hematologic malignancies, such as acute myeloid leukemia (AML), non-Hodgkin's lymphoma (NHL), MM, and so forth, not just in CLL [12,13,14,15]. Clinical studies showed venetoclax to be effective in patients with NHL and MM, with the efficacy of monotherapy ranging from 10 to 50% in patients resistant or intolerant to conventional chemotherapy or immunochemotherapy [8, 13]. In patients with AML, venetoclax monotherapy could benefit patients with refractory/recurrence or who were not suitable for standard chemotherapy-induced therapy; the efficiency of venetoclax could be further improved by combining with hypomethylating agents (HMAs) or low-dose cytarabine (LDAC) [16,17,18].
Nevertheless, venetoclax could still cause therapeutic resistance. The overexpression of the other anti-apoptotic proteins in the BCL2 family, such as myeloid cell lymphoma-1 (MCL1) and BCL2 like 1 (BCL2L1, also known as BCL-XL), mediated by various mechanisms has been experimentally demonstrated to lead to resistance to venetoclax [17, 19]. Therefore, some studies suggested that the ratio of BCL2 family protein expression rather than BCL2 expression could predict the efficacy of venetoclax [5]. According to the mechanism of venetoclax resistance in hematologic malignancies, drugs with different mechanisms are selected in hopes of overcoming the resistance to venetoclax, which can be used in combination with venetoclax. Common combination drugs include small-molecule kinase inhibitors (ibrutinib, sunitinib, idelalisib, and so forth), HAMs (azacytidine and decitabine), anti-CD20 monoclonal antibodies (rituximab and obinutuzumab), and highly selective MCL1 and BCL-XL inhibitors. This study aimed to summarize the possible mechanisms of resistance to venetoclax in a variety of hematologic malignancies and combination strategies for overcoming resistance to venetoclax in recent years.
Resistance mechanisms of venetoclax
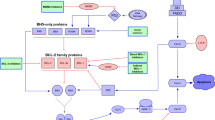
The regulation of apoptosis by BCL2 family proteins is achieved by the balance between pro-apoptotic proteins and anti-apoptotic proteins [4, 20]. The anti-apoptotic proteins include BCL2, BCL-XL, MCL1, BCL-w, and so forth. The pro-apoptotic proteins can be further divided into two subtypes according to their structure: the multidomain proteins, such as BAX and BAK; and the BH3-only proteins, such as BID, BIK, NOXA, PUMA, BAD, BIM (also known as BCL-XL1), and so forth [7]. The BH3-only protein is an apoptosis activator that can initiate cell apoptosis by inhibiting the anti-apoptotic proteins or directly activating the pro-apoptotic proteins [7]. BCL2 and its anti-apoptotic family members can bind to and inactivate BAX or BAK, or directly bind to BH3-only proteins to block apoptosis, so as to maintain cell survival and proliferation [21]. Under the influence of the death signal, BAX or BAK dissociates from BCL2, and the activated free BAX/BAK undergoes oligomerization and forms holes in the outer mitochondrial membrane, leading to the entry of cytochrome c and the other pro-apoptotic molecules in the mitochondria into the cytoplasm, and then activating the caspases, leading to the occurrence of apoptosis [20,21,22,23]. On the contrary, the overexpression of anti-apoptotic proteins MCL1 and BCL-XL blocks the activation of BAX/BAK by binding to BH3-only proteins, thereby blocking cell apoptosis. Several previous studies showed that the ratio of the expression levels of anti-apoptotic proteins and pro-apoptotic proteins often indicated sensitivity to venetoclax [5, 24, 25]. Therefore, the main molecular mechanism of venetoclax resistance is dysregulating of BCL2 family proteins and other non-BCL2 family proteins [20, 26, 27] caused by different reasons. This review aimed to summarize the mechanisms according to the biological processes and the combination therapy strategies to improve the efficacy of venetoclax. Figure 1 depicts potential mechanisms of venetoclax resistance.

Potential mechanisms of venetoclax resistance. Ventoclax recognizes the BH3-biding groove of BCL2 and eases BAK/BAX complex formation, releasing cytochrome c from mitochondria and promote tumor cell apoptosis. Mutation of BCL2 changes protein conformation and impedes venetoclax binding to its target thus against its pro-apoptic effect. Also, genetic alterations such as mutation of TP53 and amplification of 1q23 combined with running out of ATP on mitochondria membrane lead AMPK/PKA pathway aberrantly activating, diminishing the permeability of mitochondria membrane and inducing venetoclax resistance. Interact with non-tumor cells in surrounding microenvironment though membrane molecules activate multiple signaling pathways including NF-κB and PI3K/AKT, which upregulating the anti-apoptic proteins and relseasing varity of inflammatory cytokines. Gene mutation and immune phenotype alteration promote clonal evolution, dysregulate cancer signaling pathways activation and proteins expression, finally lead to venetoclax resistance
Mutation of target proteins
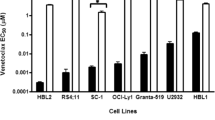
Mutation of the target proteins is pivotal in the failed treatment of cancers, which may change the binding sites of protein inhibitors and impeding functions of the affected proteins [28]. The BH3-binding groove of BCL2 protein is a venetoclax-binding site, whose mutations lead to the change in protein conformation, impeding venetoclax binding to BCL2 or downregulating the binding affinity, thereby inducing venetoclax resistance [26, 29,30,31]. After long-term in vitro induction, lymphoma cell lines LyBCL2-6, LyBCL2-9, and SC-1 were resistant to venetoclax. Sequencing of the BCL2 family protein gene showed that some missense mutations (F101C, F101L, or F104L) in the same codon within the BH3 domain of BCL2 were detected in the resistant cells, which were different from their parents’ sensitive cell lines [29, 30]. In patients with venetoclax-resistant CLL, whole-exome sequencing revealed acquiring variants of BCL2-G101V and BCL2-D103Y at different time points [26, 32]. Another advanced study showed BCL2-G101 mutation in venetoclax-resistant patients and confirmed it as a de novo acquisition mutation compared with pre-treatment samples [31, 33]. However, the frequency of mutation was relatively as low as 1%, implying other mechanisms of clonal shifts and resistance to venetoclax [33].
Clonal evolution
Numerous cytogenetic and molecular genetic abnormalities in tumor cells often lead to resistance to venetoclax, although part of the pathological mechanism is still unclear. Previous studies on patients with AML treated with venetoclax combined with HMAs or LDAC showed that patients with nucleophosmin1 (NPM1) or isocitrate dehydrogenase 2 (IDH2) mutations had better responsiveness to venetoclax. Especially, patients with NPM1 mutations often have longer molecular remission time. In addition, in patients with fms-like tyrosine kinase 3 (FLT3) internal tandem duplication gain, the clonal activation of the RAS pathway or tumor protein 53 (TP53) biallelically perturbation suggests resistance to venetoclax combined with HMAs or LDAC [34]. The upregulation of BCL2 anti-apoptotic gene expression caused by the activation of signaling pathways [mitogen-activated protein kinase (MAPK), extracellular-signal-regulated kinase (ERK), or phosphatidylinositol 3-kinase (PI3K)/AKT] in cells may be one of the mechanisms of drug resistance [35, 36]. Among patients with MCL treated with venetoclax combined with ibrutinib, patients with SWI-SNF chromatin-remodeling complex often have primary drug resistance or early recurrence. SWI-SNF complex-induced BCL-XL expression may be a mechanism of drug resistance [37]. A consensus has been formed regarding the existence of many subclones of tumor cells, and clonal evolution inevitably occurs during the development of the disease. Genetic abnormalities can appear in the early stages of the disease or during relapse and drug resistance.
Clonal evolutions are the final results of genome alterations. Ongoing clonal evolution of CLL cells under venetoclax treatment pressure is a crucial step in drug resistance. Hence, deciphering the detailed mechanisms can help gain an insight into the failure of venetoclax therapy and disease progression [38, 39]. Whole-exome sequencing and methylation profiles were used with the primary CLL samples collected from patients before venetoclax therapy to reveal the genome alterations and clonal shifts. The baseline of genome alteration was about 25.5 mutations before venetoclax treatment [38]. After exposure to venetoclax, nearly 12.5% of the genome changed when drug resistance occurred, indicating the CLL clonal evolution.
The patterns of clonal evolution are heterogeneous, including linear, convergent, and divergent. Somatic genome alterations showed abundant mutations and recurrent alterations. Genome focal amplification related to programmed death ligand 1 (PD-L1) expression was observed when patients developed venetoclax resistance, the site of amplification also including CD274-encoding region, which, after translation, induced a prominent infiltration of CD3-positive lymphocytes, indicating that the new subclone might be susceptible to immune therapy [38, 40]. Further, 3.8% of patients with venetoclax resistance acquired v-raf murine sarcoma viral oncogene homolog B1 (BRAF) mutation. BRAF mutation has been confirmed as oncogenic. Inducing mutant BRAF expression in CLL cell line OCY-LY19 upregulated MCL1 protein synthesis and increased the IC50 of venetoclax, indicating that it might be a driver mutation of CLL clonal evolution [38, 41].
A recent study reported that CLL clonal shifts drove the drug resistance, and the BCL2 family took part in the pathogenesis. Amplification of BCL2, MCL1, and BCL-XL in CLL when venetoclax therapy failed, and the overexpression of these genes in CLL cell line increased the IC50 of venetoclax [39]. In MCL, clonal evolution also drove drug resistance [42]. In patients resistant to ibrutinib, a combination of ibrutinib with venetoclax showed a synergistic effect; however, drug resistance was inevitable. Whole-exome sequencing showed SMARCA4 and KMT2C/D mutations as new genome alterations at progression. Harboring these two gene mutations might initiate de novo clone formation and venetoclax resistance [42].
Tumor microenvironment
Chemo-resistance mediated by the tumor microenvironment is another major problem in cancer treatment [25, 43,44,45]. The lymph nodes and bone marrow are the main shelters of hematologic malignant cells, where a variety of cell components activate the signaling pathways on tumor cells and promote cancer progression [25, 43]. The activation of multiple signaling pathways in tumor cells that promote cell proliferation and survival, such as the activation of B-cell receptor (BCR) and its downstream pathway signaling molecules [45, 46] and the activation of the ERK pathway caused by the activation of extracellular receptors [36], can induce the upregulation of BCL2 family anti-apoptotic proteins, which is one of the mechanisms of venetoclax resistance caused by the microenvironment. Therefore, this may be the intracellular molecular mechanism of small-molecule inhibitors of various kinases combined with venetoclax in the treatment of hematologic tumors.
Activated T cells in the microenvironment can produce cytokines such as IL-4 and IL-21, which can stimulate CD40 on lymphoma cells and increase the expression of MCL1, BCL-XL, and BFL1. On co-culturing CLL with control 3T3 cells (transfected with empty vector) or 3T40L cells (CD-40 ligand-transfected NIH3T3 cells) in the presence of IL-4 or IL-21, CLL showed resistance to venetoclax compared with the control 3T3. In addition, knocking down BCL-XL in 3T40L cells could reverse the resistance phenomenon of venetoclax, thus confirming the role of BCL-XL in venetoclax treatment failure. Furthermore, on combining venetoclax with dasatinib, CD40-mediated venetoclax resistance was perturbed, indicating the synergistic effect of venetoclax and dasatinib [44].
In CLL, human bone marrow mesenchymal stem cells (HBMSCs) released extracellular vesicles to induce venetoclax by regulating gene expression profiles [47]. Co-culturing CLL cells with the supernatant of BMSCs could protect tumor cells from venetoclax-induced apoptosis [47]. A previous study reported that venetoclax and ibrutinib increased the expression of cleaved Poly (ADP-Ribose) Polymerase 1 (PARP1) and induced cell apoptosis synergistically [43]. However, in the presence of HBMSCs or microenvironment cytokines such as IL-10, CD40L, and CPG-ODN, the synergistic effect was reversed and drug resistance developed. Further molecular mechanisms were revealed; the activation of the NF-κB signaling pathway upregulated the expression of the BCL2 family members, which was proved to be the cornerstone of venetoclax resistance [48].
Dysregulation of mitochondrial energy metabolism
Dysregulation of mitochondrial energy metabolism also takes part in venetoclax resistance. In CLL cell line OCI-Ly1 with amplification of 1q23, AMPK/PKA pathway was aberrantly activated, which perturbed cytochrome c release and finally led to venetoclax resistance [39]. Also, running out of ATP in the inner layer of mitochondrion could stimulate the AMPK/PKA pathway, showing a synergistic effect with the amplification of 1q23 [39]. Using a genome-wide CRISPR knockout screen, Sharon et al. [49] found that gene inactivation involving mitochondrial translation sensitized AML cells with drug resistance to venetoclax. The inhibition of mitochondrial respiration by inhibiting the electron transport chain (ETC) complex 1 is one of the mechanisms by which venetoclax kills AML cells, suggesting that mitochondrial energy metabolism disorders are involved in the resistance of venetoclax. The combined use of drugs, such as tedizolid, which target the mitochondrial respiratory chain through different mechanisms, can further enhance the anti-AML effect of venetoclax combined with azacitidine in vivo and in vitro [49, 50]. In leukemic stem cells, mutant TP53 perturbed mitochondrial homeostasis by dysregulating transcription factor DP-1 (TFDP1) activation and translocation of phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1) into the mitochondria, impairing the effector function of BAX/BAK [51]. Moreover, TP53 mutation also impedes BCL2 expression, decreasing the target of venetoclax directly and leading to drug resistance [51].
Combination strategies
In view of the drug resistance mechanisms of venetoclax, researchers have designed a variety of combination treatment strategies to improve the clinical efficacy of venetoclax. In this section, the clinical research results of combined treatment in a variety of hematologic malignancies have been summarized and are also listed in Table 1. The ongoing clinical trials are listed in Table 2.
Acute myeloid leukemia
AML is a disease caused by the blocked differentiation of myeloid hematopoietic stem cells and the clonal proliferation of primitive or immature myeloid cells in the bone marrow. A phase II trial confirmed that the ORR of venetoclax monotherapy was only 19% (6/32) in patients with RR AML or previously untreated AML who were not suitable for intensive chemotherapy [15]. However, patients with AML having IDH1/2 mutations may have better efficacy, with complete remission or complete remission with incomplete count recovery (CR/CRi) of 33% [52].
Previous studies showed that the efficacy of venetoclax clearly improved when combined with drugs that downregulated MCL1 and/or BCL-XL. HMAs and cytarabine can downregulate the expression of MCL1, and may exert a synergistic effect with venetoclax to interfere with the energy metabolism of AML stem cells and kill tumor cell [50, 53, 54]. A phase Ib/II trial investigated the efficacy of venetoclax in combination with LDAC in 82 elderly patients with AML who were not eligible for intensive chemotherapy. The CR/CRi was 54%, and the patients with de novo AML, intermediate-risk cytogenetic features, and without prior HMA exposure had the highest rates of CR/CRi (71%, 63%, and 62%, respectively). The median overall survival (OS) for all patients was 10.1 months, and the median duration of remission (DOR) was 8.1 months. Bone marrow suppression was the most common adverse event (AE) [17] (Table 1). In a phase Ib study, 57 patients with newly diagnosed AML who were not suitable for intensive chemotherapy were randomly divided into three groups. Group A received combined treatment with venetoclax and decitabine, and group B received combined treatment with venetoclax and azacitidine; the total ORR of the two groups was 62%, and the CR/CRi was 60%. The ORR of groups A and B was 65% and 59%, respectively. The median DOR of the two groups was 11.0 months, and the median OS was 15.2 months. Hematologic toxicity was the most common adverse reaction [55] (Table 1). In a phase Ib trial, the efficacy of venetoclax in combination with decitabine or azacitidine was evaluated in 145 treatment-naïve AML ineligible for intensive chemotherapy; 49% of the patients had cytogenetic abnormalities with poor prognosis. After a median of 8.9 months of treatment, 67% of the patients achieved CR/CRi; among the patients with CR/CRi, 29% of patients were minimal residual disease (MRD) negative. The median OS was 17.5 months, and the estimated 2-year OS was 46%. Hematological and gastrointestinal AEs were the most common toxicities observed [16] (Table 1).
Small-molecule kinase inhibitors can downregulate the expression of BCL2 family apoptosis inhibitors through the regulation of cell signaling pathways [35, 36]. Cobimetinib (GDC-0973), an allosteric MEK inhibitor, was demonstrated to have synergistic anti-leukemic effect with venetoclax in vitro by inhibiting the proliferation of AML cell lines and primary AML cells and reducing the leukemic burden in xenograft mouse models [35]. A phase I clinical trial of the combination of venetoclax and cobimetinib is currently being conducted on patients with RR AML (NCT02670044) (Table 2). In patients with AML having FLT3 mutations, the corresponding kinase inhibitors can also be used in combination [20]. For example, sorafenib inhibits FLT3 and downregulates MCL1 expression. It can synergize anti-AML with venetoclax. However, clinical trials are still needed to confirm its efficacy [56, 57]. The safety and scientific validity of venetoclax in combination with FLT3 inhibitors gilteritinib (NCT03625505) and quizartinib (NCT03735875) are currently being tested in RR AML with FLT3 mutation (Table 2).
The TP53 gene is an important tumor suppressor gene that regulates apoptosis and cyclin expression. The mutations or deletions of the TP53 gene are associated with the occurrence and development of a variety of tumors, which can downregulate the expression of BCL2 family pro-apoptotic proteins in AML cells, leading to the inactivation of the mitochondrial apoptotic pathway [8, 58]. Murine double minute homolog 2 (MDM2) is the most important negative regulator of TP53. The combination of MDM2 inhibitor idasanutlin and venetoclax may significantly inhibit the proliferation and induce apoptosis of wild-type AML cells (OCI-AML3) in vitro and in vivo. The cell line has high expression of MCL1 and is resistant to both idasanutlin and venetoclax [59]. A phase I clinical trial is currently testing the tolerability and safety of venetoclax in combination with cobimetinib or idasanutlin in patients with RR AML who are not suitable for cytotoxic therapy (NCT02670044) (Table 2).
A direct and effective treatment strategy to overcome venetoclax resistance is to combine it with specific MCL1 inhibitors [23, 60, 61]. A-1210477 is the first high-affinity, selective MCL1 inhibitor. It can synergistically inhibit the proliferation of BCL2/MCL1-dependent AML cell lines and induce apoptosis when combined with venetoclax [61]. VU661013, a novel, potent, selective MCL1 inhibitor, which destabilizes the BIM/MCL1 association, leads to apoptosis in AML and is active in venetoclax-resistant cells and patient-derived xenografts. It can synergize with venetoclax to kill AML cells [23]. S63845 is a selective MCL1 inhibitor that can selectively bind to the BH3-binding groove of MCL1, thereby effectively killing MCL1-dependent tumor cells, including MM, leukemia, and lymphoma cells [60]. A phase Ib clinical trial (NCT03672695) combining venetoclax with the selective MCL1 inhibitor S64315 is currently underway on patients with AML (Table 2).
Acute lymphoblastic leukemia
Acute lymphoblastic leukemia (ALL) is a malignant neoplastic disease in which lymphocytic B or T cells proliferate abnormally in the bone marrow, and can also invade extramedullary tissues. In vitro studies indicated that MCL1-specific inhibitor S63845 and venetoclax could synergistically suppress T-ALL cells, but the two drugs had no independent killing effect on T-ALL cells, which might be due to the high expression of MCL1 and BCL2 in T-ALL cells [62]. Animal experiments on ALL cell xenotransplantation confirmed the killing effect of venetoclax on ALL cells, especially in patients with mixed-lineage leukemia (MLL)-rearranged leukemia. However, in ALL cells, the expression level of BCL-XL largely determines the sensitivity of ALL cells to venetoclax [63]. Animal experiments on ALL cell xenotransplantation confirmed the killing effect of venetoclax on ALL cells, especially in patients with mixed-lineage leukemia (MLL)-rearranged leukemia. However, in ALL cells, the expression level of BCL-XL largely determines the sensitivity of ALL cells to venetoclax [64]. In Philadelphia chromosome-positive ALL, tyrosine kinase inhibitor (dasatinib and ponatinib) can upregulate BIM and inhibit the expression of MCL1, thereby cooperating with venetoclax to inhibit ALL cells [65]. However, the aforementioned conclusions still need to be carefully verified in patients.
Chronic lymphocytic leukemia
CLL is a disease involving the clonal proliferation of mature B lymphocytes. Previous studies showed that almost all CLL cells overexpressed BCL2 [66]. An exploratory clinical study showed that venetoclax monotherapy had a certain therapeutic effect in patients with CLL, with an ORR of 80% and a CR of 40% in patients with newly diagnosed CLL [67]. Among patients with high-risk RR CLL, the CR/CRi was only 8%, although an ORR of 79% was achieved [11], suggesting that some CLL cells were resistant to venetoclax. Previous studies showed that the combination of anti-CD20 monoclonal antibodies (rituximab and obinutuzumab) and some kinase inhibitors, such as the combined use of TKIs, spleen tyrosine kinase inhibitors, BTK inhibitors, and phosphatidylinositol 3-kinase (PI3K) inhibitors, can overcome resistance to venetoclax, which can effectively eliminate the upregulation of anti-apoptotic genes in the BCL2 family, such as MCL1, BCL-XL, and BFL-1/A1, caused by the activation of microenvironment-mediated and intracellular signaling pathways, thereby overcoming venetoclax resistance [44,45,46, 68,69,70,71].
Venetoclax and the BTK inhibitor ibrutinib have a synergistic effect [69, 70]. Ibrutinib-mediated BTK inhibition can lead to a decrease in MCL1 protein expression, while it increases or has no effect on BCL2 levels. In addition, ibrutinib can effectively mobilize CLL cells from the microenvironment that provides growth signals, avoiding venetoclax resistance mediated by the tumor microenvironment [44, 71]. A phase II study explored the effect of ibrutinib combined with venetoclax, involving previously untreated high-risk and older patients with CLL. After 18 cycles, CR/CRi was as high as 96%, and 69% of patients had remission with undetectable MRD in the bone marrow. The estimated 1-year PFS and OS were 98% and 99%, respectively [72] (Table 1), suggesting that venetoclax combined with ibrutinib had a better effect in patients with high-risk and early-stage CLL.
Venetoclax in combination with an anti-CD20 monoclonal antibody has been shown to overcome microenvironment-mediated resistance to venetoclax [44]. Rituximab can reduce the expression of MCL1 protein and increase the sensitivity of CLL cells to venetoclax-induced apoptosis [73]. In a phase I study, venetoclax in combination with rituximab was administered to patients with RR CLL. Overall, 86% of the patients achieved ORR, including 51% with CR/CRi. Negative MRD in the bone marrow was achieved in 80% of CR/CRi responders and 57% of all patients. The estimated 2-year PFS and OS were 82% and 89%, respectively. AEs appeared to be similar to venetoclax in monotherapy [74] (Table 1). Another phase III MURANO trial compared the efficacy of venetoclax combined with rituximab (194) and bendamustine combined with rituximab (195) in patients with RR CLL. The results suggested that the ORR was 92.3% and CR/CRi was 26.8% in the venetoclax group, which were significantly higher than those (72.3% and 8.2%, respectively) in the bendamustine group. The evaluable peripheral blood MRD-negative rate in the venetoclax and bendamustine groups was 62.4% and 13.3%, respectively. In addition, the 2-year PFS rate (84.9%) and the 2-year OS rate (97.9%) were also significantly higher in the venetoclax group than in the bendamustine group. Even in the high-risk group of del17p patients, venetoclax had obvious advantages, and the 2-year PFS was 81.5% and 27.8%, respectively [75] (Table 1), suggesting that the combination of venetoclax and rituximab was effective in patients with RR CLL.
Obinutuzumab is an artificial, glycosylated type II anti-CD20 monoclonal antibody with excellent single-agent activity in CLL [76, 77],, and its efficacy is superior to that of rituximab [78]. In a phase Ib trial, venetoclax combined with obinutuzumab was administered for six cycles, followed by venetoclax monotherapy until disease progression (R/R) or two-drug combination treatment was administered for a fixed duration of 1 year (1L). Overall, ORR was 95% and 100% in R/R and 1L patients, respectively, and 37% of the R/R patients and 78% of the 1L patients achieved CR/CRi. The rate of undetectable MRD in the bone marrow for R/R and 1L patients was 64% and 78%, respectively. The estimated 24-month PFS was 85.4% and 90.6% in R/R and 1L patients, and the median DOR was 40.9 months and not reached, respectively. The most common grade 3–4 AE was neutropenia [79]. This experiment confirmed that the combination of venetoclax and obinutuzumab was effective and had acceptable therapeutic toxicity.
The combination of venetoclax with kinase inhibitors and anti-CD20 monoclonal antibodies may lead to better outcomes. A phase Ib trial investigated the efficacy and safety of the combined use of venetoclax, ibrutinib, and obinutuzumab in patients with RR CLL. Despite only 12 patients with RR CLL, all patients completed 14 cycles of treatment. ORR was up to 92% in the early evaluation, with 42% of the patients achieving CR/CRi. All patients achieved MRD undetectable in either the blood or the bone marrow, with 50% of the patients achieving MRD undetectable in both the blood and the bone marrow. The estimated 24-month PFS was 92%. Hematologic toxicity was the most frequent AE [76] (Table 1).
Diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma (DLBCL) is the most common aggressive NHL [80]. Rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone (R-CHOP) is the first-line immunochemotherapeutic regimen for patients with newly diagnosed DLBCL. Although its CR could reach 76%, still 20–40% of patients failed to treatment or relapsed [81, 82] no effective regimen was recommended for these patients. In phase I clinical trial, venetoclax monotherapy was administered to patients with RR DLBCL. The ORR was only 18%, the CR was only 12%, and the estimated median PFS was only 1 month [13], suggesting that venetoclax monotherapy had limited activity and short-term effects in DLBCL. This might be related to the activation of multiple signaling pathways, such as Akt, in DLBCL tumor cells, leading to the overexpression of BCL2 family anti-apoptotic proteins [83,84,85]. Previous studies showed that a variety of kinase inhibitors, such as BTK inhibitors (ibrutinib), SYK inhibitors (R406), PI3K/mTOR dual inhibitors (NVP-BEZ235), PI3K inhibitors (idelalisib, copanlisib, ACP-319, and KA2237) or ATK inhibitors (MK-2206), and CDK9 inhibitor (dinaciclib) [86, 87], alone or in combination with venetoclax, significantly reduced the expression of anti-apoptotic proteins BCL2 tumor cells in vitro and then exerted synergistic killing effects on lymphoma cells. This conclusion was further confirmed in patient-derived xenograft (PDX) animal models [83, 86, 88]. Some of them are undergoing clinical trials to evaluate the effect of combined application with venetoclax.
Homoharringtonine (HHT) is a commonly used anti-leukemia drug with multiple mechanisms of action, including the downregulation of MCL1 expression [89]. Previous studies showed that HHT combined with venetoclax in DLBCL cell lines and mouse models of xenografts produced synergistic effects, suggesting that this combination might be a promising combination therapy [90]. However, corresponding clinical application data are still lacking.
Preclinical studies confirmed that venetoclax in combination with bendamustine and rituximab (BR) could exhibit synergistic effects. A phase Ib clinical trial enrolled 60 patients with RR NHL [32 with follicular lymphoma (FL), 22 with DLBCL, 6 with MZL] receiving venetoclax and BR combination therapy. After the median follow-up of 7.5 months, the ORR of all patients was 65% and the CR was 30%. Among them, the ORR of patients with DLBCL was 41% and the CR was 14%. Despite having a slightly lower effect in patients with FL and MZL, the combination was better than venetoclax as a single agent [91] (Table 1). Previous studies confirmed that the R-CHOP treatment regimen was less beneficial against BCL2-positive DLBCL [92]. Therefore, the use of BCL2 inhibitors in combination with R-CHOP in such patients may have beneficial effects. A phase Ib/II CAVALLI rial enrolled 56 patients with newly diagnosed DLBCL (including 43% FL and 32% DLBCL) receiving venetoclax in combination with R/G-CHOP therapy. The total ORR was 87.5%, of which the ORR of patients with DLBCL was 88.9%. These patients achieved CR, and the CR of patients with DLBCL having a dual expression of BCL2 and MYC was 87.5% [93] (Table 1).
Follicular lymphoma
FL is one of the most common indolent lymphomas. Studies showed that 85–90% of patients with FL had a t(14;18) translocation, resulting in the overexpression of BCL2 [94, 95]. However, in a phase I clinical trial, the ORR of venetoclax monotherapy in patients with RR FL was 38%, and the CR was only 14%. The estimated median PFS was 11 months [13]. Therefore, even if FL is a disease characterized by high expression of BCL2, the effect of venetoclax monotherapy is still limited. The overexpression of BCL2 family anti-apoptotic proteins caused by the activation of multiple signaling pathways, such as JNK, AKT and ERK1/2 pathways in tumor cells, is the main mechanism of drug resistance [24]. Therefore, a combination with specific ERK inhibitors, pan-PI3K inhibitors, rituximab, and so forth, could significantly enhance venetoclax-induced FL cell apoptosis and overcome venetoclax resistance [24].
Preclinical studies confirmed that venetoclax combined with BR could lead to 100% complete tumor regression in NHL xenograft models with t(14;18) translocation [91]. In a phase Ib clinical trial, the use of venetoclax in combination with BR was investigated in patients with RR FL. After a 7.5-month follow-up period, the ORR was 75%, the CR was 38%, and the PR was 38%, which was significantly higher than that in DLBCL [91] (Table 1). In the aforementioned CAVALLI study, 24 patients with FL received venetoclax combined with R/G-CHOP and achieved an ORR of 83.3%; CR was 75%, PR was 8.3%, and the 1-year PFS was more than 90% [93] (Table 1).
Mantle cell lymphoma
MCL is an aggressive small B-cell lymphoma, usually overexpressing BCL-2 [67]. In a phase I clinical trial, patients with RR MCL were treated with venetoclax monotherapy. The ORR was 75% and CR was only 14%; it was one of the best responding groups among patients with NHL [13]. Although venetoclax monotherapy is effective in patients with MCL, resistance to venetoclax is inevitable. Previous studies confirmed that the expression of MCL1 and BCL-XL could determine the sensitivity of MCL cells to venetoclax resistance [25, 96, 97]. Obinutuzumab has been proved to block the expression of BCL-XL by inhibiting NF-κB signaling in vitro, thereby counteracting the protective effect of the microenvironment and overcoming resistance to venetoclax in MCL cell lines [98].
Ibrutinib has been approved by the FDA for treating MCL [99]. It promotes MCL cells to enter the peripheral circulation by inhibiting chemokine and BCR signals and downregulating the expression of MCL1 in MCL cells, thereby overcoming tumor microenvironment-related drug resistance [100, 101]. A phase II clinical study evaluated the efficacy of venetoclax combined with ibrutinib in patients with RR or previously untreated MCL. Half of the patients had abnormal TP53, and 75% of the patients were high-risk patients. After 16 weeks of treatment, PET/CT assessment showed a CR of 67%, and 67% of the patients tested negative for MRD using flow cytometry. The median PFS was not reached, and the 12-month estimated PFS and OS rates were 75% and 79%, respectively. The most common AE was gastrointestinal toxicity [102] The combination of venetoclax with obinutuzumab and ibrutinib might be more effective than other combinations. Phase I/II trials are underway to assess the efficacy and safety of this combination in patients with RR MCL (NCT02558816) (Table 2).
Previous studies showed that venetoclax combined with the selective MCL1 inhibitor S63845 had a significant synergistic killing effect on MCL cell lines in vitro and induced long-term lymphoma-free survival in MCL xenograft models [103]. Therefore, venetoclax in combination with highly selective MCL1 inhibitors might be a promising treatment option in the future.
Multiple myeloma
MM is a malignant proliferative disease of plasma cells. Previous studies showed that MM cells depended on the anti-apoptotic proteins BCL2, MCL1, and BCL-XL to survive [104, 105]. Venetoclax therapy is considered to be limited to subtypes of t(11;14) [14]. In a phase I clinical trial, venetoclax monotherapy was administered to patients with RR MM; the ORR was 21%, with 15% reaching VGPR or better. In the subset of patients with t(11;14) translocation, the ORR was 40%, and 27% achieved VGPR or better [14, 106].
Venetoclax monotherapy has limited efficacy in patients with MM. The overexpression of MCL1 is one of the important reasons for the inherent resistance of MM cells to venetoclax [105]. Therefore, the inhibition of MCL1 could increase the sensitivity of MM cells to venetoclax [107]. The proteasome inhibitor bortezomib can inhibit MCL1 indirectly by stabilizing the MCL1-neutralizing protein NOXA and overcoming the resistance of MM cells to venetoclax in a xenograft model [108]. Dexamethasone can upregulate the expression of BIM to increase the dependence of MM cells on BCL2, thereby increasing the sensitivity to venetoclax [109]. phase I clinical trial showed that the combined treatment of venetoclax and dexamethasone for patients with RR MM resulted in a significantly higher ORR (65%) compared with venetoclax monotherapy (40%) [7]. A phase Ib trial studied the efficacy of venetoclax in combination with bortezomib and dexamethasone in patients with RR MM. The ORR was 67%, with 42% achieving VGPR or better. In patients with and without t(11;14) translocation, the ORR rate was 78% and 65%, respectively. The ORR in patients with high and low expression of BCL2 was 94% and 59%, respectively. The median time to progression and DOR were 9.5 months and 9.7 months, respectively. The common AEs included mild gastrointestinal toxicities and grade 3/4 cytopenias [110] (Table 1), indicating that this three-drug combination regime had a good effect and mild adverse reactions.
CDK9 inhibitors, such as flavopiridol [111], seliciclib [112], and so forth, can also downregulate the expression of MCL1. Hence, when combined with venetoclax, these inhibitors can also produce a synergistic effect to overcome the resistance of MM to venetoclax. The efficacy of venetoclax in combination with the CDK9 inhibitors dinaciclib (NCT03484520) and alvocidib (NCT03441555) is currently being evaluated in patients with RR AML (Table 2).
Conclusion
Venetoclax showed significant activity in hematologic malignancies, such as CLL, AML, MM, DLBCL, MCL, FL, and so forth. The occurrence and development of malignant tumors often involved multiple signaling pathways. Hematologic malignancies often developed acquired or inherent resistance to venetoclax. The most common mechanism of venetoclax resistance was the overexpression of the BCL2 family anti-apoptotic proteins, such as MCL1 and BCL-XL, for a variety of reasons. Based on this resistance mechanism, various clinical trials have been conducted to overcome resistance to venetoclax in recent years. Venetoclax-based combination regimens are important treatment options for the treatment of hematologic malignancies.
Availability of data and materials
Not applicable.
Abbreviations
- AEs:
-
Adverse events
- ALL:
-
Acute lymphocytic leukemia
- AML:
-
Acute myeloid leukemia
- BCL2:
-
B-cell lymphoma-2
- BCR:
-
B-cell receptor
- BM:
-
Bone marrow
- BRAF:
-
V-raf murine sarcoma viral oncogene homolog B1
- BTK:
-
Bruton’s tyrosine kinase
- CDK:
-
Cyclin-dependent kinase
- CLL:
-
Chronic lymphocytic leukemia
- CR:
-
Complete remission
- CRi:
-
Complete remission with incomplete count recovery
- DLBCL:
-
Diffuse large B-cell lymphoma
- DOR:
-
Duration of remission
- ERK:
-
Extracellular-signal-regulated kinase
- ETC:
-
Electron transport chain
- FDA:
-
Food and Drug Administration
- FL:
-
Follicular lymphoma
- FLT3:
-
FMS-like tyrosine kinase 3
- HBMSCs:
-
Human bone marrow mesenchymal stem cells
- HMAs:
-
Hypomethylating agents
- IDH2:
-
Isocitrate dehydrogenase 2
- LDAC:
-
Low-dose cytarabine
- MAPK:
-
Mitogen-activated protein kinase
- MCL1:
-
Myeloid cell leukemia-1
- MDM2:
-
Murine double minute homolog 2
- MEK:
-
Methyl ethyl ketone
- MLL:
-
Mixed-lineage leukemia
- MM:
-
Multiple myeloma
- MRD:
-
Minimal residual disease
- NHL:
-
Non-Hodgkin's lymphoma
- NPM1:
-
Nucleophosmin1
- ORR:
-
Objective remission rate
- OS:
-
Overall survival
- PD-L1:
-
Programmed death ligand 1
- PDX:
-
Patient-derived xenograft
- PFS:
-
Progression-free survival
- PI3K:
-
Phosphatidylinositide 3-kinase
- PMAIP1:
-
Phorbol-12-myristate-13-acetate-induced protein 1
- RR:
-
Relapsed/refractory
- R-CHOP:
-
Rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone
- SYK:
-
Spleen tyrosine kinase
- TFDP1:
-
Transcription DP-1
- TLS:
-
Tumor lysis syndrome
- TKI:
-
Tyrosine kinase inhibitor
- TP53:
-
Tumor protein 53
- VGPR:
-
Very good partial remission
References
Reed JC. Apoptosis-based therapies. Nat Rev Drug Discov. 2002;1(2):111–21.
Adams JM, Cory S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2017;25(1):27–36.
Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16(2):99–109.
Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7(12):989–1000.
Tessoulin B, Papin A, Gomez-Bougie P, Bellanger C, Amiot M, Pellat-Deceunynck C, et al. BCL2-family dysregulation in B-cell malignancies: from gene expression regulation to a targeted therapy biomarker. Front Oncol. 2018;8:645.
Huang J, Fairbrother W, Reed JC. Therapeutic targeting of Bcl-2 family for treatment of B-cell malignancies. Expert Rev Hematol. 2015;8(3):283–97.
Perini GF, Ribeiro GN, Pinto Neto JV, Campos LT, Hamerschlak N. BCL-2 as therapeutic target for hematological malignancies. J Hematol Oncol. 2018;11(1):65.
Ruefli-Brasse A, Reed JC. Therapeutics targeting Bcl-2 in hematological malignancies. Biochem J. 2017;474(21):3643–57.
Pekarsky Y, Croce CM. Role of miR-15/16 in CLL. Cell Death Differ. 2015;22(1):6–11.
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2006;102(39):13944–21394.
Stilgenbauer S, Eichhorst B, Schetelig J, Coutre S, Seymour JF, Munir T, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17(6):768–78.
Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311–22.
Davids MS, Roberts AW, Seymour JF, Pagel JM, Kahl BS, Wierda WG, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma. J Clin Oncol. 2017;35(8):826–33.
Kumar S, Kaufman JL, Gasparetto C, Mikhael J, Vij R, Pegourie B, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017;130(22):2401–9.
Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106–17.
DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17.
Wei AH, Strickland SA Jr, Hou JZ, Fiedler W, Lin TL, Walter RB, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J Clin Oncol. 2019;37(15):1277–84.
Mei M, Aldoss I, Marcucci G, Pullarkat V. Hypomethylating agents in combination with venetoclax for acute myeloid leukemia: update on clinical trial data and practical considerations for use. Am J Hematol. 2019;94(3):358–62.
Punnoose EA, Leverson JD, Peale F, Boghaert ER, Belmont LD, Tan N, et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol Cancer Ther. 2016;15(5):1132–44.
Bose P, Gandhi V, Konopleva M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma. 2017;58(9):1–17.
Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol. 2008;70:73–91.
Zhang L, Ming L, Yu J. BH3 mimetics to improve cancer therapy; mechanisms and examples. Drug Resist Updates. 2007;10(6):207–17.
Ramsey HE, Fischer MA, Lee T, Gorska AE, Arrate MP, Fuller L, et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 2018;8(12):1566–81.
Bodo J, Zhao X, Durkin L, Souers AJ, Phillips DC, et al. Acquired resistance to venetoclax (ABT-199) in t(14;18) positive lymphoma cells. Oncotarget. 2016;7(43):70000–10.
Chiron D, Dousset C, Brosseau C, Touzeau C, Maïga S, Moreau P, et al. Biological rational for sequential targeting of Bruton tyrosine kinase and Bcl-2 to overcome CD40-induced ABT-199 resistance in mantle cell lymphoma. Oncotarget. 2015;6(11):8750–9.
Birkinshaw RW, Gong JN, Luo CS, Lio D, White CA, Anderson MA, et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat Commun. 2019;10(1):2385.
Agarwal R, Dawson MA, Dreyling M, Tam CS. Understanding resistance mechanisms to BTK and BCL2 inhibitors in mantle cell lymphoma: implications for design of clinical trials. Leuk Lymphoma. 2018;59(12):2769–81.
Tausch E, Close W, Dolnik A, Bloehdorn J, Chyla B, Bullinger L, et al. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica. 2019;9(104):e434–7.
Fresquet V, Rieger M, Carolis C, Garcia-Barchino MJ, Martinez-Climent JA. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood. 2014;123(26):4111–9.
Tahir SK, Smith ML, Hessler P, Rapp LR, Idler KB, Park CH, et al. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer. 2017;17(1):399.
Thangavadivel S, Byrd JC. Gly101Val BCL2 mutation: one step closer to understanding venetoclax resistance in CLL. Cancer Discov. 2019;9(3):320–2.
Blombery P, Anderson MA, Gong JN, Thijssen R, Birkinshaw RW, Thompson ER, et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. 2019;9(3):342–53.
Weiss J, Peifer M, Herling CD, Frenzel LP, Hallek M. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia (comment to Tausch et al.). Haematologica. 2019;104(11):e540.
DiNardo CD, Tiong IS, Quaglieri A, MacRaild S, Loghavi S, Brown FC, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135(11):791–803.
Han L, Zhang Q, Dail M, Shi C, Cavazos A, Ruvolo VR, et al. Concomitant targeting of BCL2 with venetoclax and MAPK signaling with cobimetinib in acute myeloid leukemia models. Haematologica. 2020;105(3):697–707.
Konopleva M, Milella M, Ruvolo P, Watts JC, Ricciardi MR, Korchin B, et al. MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia. 2012;26(4):778–87.
Agarwal R, Chan YC, Tam CS, Hunter T, Vassiliadis D, Teh CE, et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med. 2019;25(1):119–29.
Herling CD, Abedpour N, Weiss J, Schmitt A, Jachimowicz RD, Merkel O, et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat Commun. 2018;9(1):727.
Guieze R, Liu VM, Rosebrock D, Jourdain AA, Hernandez-Sanchez M, Martinez Zurita A, et al. Mitochondrial reprogramming underlies resistance to BCL-2 inhibition in lymphoid malignancies. Cancer Cell. 2019;36(4):369–84.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65.
Bowyer SE, Rao AD, Lyle M, Sandhu S, Long GV, McArthur GA, et al. Activity of trametinib in K601E and L597Q BRAF mutation-positive metastatic melanoma. Melanoma Res. 2014;24(5):504–8.
Zhao S, Kanagal-Shamanna R, Navsaria L, Ok CY, Zhang S, Nomie K, et al. Efficacy of venetoclax in high risk relapsed mantle cell lymphoma (MCL)—outcomes and mutation profile from venetoclax resistant MCL patients. Am J Hematol. 2020;95(6):623–9.
Jayappa KD, Portell CA, Gordon VL, Capaldo BJ, Bekiranov S, Axelrod MJ, et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017;1(14):933–46.
Thijssen R, Slinger E, Weller K, Geest CR, Beaumont T, Oers MHJV, et al. Resistance to ABT-199 induced by microenvironmental signals in chronic lymphocytic leukemia can be counteracted by CD20 antibodies or kinase inhibitors. Haematologica. 2015;100(8):302–6.
Ten Hacken E, Burger JA. Molecular pathways targeting the microenvironment in chronic lymphocytic leukemia focus on the B cell receptor. Clin Cancer Res. 2014;20(3):548–57.
Bojarczuk K, Sasi BK, Gobessi S, Innocenti I, Pozzato G, Laurenti L, et al. BCR signaling inhibitors differ in their ability to overcome Mcl-1-mediated resistance of CLL B cells to ABT-199. Blood. 2016;127(25):3192–201.
Crompot E, Van Damme M, Pieters K, Vermeersch M, Perez-Morga D, Mineur P, et al. Extracellular vesicles of bone marrow stromal cells rescue chronic lymphocytic leukemia B cells from apoptosis, enhance their migration and induce gene expression modifications. Haematologica. 2017;102(9):1594–604.
Asslaber D, Wacht N, Leisch M, Qi Y, Maeding N, Hufnagl C, et al. BIRC3 expression predicts CLL progression and defines treatment sensitivity via enhanced NF-kappaB nuclear translocation. Clin Cancer Res. 2019;25(6):1901–12.
Sharon D, Cathelin S, Mirali S, Di Trani JM, Yanofsky DJ, Keon KA, Rubinstein JL, Schimmer AD, Ketela T, Chan SM. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci Transl Med. 2019;11(516):eaax2863.
Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24(12):1859–66.
Nechiporuk T, Kurtz SE, Nikolova O, Liu T, Jones CL, D’Alessandro A, et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov. 2019;9(7):910–25.
Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21(2):178–84.
Teh TC, Nguyen NY, Moujalled DM, Segal D, Pomilio G, Rijal S, et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia. 2018;32(2):303–12.
Niu X, Zhao J, Ma J, Xie C, Edwards H, Wang G, et al. Binding of released bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res. 2016;22(17):4440–51.
DiNardo CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;19(2):216–28.
Rahmani M, Davis EM, Crabtree TR, Habibi JR, Nguyen TK, Dent P, et al. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol. 2007;27(15):5499–513.
Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J Biol Chem. 2005;280(42):35217–27.
Roberts AW, Huang D. Targeting BCL2 with BH3 mimetics: basic science and clinical application of venetoclax in chronic lymphocytic leukemia and related B cell malignancies. Clin Pharmacol Ther. 2017;101(1):89–98.
Lehmann C, Friess T, Birzele F, Kiialainen A, Dangl M. Superior anti-tumor activity of the MDM2 antagonist idasanutlin and the Bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J Hematol Oncol. 2016;9(1):50.
Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477–82.
Wang Q, Wan J, Zhang W, Hao S. MCL-1 or BCL-xL-dependent resistance to the BCL-2 antagonist (ABT-199) can be overcome by specific inhibitor as single agents and in combination with ABT-199 in acute myeloid leukemia cells. Leuk Lymphoma. 2019;60(9):2170–80.
Li Z, He S, Look AT. The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia. 2019;33:262–6.
Khaw SL, Suryani S, Evans K, Richmond J, Robbins A, Kurmasheva RT, et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood. 2016;128(10):1382–95.
Benito JM, Godfrey L, Kojima K, Hogdal L, Wunderlich M, Geng H, et al. MLL-rearranged acute lymphoblastic leukemias activate BCL-2 through H3K79 methylation and are sensitive to the BCL-2-specific antagonist ABT-199. Cell Rep. 2015;13(12):2715–27.
Leonard JT, Rowley JSJ, Eide CA, Traer E, Hayes-Lattin B, Loriaux M, et al. Targeting BCL-2 and ABL/LYN in Philadelphia chromosome-positive acute lymphoblastic leukemia. Sci Transl Med. 2016;8(354):354ra114.
O’Brien S, Moore JO, Boyd TE, Larratt LM, Skotnicki A, Koziner B, et al. Randomized phase III trial of fludarabine plus cyclophosphamide with or without oblimersen sodium (Bcl-2 antisense) in patients with relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol. 2007;25(9):1114–20.
Mihalyova J, Jelinek T, Growkova K, Hrdinka M, Simicek M, Hajek R. Venetoclax: a new wave in hematooncology. Exp Hematol. 2018;61:10–25.
Oppermann S, Ylanko J, Shi Y, Hariharan S, Oakes CC, Brauer PM, et al. High-content screening identifies kinase inhibitors that overcome venetoclax resistance in activated CLL cells. Blood. 2016;128(7):934–47.
Deng J, Isik E, Fernandes SM, Brown JR, Letai A, Davids MS. Bruton’s tyrosine kinase inhibition increases BCL-2 dependence and enhances sensitivity to venetoclax in chronic lymphocytic leukemia. Leukemia. 2017;31(10):2075–84.
Cervantes-Gomez F, Lamothe B, Woyach JA, Wierda WG, Keating MJ, Balakrishnan K, et al. Pharmacological and protein profiling suggests venetoclax (ABT-199) as optimal partner with ibrutinib in chronic lymphocytic leukemia. Clin Cancer Res. 2015;21(16):3705–15.
Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117(23):6287–96.
Jain N, Keating M, Thompson P, Ferrajoli A, Burger J, Borthakur G, et al. Ibrutinib and venetoclax for first-line treatment of CLL. N Engl J Med. 2019;380(22):2095–103.
Byrd JC, Kitada S, Flinn IW, Aron JL, Pearson M, Lucas D, et al. The mechanism of tumor cell clearance by rituximab in vivo in patients with B-cell chronic lymphocytic leukemia evidence of caspase activation and apoptosis induction. Blood. 2002;99(3):1038–43.
Seymour JF, Ma S, Brander DM, Choi MY, Barrientos J, Davids MS, et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: a phase 1b study. Lancet Oncol. 2017;18(2):230–40.
Seymour JF, Kipps TJ, Eichhorst B, Hillmen P, D’Rozario J, Assouline S, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378(12):1107–20.
Rogers KA, Huang Y, Ruppert AS, Awan FT, Heerema NA, Hoffman C, et al. Phase 1b study of obinutuzumab, ibrutinib, and venetoclax in relapsed and refractory chronic lymphocytic leukemia. Blood. 2018;132(15):1568–72.
Cartron G, de Guibert S, Dilhuydy MS, Morschhauser F, Leblond V, Dupuis J, et al. Obinutuzumab (GA101) in relapsed/refractory chronic lymphocytic leukemia: final data from the phase 1/2 GAUGUIN study. Blood. 2014;124(14):2196–202.
Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370(12):1101–10.
Flinn IW, Gribben JG, Dyer MJS, Wierda W, Maris MB, Furman RR, et al. Phase 1b study of venetoclax-obinutuzumab in previously untreated and relapsed/refractory chronic lymphocytic leukemia. Blood. 2019;133(26):2765–75.
Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(3):503–11.
Vitolo U, Trněný M, Belada D, Burke JM, Carella AM, Chua N, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol. 2017;35(31):3529–37.
Coiffier B, Lepage E, Brière J, Herbrecht R, Tilly H, Bouabdallah R, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235–42.
Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, et al. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015;6:e1593.
Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating antiapoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood. 2008;111(2):846–55.
Zang C, Eucker J, Liu H, Müller A, Possinger K, Scholz CW. Concurrent inhibition of PI3-Kinase and mTOR induces cell death in diffuse large B cell lymphomas, a mechanism involving down regulation of Mcl-1. Cancer Lett. 2013;339(2):288–97.
Pham LV, Huang S, Zhang H, Zhang J, Bell T, Zhou S, et al. Strategic therapeutic targeting to overcome venetoclax resistance in aggressive B-cell lymphomas. Clin Cancer Res. 2018;24(16):3967–80.
Li L, Pongtornpipat P, Tiutan T, Kendrick SL, Park S, Persky DO, et al. Synergistic induction of apoptosis in high-risk DLBCL by BCL2 inhibition with ABT-199 combined with pharmacologic loss of MCL1. Leukemia. 2015;29(8):1702–12.
Sasi BK, Martines C, Xerxa E, Porro F, Kalkan H, Fazio R, et al. Inhibition of SYK or BTK augments venetoclax sensitivity in SHP1-negative/BCL-2-positive diffuse large B-cell lymphoma. Leukemia. 2019;33(10):2416–28.
Chen R, Guo L, Chen Y, Jiang Y, Wierda WG, Plunkett W. Homoharringtonine reduced Mcl-1 expression and induced apoptosis in chronic lymphocytic leukemia. Blood. 2011;117(1):156–64.
Klanova M, Andera L, Brazina J, Svadlenka J, Benesova S, Soukup J, et al. Targeting of BCL2 family proteins with ABT-199 and homoharringtonine reveals BCL2- and MCL1-dependent subgroups of diffuse large B-cell lymphoma. Clin Cancer Res. 2016;22(5):1138–49.
de Vos S, Swinnen LJ, Wang D, Reid E, Fowler N, Cordero J, et al. Venetoclax, bendamustine, and rituximab in patients with relapsed or refractory NHL: a phase Ib dose-finding study. Ann Oncol. 2018;29(9):1932–8.
Iqbal J, Meyer PN, Smith LM, Johnson NA, Vose JM, Greiner TC, et al. BCL2 predicts survival in germinal center B-cell-like diffuse large B-cell lymphoma treated with CHOP-like therapy and rituximab. Clin Cancer Res. 2011;17(24):7785–95.
Zelenetz AD, Salles G, Mason KD, Casulo C, Le Gouill S, Sehn LH, et al. Venetoclax plus R- or G-CHOP in non-Hodgkin lymphoma: results from the CAVALLI phase 1b trial. Blood. 2019;133(18):1964–76.
Freedman A. Follicular lymphoma: 2018 update on diagnosis and management. Am J Hematol. 2018;93(2):296–305.
Hiddemann W, Cheson BD. How we manage follicular lymphoma. Leukemia. 2014;28(7):1388–95.
Burger JA, Ford RJ. The microenvironment in mantle cell lymphoma: cellular and molecular pathways and emerging targeted therapies. Semin Cancer Biol. 2011;21(5):308–12.
Kurtova AV, Tamayo AT, Ford RJ, Burger JA. Mantle cell lymphoma cells express high levels of CXCR4, CXCR5, and VLA-4 (CD49d): importance for interactions with the stromal microenvironment and specific targeting. Blood. 2009;113(19):4604–13.
Chiron D, Bellanger C, Papin A, Tessoulin B, Dousset C, Maiga S, et al. Rational targeted therapies to overcome microenvironment-dependent expansion of mantle cell lymphoma. Blood. 2016;128(24):2808–18.
Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–16.
Chang BY, Francesco M, De Rooij MF, Magadala P, Steggerda SM, Huang MM, et al. Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood. 2013;122(14):2412–24.
Zhao X, Bodo J, Sun D, Durkin L, Lin J, Smith MR, et al. Combination of ibrutinib with ABT-199 synergistic effects on proliferation inhibition and apoptosis in mantle cell lymphoma cells through perturbation of BTK, AKT and BCL2 pathways. Br J Haematol. 2015;168(5):765–8.
Tam CS, Anderson MA, Pott C, Agarwal R, Handunnetti S, Hicks RJ, et al. Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N Engl J Med. 2018;378(13):1211–23.
Prukova D, Andera L, Nahacka Z, Karolova J, Svaton M, Klanova M, et al. Cotargeting of BCL2 with venetoclax and MCL1 with S63845 Is synthetically lethal in vivo in relapsed mantle cell lymphoma. Clin Cancer Res. 2019;25(14):4455–65.
Gong JN, Khong T, Segal D, Yao Y, Riffkin CD, Garnier JM, et al. Hierarchy for targeting prosurvival BCL2 family proteins in multiple myeloma: pivotal role of MCL1. Blood. 2016;128(14):1834–44.
Touzeau C, Ryan J, Guerriero J, Moreau P, Chonghaile TN, Le Gouill S, et al. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia. 2016;30(3):761–4.
Touzeau C, Gouill SL, Mahé B, Boudreault JS, Gastinne T, Blin N, et al. Deep and sustained response after venetoclax therapy in a patient with very advanced refractory myeloma with translocation t(11;14). Haematologica. 2017;107(3):102–12.
Touzeau C, Dousset C, Le Gouill S, Sampath D, Leverson JD, Souers AJ, et al. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2014;28(1):210–2.
Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65(14):6282–93.
Matulis SM, Gupta VA, Nooka AK, Hollen HV, Kaufman JL, Lonial S, et al. Dexamethasone treatment promotes Bcl-2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia. 2016;30(5):1086–93.
Moreau P, Chanan-Khan A, Roberts AW, Agarwal AB, Facon T, Kumar S, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood. 2017;130(22):2392–400.
Zhou L, Zhang Y, Sampath D, Leverson J, Dai Y, Kmieciak M, et al. Flavopiridol enhances ABT-199 sensitivity in unfavourable-risk multiple myeloma cells in vitro and in vivo. Br J Cancer. 2018;118(3):388–97.
Raje N, Kumar S, Hideshima T, Roccaro A, Ishitsuka K, Yasui H, et al. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood. 2005;106(3):1042–7.
Acknowledgements
None.
Author information
Authors and Affiliations
Contributions
XY wrote part of the manuscript and reviewed literature. QC wrote part of the manuscript and reviewed literature. JH wrote part of the manuscript, reviewed it, and provided constructive suggestions. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors approved the final version of the manuscript and provided their consent for its publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yue, X., Chen, Q. & He, J. Combination strategies to overcome resistance to the BCL2 inhibitor venetoclax in hematologic malignancies. Cancer Cell Int 20, 524 (2020). https://doi.org/10.1186/s12935-020-01614-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-020-01614-z