
Abstract
The PRKDC gene encodes the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) protein. DNA-PKcs plays an important role in nonhomologous end joining (NHEJ) of DNA double-strand breaks (DSBs) and is also closely related to the establishment of central immune tolerance and the maintenance of chromosome stability. The occurrence and development of different types of tumors and the results of their treatment are also influenced by DNA-PKcs, and it may also predict the results of radiotherapy, chemotherapy, and therapy with immune checkpoint inhibitors (ICIs). Here, we discuss and review the structure and mechanism of action of PRKDC and DNA-PKcs and their relationship with cancer.
Similar content being viewed by others

Structure of PRKDC and DNA-PKcs
The PRKDC gene (also known as the XRCC7 gene) is located on chromosome 8q11 and has a transcript length of 12,784 bp. The PRKDC gene encodes the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), which is a member of the phosphatidylinositol 3-kinase related kinase (PIKK) protein kinase family and is a serine/threonine protein phosphorylation kinase. The DNA-PKcs protein contains 4128 amino acids; it has a relative molecular weight of 469 kDa and is the highest molecular weight kinase identified thus far. Sibanda et al. [1] analyzed the structure of DNA-PKcs and found that it folded into three large structural units: the N-terminal region (1–892 aa, N1-N4), the Circular Cradle (893–2801 aa, CC-CC5), and the C-terminal Head domain (2802–4128 aa). Existing studies have shown that a central cavity formed by the N-terminal region and the α-helical HEAT repeat sequence of the Circular Cradle is mainly used to mediate the binding to a DNA-Ku70/80 protein dimer complex and provides a passage for the DNA molecule during the DNA repair process [2,3,4]. The Circular Cradle contains two autophosphorylation clusters, namely the ABCDE cluster and the PQR cluster. The autophosphorylation of the ABCDE cluster promotes the joining of DNA ends. After DNA-PKcs is activated by binding to the DNA-Ku complex, it undergoes autophosphorylation along with phosphorylation of other substrates such as XRCC and H2AX to further complete the double-strand break (DSB) repair process. [1, 5, 6] The C-terminal Head domain contains the FAT domain (2802–3675 aa), the bilobal kinase domain (3676–4100 aa), and the FATC domain (4101–4128 aa). Among these domains, the structure of the bilobal kinase domain is similar to that of the PI3 family, with high basic kinase activity, and its main function is to activate the downstream signaling pathway of PI3K. Previous studies [7, 8] have shown that the kinase activity is not activated by the DNA-Ku protein dimer complex; therefore, this part may not play a major role in DSB repair. The FAT and FATC domains are tightly intertwined with the kinase domain and have a regulatory effect on the catalytic activity of kinases (Fig. 1) [2, 9]. As a catalytic protein, DNA-PKcs together with Ku70 and Ku80, which are two subunits that can bind to DNA and have regulatory function (Ku protein dimers), constitute a DNA-dependent protein kinase (DNA-PK) [10].

The overall structure of DNA-PKcs (PDB id: 6ZFP). The structural domains are colored according to the schematic shown below the structures. The N terminus is shown in purple, the circular cradle in orange, the FAT domain in teal, and the kinase domain in yellow. (Reference: Chaplin AK, Hardwick SW, Liang S et al. Dimers of DNA-PK create a stage for DNA double-strand break repair. Nature Structural & Molecular Biology 2021, 28(1):13–19.)
Function and mechanism of PRKDC and DNA-PKcs
PRKDC is involved in DNA DSB repair
DNA damage takes many forms, including DNA single-strand breaks (SSBs); DNA DSBs; DNA adducts; and nucleotide mutations, substitutions, deletions, and insertions [11]. Among these forms, DSB is the most cytotoxic form of DNA damage, which is mainly caused by ionizing radiation and radioactive drugs. The mechanisms of repair of DSB include homologous recombination (HR) and nonhomologous end joining (NHEJ); the former requires the same chromosome as the template and can ensure strict pairing to accurately repair DSB, but the repair efficiency is slow. On the other hand, during NHEJ, the two ends of the break are directly joined and repair occurs quickly, which can avoid further serious DNA damage. Although this process cannot ensure accurate repair of DSB, it still plays an important role in maintaining genomic stability [12].
Current results show that DNA-PK is a key protein in NHEJ, and DNA-PKcs encoded by the PRKDC gene is the most important component of DNA-PK [12, 13]. When a DSB occurs, the circular Ku70/Ku80 heterodimer recognizes and binds to the broken DNA end to initiate NHEJ, and this complex recruits DNA-PKcs. The DNA-PKcs protein acts as a linker between DNA ends and prevents degradation by exonucleases, a process termed DNA-end tethering [13,14,15,16]. DNA-PKcs recruited to the DNA ends is autophosphorylated at the ABCDE cluster [17,18,19] and subsequently recruits more core proteins of the NHEJ pathway, including XRCC4, XLF, and DNA ligase IV (LIG4) [20,21,22], which further align and join the DNA ends. As the name implies, this joining process is a direct connection without a template. From the description, it seems to be a very error-prone process, with the possibility of mutation occurrence; however, Betermier et al. [23] showed that NHEJ is an accurate and effective process for joining DNA ends.
An et al. [13] showed that DNA-PKcs can also repair DSBs through other pathways. When ionizing radiation causes DSBs, histone H2AX is phosphorylated to the histone variant H2AX (γH2AX) which plays an important role in the identification and repair of DNA damage; thus, γH2AX is an important marker for DSB repair. The same study [13] also found that DNA-PKcs plays a major role in regulating the phosphorylation of H2AX. The activated DNA-PKcs phosphorylates the 139th serine residue on γH2AX, either directly or indirectly, through the AKT/GSK3β signaling pathway, and repair factors are then recruited to the site of DSBs that coordinate DSB repair.
In the NHEJ process, the recruitment sequence of the various macromolecular proteins involved in DNA repair and the function of the linker are currently being explored; nevertheless, the importance of DNA-PKcs is unquestionable. Experiments conducted by Zhang et al. [24] showed that inhibition of DNA-PKcs directly leads to low DNA repair efficiency, and both in vivo and in vitro experimental results revealed that cells become sensitive to DSBs after exposure to ionizing radiation and cytotoxic drugs. This demonstrates the precise and important role of DNA-PKcs in the NHEJ pathway.
PRKDC involved in the regulation of transcription factors and establishment of central immune tolerance
DNA-PK also interacts with the transcription factor autoimmune regulator (AIRE) to establish central T cell immune tolerance [25, 26]. AIRE is mainly observed in the nucleolus of thymic medullary epithelial cells (MECs) where it plays an important role in the establishment and maintenance of central and peripheral immune tolerance by regulating the heterotopic expression of the tissue-specific antigen (TSA) gene in MECs and inducing the clearance of thymic autoreactive T cells [25, 26]. Abramson et al. [27] used mass spectrometry to identify a group of AIRE-related proteins, and the results showed that DNA-PK exhibited the highest correlation with AIRE. Liiv et al. [28] showed that DNA-PK can phosphorylate Thr68 and Ser156 of AIRE in vitro, thereby allowing AIRE to exert its regulatory function. In cells lacking DNA-PKcs, AIRE loses its ability to bind to DNA and activate transcription of the TSA genes; this finding indicates that DNA-PKcs can guide AIRE to the transcriptional activation site. Therefore, DNA-PKcs is one of the important cofactors for AIRE to perform its functions. DNA-PKcs interacts with AIRE to promote the transcription of TSAs and the subsequent elimination of autoreactive T cells. The abnormal function of DNA-PKcs results in the development of inflammatory disease with organ-specific autoimmunity. This also suggests an important role of DNA-PK in maintaining AIRE-dependent autoimmune tolerance [29].
PRKDC is involved in the maintenance of chromosome stability
Telomeres are structures at the ends of chromosomes that contain guanosine repeats. Their sequence and structure form a “cap” that protects telomeres from degradation. Therefore, telomeres play an important role in genome stability and prevention of the development of malignant cells. Current studies [30, 31] have shown that DNA-PK plays an important role in maintaining telomere length and stability. DNA-PK defects can accelerate telomere degradation and cell apoptosis, leading to increased chromosomal instability and tumorigenesis. DNA-PKcs is also a key regulator of cell mitosis. Lee et al. [32] showed that DNA-PKcs plays an important role in the formation of spindles and in the attachment of microtubules to chromosomes during mitosis. The depletion or functional inhibition of DNA-PKcs leads to chromosome dislocation and mitotic dysfunction, which further increases chromosomal instability. Shang et al. [33] also found that the inactivation of DNA-PKcs led to spindle destruction and mitotic catastrophe.
The effect of PRKDC on cell apoptosis
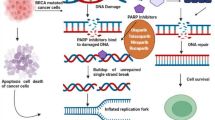
As mentioned above, when DSBs occur, DNA-PKcs and Ku70/80 work together to repair DNA damage. When a large amount of DSBs is produced in the cell, the DNA-PK complex also activates the apoptosis program. Abe et al. [34] showed that when high-dose etoposide induces a large amount of DSBs and the activated DNA-PK exceeds the level required for DNA damage repair (DDR), the excess fraction leads to phosphorylation of the Artemis protein which mediates apoptosis. Other studies have also shown that DNA-PK may activate the apoptosis program in ionizing radiation-exposed cells by phosphorylating the p53 protein (Fig. 2) [35, 36].

Function and mechanism of DNA-PKcs
Relationship between PRKDC and tumor
PRKDC mutation rate in tumors
The mutation rate of PRKDC and the expression level of DNA-PKcs in various tumors are quite different. Chen et al. [37] conducted statistical analysis on the mutation rate of PRKDC as reported in the Cancer Genome Atlas (TCGA) and the Chinese population database; the authors found that PRKDC had a high mutation rate in several tumors, such as colorectal cancer, gastric cancer, and endometrial cancer, with a high correlation with microsatellite instability. In the TCGA database, PRKDC mutations were found in 51 (9.66%) of 528 colorectal cancers, 42 (9.63%) of 436 gastric cancers, and 23 (9.27%) of 248 endometrial cancers. In contrast, PRKDC mutations were less common in some other types of tumors such as thyroid cancer (0.99%), glioblastoma (1.37%), and liver cancer (1.61%).
DNA-PKcs expression levels are different in different types of tumors
Many studies have analyzed the expression levels of DNA-PKcs in clinical samples of various types of tumors. On the one hand, these results are expected to explain some association between DNA-PKcs activity and the occurrence and development of cancer, and on the other hand, they provide some important information for predicting the tumor response to radiotherapy and chemotherapy.
In most types of tumors, the expression of DNA-PKcs in tumor tissues was found to be significantly higher than that in adjacent tissues. Lu et al. [38] extracted and quantified the total protein of 16 fresh colorectal cancer surgical specimens and normal adjacent tissues, and both immunohistochemistry (IHC) and western blot results showed that the expression of DNA-PKcs in colorectal cancer specimens was significantly higher than that in adjacent tissues (P < 0.05). Zhang et al. [39] analyzed 80 pairs of gastric cancer tissues and adjacent tissues and found that PRKDC was overexpressed in most subtypes of gastric cancer tissues and that PRKDC was the most significantly upregulated gene related to DDR. Maag et al. [40] collected samples from 17 patients with normal squamous esophagus, 22 patients with Barrett’s esophagus, and 12 patients with esophageal adenocarcinoma for immunohistochemical analysis of DNA-PKcs expression. The IHC density score showed that the expression of DNA-PKcs in esophageal adenocarcinoma tissues was significantly higher than that in Barrett’s esophagus and normal squamous esophagus tissues. A similar expression pattern was also found in renal cell carcinoma (RCC), nasopharyngeal carcinoma, and non-small cell lung cancer (NSCLC) [41,42,43,44].
Other types of tumors have shown reduced or lack of DNA-PKcs expression. For example, it was found that the expression of DNA-PKcs was decreased or absent in ovarian cancer tissues as compared to that in normal tissues. In the included 20 normal ovarian tissues, 20 benign ovarian tumors, 20 borderline ovarian tumors, and 40 malignant ovarian tumors, the positive rate of DNA-PKcs in each group was 100%, 95%, 90%, and 60%, respectively [45]. Lee et al. [46] also used IHC to analyze the expression of DNA-PKcs in 279 patients with gastric cancer, and they found that DNA-PKcs expression was negative in 23% of gastric cancer samples and that the lack of DNA-PKcs was associated with lymph node metastasis and a poor prognosis.
The difference (increase or decrease) in the expression level of DNA-PKcs in tumor tissues as compared to that in adjacent normal tissues suggests that DNA-PKcs may play a role in the process of tumorigenesis and cancer development.
The role of PRKDC in the process of tumorigenesis and cancer development
By analyzing the differential expression of DNA-PKcs in tumor tissues and normal tissues, many studies [38, 42, 47,48,49,50,51,52,53,54,55] have further shown that the expression level of DNA-PKcs is also closely related to the genesis, development, invasion, and distant metastasis of various types of tumors.
The recognized effects and possible underlying mechanisms of PRKDC and DNA-PKcs in tumorigenesis and cancer development can be summarized from the following findings. First, PRKDC mutations and DNA-PKcs expression defects cause dysfunction of DSB repair. This leads to genome instability and accumulation of mutations that increase the risk of cancer. It has been found that tumorigenesis in many types of cancers are related to DNA-PKcs defects, and its impaired activity makes the human body prone to the development of various malignant tumors. McKean-Cowdin et al. [47] proposed that co-mutations in DDR genes were associated with an increased risk of glioblastoma multiforme (GBM), and a variant of PRKDC increased the risk of glioma by 44%. The 2004 study by Wang et al. [48] also showed that another variant of PRKDC increased the risk of glioma by 1.82-fold. Teneng et al. [49] demonstrated through an in vitro study that the knockdown of DNA-PKcs significantly increased the amount of detectable DNA damage after bleomycin treatment and led to malignant transformation of bronchial epithelial cells and tumorigenesis. Second, DNA-PKcs affects tumor susceptibility and progression by regulating certain tumor suppressor genes and signaling pathways. Mori et al. [50] found that DNA-PKcs is a candidate regulatory factor for the radiation-induced apoptosis 1 (Rapop1) gene, which affects the susceptibility to radiation-induced lymphoma. As mentioned above, DNA-PKcs is a member of the PIKK protein kinase family; thus, it can act as an upstream regulator to activate mTORC2-AKT, a cell proliferation-related signaling pathway. Zheng et al. [41] found that in RCC, the overexpression of DNA-PKcs was significantly associated with the activation of the mTORC2-AKT signaling pathway. The knockdown of DNA-PKcs inhibited AKT phosphorylation, and the expression of HIF-2α (an mTORC2 regulatory gene) was also significantly downregulated. Third, the DNA-PKcs protein also participates in tumor progression by changing the tumor microenvironment. Kotula et al. [51] inoculated DNA-PKcs-deficient human melanoma cells into nude mice to generate subcutaneous tumors. They found that these tumors had significantly fewer blood vessels than non-DNA-PKcs-deficient tumors, and the proliferation index was low. The survival analysis of mice showed that the distant metastasis-free survival (MFS) of DNA-PKcs-deficient mice was significantly better than that of non-DNA-PKcs-deficient animals. Liu et al. [52] also suggested that inhibition of DNA-PKcs in glioblastoma could reduce the secretion of VEGF, thus inhibiting the migration and metastasis of tumors. Kotula et al. [51] also reported that DNA-PKcs can control a variety of metastasis-related proteins, including matrix metalloproteinases, to affect the tumor microenvironment and promote tumor migration. In other malignancies, including colorectal cancer, gastric cancer, hepatocellular carcinoma, and nasopharyngeal carcinoma, elevated DNA-PKcs protein levels were found to be significantly associated with lymphatic and distant metastasis in the late stage of tumor growth as well as with low differentiation of tumor cells (Fig. 3) [38, 42, 53,54,55].

DNA-PKcs and tumorigenesis and tumor progression
Both PRKDC and DNA-PKcs influence genesis, development, invasion, and metastasis of tumors through various mechanisms, and further prognostic analysis also suggests that DNA-PKcs expression can help to predict the prognosis of patients. After studying the influence of the DNA-PKcs expression level on the clinical staging and lymphatic and distant metastasis of colorectal cancer, Lu et al. [38] further proved that the DNA-PKcs expression level was negatively correlated with the 5-year survival rate of patients. Xing et al. [44] analyzed the gene expression profile of NSCLC cells, and the results showed that patients with high DNA-PKcs or ATM expression levels in tumor sample/normal tissue sample (T/N) ratio had a significantly increased risk of death. In other cancers such as gastric cancer, liver cancer, breast cancer, and nasopharyngeal carcinoma, the expression level of DNA-PKcs is also negatively correlated with prognosis [39, 42, 53, 56].
In addition to DNA-PKcs expression in tumor cells, its expression level in peripheral blood lymphocytes (PBLs) of tumor patients is also related to the biological behavior of tumors and patient prognosis. At present, it is generally believed that the decreased expression of DNA-PKcs in PBLs is associated with malignant progression and a poor prognosis for patients. Someya et al. [57] found that the activity of DNA-PKcs in PBLs was generally decreased in patients with advanced cancer, which was correlated with an invasive phenotype and a poor prognosis. Auckland et al. [58] reported that the decrease in DNA-PKcs in peripheral monocytes was associated with the development of lung cancer.
It was also found that in patients with cervical cancer and breast cancer, the DNA-PKcs expression level in PBL was significantly lower than that in healthy volunteers, and the reduction in DNA-PKcs expression was associated with an increased risk of cancer [59].
PRKDC and tumor treatment
PRKDC and response to chemotherapy and radiotherapy
Association of DNA-PKcs expression with response to radiotherapy and chemotherapy
Radiotherapy and chemotherapy are very important in cancer treatment, and their anticancer mechanisms are related to the induction of lethal DSBs in tumor cells [60]. As a key protein in NHEJ, DNA-PKcs plays an important role in repairing DSBs; therefore, PRKDC mutations or abnormal expression of DNA-PKcs is associated with the response to radiotherapy and chemotherapy. An increased level of DNA-PKcs enhances the repair of DSBs in cells that weakens the lethal effect of radiotherapy and cytotoxic drugs, thereby making tumors more resistant to radiotherapy and chemotherapy. Sun et al. [61] found a significant correlation between DNA-PKcs expression and the response to chemotherapy in patients with breast cancer. The expression level of DNA-PKcs in tumor cells of chemotherapy-resistant patients was significantly higher than that in tumor cells of patients who responded to chemotherapy. In addition, among the patients who received chemotherapy, the overall survival of those with high DNA-PKcs expression levels was significantly reduced compared to that of patients with low DNA-PKcs expression levels. Previous studies have also used PRKDC knockout breast cancer cell lines and untreated breast cancer cell lines to inoculate mice. In vivo experiments confirmed that the expression level of DNA-PKcs directly affects the tumor response to chemotherapy, and PRKDC knockout can sensitize the inoculated tumors to chemotherapy with significant tumor regression. Shao et al. [62] also found that the DNA-PK expression level in glioma cells had a high positive correlation with sensitivity to cisplatin in vitro, and the expression of DNA-PK was high in patients who did not respond to chemotherapy. Studies on oral squamous cell carcinoma and cervical squamous cell carcinoma also showed that the expression of DNA-PKcs was upregulated in surviving tumor cells after radiotherapy, which indicated that these cells were resistant to radiotherapy and survived selectively [63, 64].
Application of DNA-PKcs inhibitors in tumor chemoradiotherapy
The upregulation of DNA-PKcs leads to radiotherapy and chemotherapy resistance. Conversely, inhibition of DNA-PKcs can induce the programmed cell death of tumors and enhance the cell-killing effect of chemotherapy and radiotherapy by inhibiting the repair of DSBs. It has been confirmed that a variety of small molecules can inhibit the activity of DNA-PKcs, including NU7441, NU7026, SU11752, and IC87361. Attempts have been made to combine DNA-PKcs inhibitors with chemoradiotherapy or other antitumor agents for tumor treatment. Sarah et al. [65] showed that the combination of mitoxantrone and the DNA-PKcs inhibitor NU7441 can improve the treatment response of patients with high-risk chronic lymphocytic leukemia (CLL). Wojciech et al. [66] reported that NU7441 can increase the sensitivity of three types of breast cancer cell lines to radiation and adriamycin. Cindy et al. [67] showed that the phosphorylation of DNA-PKcs can accelerate the repair of damaged DNA in cells induced by ionizing radiation, while DNA-PKcs inhibitors can inhibit this process. In vivo experiments showed that the survival rate of mice treated with radiotherapy combined with a DNA-PKcs inhibitor was significantly higher than that of mice treated with radiotherapy alone. Zhang et al. [24] revealed that treatment of radiation-resistant cells with NU7441 can re-sensitize the cells to radiotherapy both in vivo and in vitro.
DNA-PKcs is also a protein kinase necessary for cell cycle progression during chromosome separation and mitosis, and the cell cycle checkpoint arrest caused by inhibiting DNA-PKcs can also lead to increased apoptosis and enhanced cell killing by radiotherapy and chemotherapy. Azad et al. [68] reported that in NSCLC cells, the inhibition of DNA-PKcs can arrest the irradiated cells in the G2/M phase and accelerate apoptosis.
Other studies have shown that the inhibition of DNA-PKcs or a decrease in DNA-PKcs expression levels can also activate autophagy in irradiated cells, thereby contributing to the efficacy of radiotherapy. Zhuang et al. [69] showed that in irradiated glioma cells, inhibition of DNA-PKcs promotes autophagy of tumor cells by inducing the expression of two autophagy markers, namely microtubule-associated protein light chain 3 (LC3) and Beclin1, and it increases the death of irradiated cells.
PRKDC and immunotherapy
Immunotherapies have received increasing attention in recent years, and among the most important ones are the immune checkpoint inhibitor (ICI) therapies [70, 71]. Currently, an important factor in predicting the response to ICIs is tumor mutation burden (TMB) [37], which refers to the number of nonsynonymous mutations per million bases of the tumor cell genome. The damage to the DDR system is closely related to the TMB, and a functional defect of the DDR system leads to increased genomic instability and accumulation of mutations in cells [37]. PRKDC is a key gene in the NHEJ pathway and in DSB repair, and its relationship with immunotherapy is being gradually explored. Several current studies have shown that mutations in the PRKDC gene are associated with a better response to ICIs, mainly due to the increased TMB of PRKDC mutant cells and the phenotype of immune cell infiltration. Rizvi et al. [72] performed whole-genome sequencing on 34 cases of NSCLC where patients were treated with pembrolizumab to detect genomic biomarkers that can predict the response to PD-1 monoclonal antibody treatment. They showed that, of the 14 patients with a durable clinical benefit (DCB), two had PRKDC mutations, while out of 20 patients with nondurable clinical benefit (NDCB), none of them had PRKDC mutations. Chen et al. [37] comprehensively analyzed the relationship between PRKDC mutations and TMB, tumor microenvironment, and treatment outcomes of pan-cancer patients after ICI therapy. They found that PRKDC mutations were significantly correlated with elevated TMB and an inflammatory tumor microenvironment. In the clinical cohorts of NSCLC and melanoma patients treated with ICIs, they reported that PRKDC mutations are confirmed to be associated with a better prognosis. Tan et al. [73] analyzed previous immunotherapy-related studies and found that, in melanoma, NSCLC, and kidney cancer, the ICI response rates of patients with PRKDC mutations reached 53.8%, 75%, and 50%, respectively (Table 1). Moreover, the CT26 animal model was also used in this study to confirm that the PRKDC knockout cells had a better response to ICIs as shown by a significant tumor volume reduction.
Conclusion
Current evidence shows that PRKDC and its encoded protein DNA-PKcs perform a variety of important functions in cells, including DNA DSB repair, participation in the establishment of immune tolerance, and maintenance of chromosome and genome stability. Changes in the expression level of DNA-PKcs also affect the occurrence and development of tumors. Regarding tumorigenesis, DNA-PKcs defects lead to genome instability and increased tumor susceptibility. In the process of tumor development, the enhanced activity of DNA-PKcs affects the regulation of some tumor suppressor genes, leading to malignant progression characterized by poor differentiation and increased aggressiveness. Regarding tumor treatment, the elevated DNA-PKcs levels will enhance the resistance of cancer cells to DNA damage and chemoradiotherapy, thereby leading to a poor prognosis for patients. Moreover, several studies have shown that PRKDC mutations are associated with a higher TMB and a better response to ICIs; thus, PRKDC is also expected to become another predictive biomarker for ICI therapy.
Future research studies need to consider the following aspects to improve outcome: (1) several key amino acid positions in the domains of the DNA-PKcs protein are particularly important for DSB repair, such as S2023, S2029, S2041, S2053, and S2056 [1], and there is still a lack of research on the effects of mutations at these sites on protein function; and (2) because the main mechanism of the association between DNA-PKcs and tumor therapy (radiotherapy, chemotherapy, and immunotherapy) is based on the integrity or defect of the former’s DSB repair function, the exploration of key site mutations in DNA-PKcs can help to identify biomarkers for chemoradiotherapy and immunotherapy and more accurately predict therapeutic outcomes.
Search strategy and selection criteria
Data for this review were obtained by searching PubMed and references from relevant articles using the search terms “DNA-PKcs” and “PRKDC”. Articles published in English between 1992 and 2020 were included in this review.
Availability of data and materials
Data for this Review were identified by searches of PubMed.
References
Sibanda BL, Chirgadze DY, Ascher DB, Blundell TL. DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science. 2017;355(6324):520–4.
Davis AJ, Lee KJ, Chen DJ. The N-terminal region of the DNA-dependent protein kinase catalytic subunit is required for its DNA double-stranded break-mediated activation. J Biol Chem. 2013;288(26):7037–46.
Sibanda BL, Chirgadze DY, Blundell TL. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature. 2010;463(7277):118–21.
Williams DR, Lee KJ, Shi J, Chen DJ, Stewart PL. Cryo-EM structure of the DNA-dependent protein kinase catalytic subunit at subnanometer resolution reveals α helices and insight into DNA binding. Structure. 2008;16(3):468–77.
Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417(3):639–50.
Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K, Botvinick E, Qin J, Chen DJ. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem. 2005;280(15):14709–15.
Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402(6759):313–20.
Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature. 2013;497(7448):217–23.
Lempiäinen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009;28(20):3067–73.
Goodwin JF, Knudsen KE. Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 2014;4(10):1126–39.
Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7(7):675–93.
Mao Z, Bozzella M, Seluanov A, Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair. 2008;7(10):1765–71.
An J, Huang YC, Xu QZ, Zhou LJ, Shang ZF, Huang B, Wang Y, Liu XD, Wu DC, Zhou PK. DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Mol Biol. 2010;11:18.
Yoo S, Dynan WS. Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res. 1999;27(24):4679–86.
Neal JA, Meek K. Choosing the right path: does DNA-PK help make the decision? Mutat Res. 2011;711(1–2):73–86.
Graham TGW, Walter JC, Loparo JJ. Two-stage synapsis of DNA ends during non-homologous end joining. Mol Cell. 2016. https://doi.org/10.1016/j.bpj.2015.11.167.
Dvir A, Peterson SR, Knuth MW, Lu H, Dynan WS. Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc Natl Acad Sci USA. 1992;89(24):11920–4.
Gell D, Jackson SP. Mapping of protein-protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Res. 1999;27(17):3494–502.
Radhakrishnan SK, Lees-Miller SP. DNA requirements for interaction of the C-terminal region of Ku80 with the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). DNA Repair. 2017;57:17.
Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124(2):301–13.
Buck D, Malivert L, de Chasseval R, Barraud A, Fondanèche M-C, Sanal O, Plebani A, Stéphan J-L, Hufnagel M, le Deist F. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124(2):287–99.
Critchlow SE, Bowater RP, Jackson SP. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr Biol. 1997;7(8):588–98.
Bétermier M, Bertrand P, Lopez BS. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014;10(1): e1004086.
Zhang B, Wu H, Hao J, Wu Y, Yang B. Inhibition of DNA-PKcs activity re-sensitizes uveal melanoma cells to radio- and chemotherapy. Biochem Biophys Res Commun. 2020;522(3):639–46.
Goodwin JF, Knudsen KE. Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 2014. https://doi.org/10.1158/2159-8290.CD-14-0358.
Mathieu AL, Verronese E, Rice GI, et al. PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator-dependent autoimmunity. J Allergy Clin Immunol. 2015;135:1578-88.e1575.
Abramson J, Giraud M, Benoist C, Mathis D. Aire’s partners in the molecular control of immunological tolerance. Cell. 2010;140(1):123–35.
Liiv I, Rebane A, Org T, Saare M, Maslovskaja J, Kisand K, Juronen E, Valmu L, Bottomley MJ, Kalkkinen N, et al. DNA-PK contributes to the phosphorylation of AIRE: importance in transcriptional activity. Biochem Biophys Acta. 2008;1783(1):74–83.
O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4(8): e1000155.
Wang H, Perrault AR, Takeda Y, Qin W, Wang H, Iliakis G. Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res. 2003;31(18):5377–88.
Hsu HL, Gilley D, Galande SA, Hande MP, Allen B, Kim SH, Li GC, Campisi J, Kohwi-Shigematsu T, Chen DJ. Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev. 2000;14(22):2807–12.
Lee KJ, Lin YF, Chou HY, Yajima H, Fattah KR, Lee SC, Chen BP. Involvement of DNA-dependent protein kinase in normal cell cycle progression through mitosis. J Biol Chem. 2011;286(14):12796–802.
Shang ZF, Huang B, Xu QZ, Zhang SM, Fan R, Liu XD, Wang Y, Zhou PK. Inactivation of DNA-dependent protein kinase leads to spindle disruption and mitotic catastrophe with attenuated checkpoint protein 2 Phosphorylation in response to DNA damage. Can Res. 2010;70(9):3657–66.
Abe T, Ishiai M, Hosono Y, Yoshimura A, Tada S, Adachi N, Koyama H, Takata M, Takeda S, Enomoto T, et al. KU70/80, DNA-PKcs, and Artemis are essential for the rapid induction of apoptosis after massive DSB formation. Cell Signal. 2008;20(11):1978–85.
Woo RA, Jack MT, Xu Y, Burma S, Chen DJ, Lee PW. DNA damage-induced apoptosis requires the DNA-dependent protein kinase, and is mediated by the latent population of p53. EMBO J. 2002;21(12):3000–8.
Woo RA, McLure KG, Lees-Miller SP, Rancourt DE, Lee PW. DNA-dependent protein kinase acts upstream of p53 in response to DNA damage. Nature. 1998;394(6694):700–4.
Chen Y, Li Y, Guan Y, Huang Y, Lin J, Chen L, Li J, Chen G, Pan LK, Xia X, et al. Prevalence of PRKDC mutations and association with response to immune checkpoint inhibitors in solid tumors. Mol Oncol. 2020;14(9):2096–110.
Lü Y, Zhang HL, Li YZ, Zhao P. Clinicopathological significance of expressions of DNA dependent protein kinase catalytic subunit and P16 in colorectal carcinoma. Zhonghua Yi Xue Za Zhi. 2008;88(29):2025–9.
Zhang Y, Wen GM, Wu CA, Jing ZL, Li DZ, Liu GL, Wei XX, Tang MS, Li YH, Zhong Y, et al. PRKDC is a prognostic marker for poor survival in gastric cancer patients and regulates DNA damage response. Pathol Res Pract. 2019;215(8): 152509.
Maag JLV, Fisher OM, Levert-Mignon A, Kaczorowski DC, Thomas ML, Hussey DJ, Watson DI, Wettstein A, Bobryshev YV, Edwards M, et al. Novel aberrations uncovered in Barrett’s esophagus and esophageal adenocarcinoma using whole transcriptome sequencing. Mol Cancer Res. 2017;15(11):1558–69.
Zheng B, Mao JH, Li XQ, Qian L, Zhu H, Gu DH, Pan XD. Over-expression of DNA-PKcs in renal cell carcinoma regulates mTORC2 activation, HIF-2α expression and cell proliferation. Sci Rep. 2016;6:29415.
Yan SS, Liu L, Liu ZG, Zeng MS, Song LB, Xia YF. Expression and clinical significance of DNA-PKcs in nasopharyngeal carcinoma. Ai Zheng. 2008;27(9):979–83.
Pan H, Zuo C, Mao N, Chen J, Cao J, Tang B. Expression and clinical significance of Ku70, Ku80 and DNA-PKcs proteins in patients with stageI-II non-small cell lung cancer by tissue microarray. Zhongguo Fei Ai Za Zhi. 2007;10(3):203–5.
Xing J, Wu X, Vaporciyan AA, Spitz MR, Gu J. Prognostic significance of ataxia-telangiectasia mutated, DNA-dependent protein kinase catalytic subunit, and Ku heterodimeric regulatory complex 86-kD subunit expression in patients with nonsmall cell lung cancer. Cancer. 2008;112(12):2756–64.
Shao SL, Cai Y, Wang QH, Yan LJ, Zhao XY, Wang LX. Expression of GLUT-1, p63 and DNA-Pkcs in serous ovarian tumors and their significance. Zhonghua Zhong Liu Za Zhi. 2007;29(9):697–700.
Lee HS, Yang HK, Kim WH, Choe G. Loss of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) expression in gastric cancers. Cancer Res Treat. 2005;37(2):98–102.
McKean-Cowdin R, Barnholtz-Sloan J, Inskip PD, Ruder AM, Butler M, Rajaraman P, Razavi P, Patoka J, Wiencke JK, Bondy ML, et al. Associations between polymorphisms in DNA repair genes and glioblastoma. Cancer Epidemiol Biomarkers Prev. 2009;18(4):1118–26.
Wang LE, Bondy ML, Shen H, El-Zein R, Aldape K, Cao Y, Pudavalli V, Levin VA, Yung WK, Wei Q. Polymorphisms of DNA repair genes and risk of glioma. Can Res. 2004;64(16):5560–3.
Teneng I, Picchi MA, Leng S, Dagucon CP, Ramalingam S, Tellez CS, Belinsky SA. DNA-PKc deficiency drives pre-malignant transformation by reducing DNA repair capacity in concert with reprogramming the epigenome in human bronchial epithelial cells. DNA Repair. 2019;79:1–9.
Mori N, Matsumoto Y, Okumoto M, Suzuki N, Yamate J. Variations in Prkdc encoding the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) and susceptibility to radiation-induced apoptosis and lymphomagenesis. Oncogene. 2001;20(28):3609–19.
Kotula E, Berthault N, Agrario C, Lienafa MC, Simon A, Dingli F, Loew D, Sibut V, Saule S, Dutreix M. DNA-PKcs plays role in cancer metastasis through regulation of secreted proteins involved in migration and invasion. Cell Cycle. 2015;14(12):1961–72.
Liu Y, Zhang L, Liu Y, Sun C, Zhang H, Miao G, Di CX, Zhou X, Zhou R, Wang Z. DNA-PKcs deficiency inhibits glioblastoma cell-derived angiogenesis after ionizing radiation. J Cell Physiol. 2015;230(5):1094–103.
Evert M, Frau M, Tomasi ML, Latte G, Simile MM, Seddaiu MA, Zimmermann A, Ladu S, Staniscia T, Brozzetti S, et al. Deregulation of DNA-dependent protein kinase catalytic subunit contributes to human hepatocarcinogenesis development and has a putative prognostic value. Br J Cancer. 2013;109(10):2654–64.
Hosoi Y, Watanabe T, Nakagawa K, Matsumoto Y, Enomoto A, Morita A, Nagawa H, Suzuki N. Up-regulation of DNA-dependent protein kinase activity and Sp1 in colorectal cancer. Int J Oncol. 2004;25(2):461–8.
Wang FR, Wei YC, Han ZJ, He WT, Guan XY, Chen H, Li YM. Aberrant DNA-PKcs and ERGIC1 expression may be involved in initiation of gastric cancer. World J Gastroenterol. 2017;23(33):6119–27.
Zhang Y, Yang WK, Wen GM, Tang H, Wu CA, Wu YX, Jing ZL, Tang MS, Liu GL, Li DZ, et al. High expression of PRKDC promotes breast cancer cell growth via p38 MAPK signaling and is associated with poor survival. Mol Genet Genomic Med. 2019;7(11): e908.
Someya M, Sakata KI, Matsumoto Y, Kamdar RP, Kai M, Toyota M, Hareyama M. The association of DNA-dependent protein kinase activity of peripheral blood lymphocytes with prognosis of cancer. Br J Cancer. 2011;104(11):1724–9.
Auckley DH, Crowell RE, Heaphy ER, Stidley CA, Lechner JF, Gilliland FD, Belinsky SA. Reduced DNA-dependent protein kinase activity is associated with lung cancer. Carcinogenesis. 2001;22(5):723–7.
Someya M, Sakata K, Matsumoto Y, Yamamoto H, Monobe M, Ikeda H, Ando K, Hosoi Y, Suzuki N, Hareyama M. The association of DNA-dependent protein kinase activity with chromosomal instability and risk of cancer. Carcinogenesis. 2006;27(1):117–22.
Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129–43.
Sun G, Yang L, Dong C, Ma B, Shan M, Ma B. PRKDC regulates chemosensitivity and is a potential prognostic and predictive marker of response to adjuvant chemotherapy in breast cancer patients. Oncol Rep. 2017;37(6):3536–42.
Shao CJ, Fu J, Shi HL, Mu YG, Chen ZP. Activities of DNA-PK and Ku86, but not Ku70, may predict sensitivity to cisplatin in human gliomas. J Neurooncol. 2008;89(1):27–35.
Shintani S, Mihara M, Li C, Nakahara Y, Hino S, Nakashiro K, Hamakawa H. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci. 2003;94(10):894–900.
Beskow C, Skikuniene J, Holgersson A, Nilsson B, Lewensohn R, Kanter L, Viktorsson K. Radioresistant cervical cancer shows upregulation of the NHEJ proteins DNA-PKcs, Ku70 and Ku86. Br J Cancer. 2009;101(5):816–21.
Elliott SL, Crawford C, Mulligan E, Summerfield G, Newton P, Wallis J, Mainou-Fowler T, Evans P, Bedwell C, Durkacz BW, et al. Mitoxantrone in combination with an inhibitor of DNA-dependent protein kinase: a potential therapy for high risk B-cell chronic lymphocytic leukaemia. Br J Haematol. 2011;152(1):61–71.
Ciszewski WM, Tavecchio M, Dastych J, Curtin NJ. DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res Treat. 2014;143(1):47–55.
Timme CR, Rath BH, O’Neill JW, Camphausen K, Tofilon PJ. The DNA-PK inhibitor VX-984 enhances the radiosensitivity of glioblastoma cells grown in vitro and as orthotopic xenografts. Mol Cancer Ther. 2018;17(6):1207–16.
Azad A, Jackson S, Cullinane C, Natoli A, Neilsen PM, Callen DF, Maira SM, Hackl W, McArthur GA, Solomon B. Inhibition of DNA-dependent protein kinase induces accelerated senescence in irradiated human cancer cells. Mol Cancer Res. 2011;9(12):1696–707.
Zhuang W, Li B, Long L, Chen L, Huang Q, Liang ZQ. Knockdown of the DNA-dependent protein kinase catalytic subunit radiosensitizes glioma-initiating cells by inducing autophagy. Brain Res. 2011;1371:7–15.
Hodi FS, O’Day SJ, Mcdermott DF, Weber RW, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889–94.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8.
Tan KT, Yeh CN, Chang YC, Cheng JH, Fang WL, Yeh YC, Wang YC, Hsu DS, Wu CE, Lai JI, et al. PRKDC: new biomarker and drug target for checkpoint blockade immunotherapy. J Immunother Cancer. 2020. https://doi.org/10.1136/jitc-2019-000485.
Acknowledgements
We appreciate all subjects who participated in this study and wish to thank Dr. Jian-ping Lu and Dr. Wei-feng Zhu (Department of Pathology, Fujian Medical University Cancer Hospital & Fujian Cancer Hospital, Fuzhou, Fujian Province, China) and all colleagues (Cancer Bio-immunotherapy Center, Fujian Medical University Cancer Hospital) for technical support.
Funding
This project was supported in part by the National Natural Science Foundation of China (Grant No. U1705282), Fujian provincial health and family planning research talent training program (Grant No. 2018-ZQN-13).
Author information
Authors and Affiliations
Contributions
CC, YC and YL concept and design the study. JL, XZ, JL and ZF search the literatures. JX, BL and XW screened the literatures. All authors write the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, Y., Li, Y., Xiong, J. et al. Role of PRKDC in cancer initiation, progression, and treatment. Cancer Cell Int 21, 563 (2021). https://doi.org/10.1186/s12935-021-02229-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-02229-8