
Abstract
Oncoviruses, known as cancer-causing viruses, are typically involved in cancer progression by inhibiting tumor suppressor pathways and uncontrolled cell division. Myeloid cells are the most frequent populations recruited to the tumor microenvironment (TME) and play a critical role in cancer development and metastasis of malignant tumors. Tumor-infiltrating myeloid cells, including tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), tumor-associated dendritic cells (TADCs), and tumor-associated neutrophils (TANs) exert different states from anti-tumorigenic to pro-tumorigenic phenotypes in TME. Although their role in the anti-tumorigenic state is well introduced, their opposing roles, pro-tumorigenic activities, such as anti-inflammatory cytokine and reactive oxygen species (ROS) production, should not be ignored since they result in inflammation, tumor progression, angiogenesis, and evasion. Since the blockade of these cells had promising results against cancer progression, their inhibition might be helpful in various cancer immunotherapies. This review highlights the promoting role of tumor-associated myeloid cells (TAMCs) in the pathophysiology of human virus tumorigenesis.
Similar content being viewed by others

Background
Human oncogenic viruses, known as oncoviruses, potentially contribute to an estimated 12–20% of human cancers, accounting for a large fraction of the global cancer burden [1]. Recently, several oncoviruses with DNA or RNA genomes such as human papillomavirus (HPV), Epstein-Barr virus (EBV), hepatitis B virus (HBV), hepatitis C virus (HCV), human herpesvirus-8 (HHV-8), human T-cell lymphotropic virus-1 (HTLV-1), and Merkel cell polyomavirus (MCV) have been recognized as the primary contributors to cancer development [2]. Viral carcinogenesis is a complex process associated with viral factors and immune escape mechanisms. There is interesting crosstalk between different viral and host factors which mediate the signaling pathways and cellular process. In general, oncoviruses can inhibit the tumor suppressor pathway p53, which supports primary tumor growth and progression [3]. It has also been demonstrated that viral factors potentially activate PI3K-Akt-mTOR, Notch, and Wnt pathways leading to cell overgrowth, tumor invasion, and angiogenesis [4]. On the other hand, oncoviruses establish an infection-associated chronic inflammation that could mediate cancer development through different mechanisms including tissue remodeling, angiogenesis, and production of growth factors [5]. The tumor microenvironment (TME) consists of different immune cells which play a prominent role in the tumor progression. Myeloid cells are the heterogeneous population of the innate immune system, which is considered the first line of defense. These cells include tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), tumor-associated dendritic cells (TADCs), and tumor-associated neutrophils (TANs) that predominantly infiltrate the TME [6]. Despite the central role of myeloid cells in regulating anti-tumor immune responses, tumor-associated myeloid cells (TAMCs) can promote tumorigenesis mechanisms [7]. It is noteworthy that TAMCs exert crucial pro-tumorigenic functions in regulating cancer-related inflammation, expression of pro-angiogenic factors, tumor angiogenesis, tumor progression, and promotion of immune evasion [8]. The pro-tumorigenic functions of TAMCs, including anti-inflammatory cytokine secretion and chronic ROS production, have been considered significant obstacles to developing effective cancer treatments. Therefore, TAMCs are considered a double-edged sword of immune effectors in cancer progression. Given the dual role of TAMCs in cancer development and their therapeutic potential, this review highlights the role of tumor-promoting myeloid cells in the pathogenesis of human oncoviruses and provides new insights into cancer immunotherapy.
Anti- and pro-tumorigenic function of myeloid cells in cancer pathogenesis
Myeloid cells exert an immunosuppressive activity to combat the proliferating tumor cells; however, it has been demonstrated that they represent opposing functions from anti-tumorigenic to pro-tumorigenic phenotypes in the TME. Hence, we briefly describe the mechanism of the anti- and pro-tumorigenic function of TAMCs in the immune escape and cancer pathogenesis.
Tumor-associated regulatory dendritic cells (TAR-DCs)
Dendritic cells (DCs) are a double-edged sword population in the TME. Plasmacytoid (pDC), conventional (cDC1 or cDC2), and inflammatory DC (moDC) are three phenotypically and functionally distinct subsets of DCs [9]. These immune cells play a crucial role in various cancer types, including breast, lung, colorectal, ovarian, head and neck, bladder, gastric, and renal cancer [10]. Although, DCs mediate antigen trafficking and stimulation of CD8+ T-cell responses, however, TAR-DCs exhibited immunosuppressive properties by low expression of costimulatory molecules and high expression of regulatory molecules. Stromal-cell derived factor-1 (SDF-1) which is also known as CXCL12, in the TME of malignant tumors and high expression of CXCL4 ligand results in the accumulation of DCs in TME. Immunoglobulin-like transcript 7 (ILT7) recognizes bone marrow stromal cell antigen 2 (BST2), which is highly expressed on tumor cells, resulting in negative regulation of the interferon responses [11]. It has been demonstrated that IL-10 produced by TAMs potentially suppresses the secretion of IL-12, which mediates immune escape and metastatic progression. The inhibition of IL-10 could restore the functionality and cytokine production of DCs [12].
Tumor-associated macrophages (TAMs)
Tumor-associated macrophages (TAMs) are abundant myeloid cells in the TMEwith anti-tumorigenic or strongly pro-tumorigenic phenotypes. Macrophage colony-stimulating factor (M-CSF) is highly expressed in the TME, which recruits the macrophages from the bone marrow or spleen [13]. TAMs are classified as classically activated-M1 and alternatively activated-M2 macrophages which induce anti-tumorigenic Th1 immune responses and pro-tumorigenic functions such as tumor growth and invasion, immune suppression, and, angiogenesis which is mediated by cytokine and chemokine production respectively [14]. M1 macrophages exert anti-tumor activity by direct cytotoxic effects mediated by ROS production, and antibody-dependent cell-mediated cytotoxicity (ADCC) to eliminate tumor cells [15]. M2 macrophages are predominantly the vast majority of non-malignant TAMs associated with the production of immunosuppressive chemokines and factors including TGF-β and IL-10. Furthermore, TAMs are related to angiogenesis by producing pro-angiogenic factors, including vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), and matrix metalloproteinase (MMP) [13]. TAM can enhance tumor proliferation and invasion mediated by activation of NF-κB and STAT3 and expression of pro-inflammatory cytokines [16]. Elevated levels of TAMs are correlated with poor prognosis of diverse types of cancers [17, 18].
Myeloid-derived suppressor cells (MDSCs)
MDSCs are developmentally immature non-macrophage cells with an immunosuppressive function. These cells potentially prevent the activation of CD4+ and CD8+ T-cells. Also, it has been suggested that MDSCs suppress NK cells, which may disturb anti-tumor immunity [19]. Therefore, MDSCs are considered a serious hurdle against cancer immunotherapy. There are distinct subsets of MDSCs that express heterogeneous markers, including Siglec-3/CD33, CD14, CD15, and CD66b. However, CD11b is expressed by all types of human MDSCs. MDSCs exert immunosuppressive function through the production of IL-10, TGF-β, ARG1, IDO, and CD40 [12]. MDSCs inhibit T lymphocytes via the ROS or the depletion of L-arginine (L-arg) [20]. MDSCs suppress NK cells by expressing transforming growth factor β (TGF-β) and decreasing the expression of the NK-cell activating receptor NKp30 [21]. MDSCs also inhibit myeloid cell differentiation via a ROS-dependent mechanism [22].
Tumor-associated neutrophils (TANs)
As the first line of immune defense, neutrophils are a substantial population that infiltrates the TME. TANs have a dual function of anti- and pro-tumor activities, modulating anti-tumor immunity [23]. Interestingly, TANs are classified as two major types, N1 and N2, with anti-tumor and pro-tumor functions, respectively [23]. N1 mediates direct and indirect anti-tumor activity by ROS production and H2O2 and ADCC that could effectively kill tumor cells [24]. TANs actively contribute to tumor proliferation, angiogenesis, tumor progression, and metastasis through the high-level expression of neutrophil elastase and matrix metalloproteinase 9 (MMP9) [25]. Moreover, upregulation of TANs in the TME strongly predicts the poor survival rate in patients with cancer [26]. TANs could modulate innate and adaptive immune responses by different mechanisms. As an instance, they decrease the CTL response by upregulation of arginase-1. Furthermore, the production of neutrophil-secreted neutrophil elastase (NE) leads to tumor cellular proliferation. TANs mediate angiogenesis by secretion of VEGF and hepatocyte growth factor (HGF) [23].
Tumor-promoting myeloid cells and human oncoviral infection
Here we highlight the mechanistic strategies by oncoviral infection in immune disturbances (Table 1).
Epstein-Barr virus (EBV)
Epstein-Barr virus (EBV), first identified in the tumor cells of Burkitt lymphoma, is now associated with a strikingly diverse variety of lymphoproliferative lesions and malignant lymphomas of B, T, and NK cell origin [27]. Here, we highlight the association between EBV and tumor-promoting myeloid cells such as MDSCs and TAMs.
MDSCs
The latent membrane protein-1 (LMP1) is the primary oncogene of EBV that plays a critical role in the MDSCs proliferation and tumor immunosuppression. A large fraction of MDSCs is found in patients with EBV-associated T/NK cell lymphoproliferative diseases, which may dampen the antiviral T-cell responses [28]. LMP1-mediated glycolysis enhances the production of IL-1β, IL-6, and GM-CSF, the proliferation of tumor-associated MDSCs, and the inhibition of T-cells and NK cells, which lead to tumor immunosuppression [29, 30]. The accumulation of PMN-MDSCs in nasopharyngeal cancer survivors with persistent hepatitis B may suppress the host immune response [31] to the Epstein-Barr virus and be linked to tumor recurrence via ER stress/ROS pathway.
TAMs
In gastric cancer, the EBV-encoded miR-BART11 targets FOXP1 to enhance the tumor-associated macrophage-induced epithelial-mesenchymal transition [32]. Zhang et al. revealed that in nasopharyngeal carcinoma (NPC) cells, EBV induced M2 phenotype in TAMs and elevated the p-ATR expression. These two inductions were highly connected and linked to higher tumor staging, lymph node metastases, and poor patient prognosis [33]. Activation of ATR triggered by EBV increased subcutaneous tumor development, elevated Ki67 production, and lung metastasis in nude mice through the M2-type TAMs recruitment [33]. CD68 as a TAMs marker was higher in EBV-positive NPC. However, between EBV-positive and EBV-negative NPC, there was no variation in M2 macrophage number [34]. The survival of EBV+ tumor cells is dependent on TAMs in the EBV-positive TME [35]. The EBV status of lymphoma cells affected TAMs by up-regulation of CXCR10 and VEGF, causing angiogenesis and tumor survival. The in vivo reduction of macrophages revealed that they are required to survive EBV-positive tumor cells [35].
EBV expression has been identified in lesional macrophages of different cancers, ranging from thyroid to uterine carcinoma and some types of lymphoma that possibly elicit EBV lytic infection of macrophages in many tumor-associated macrophages in EBV-related malignancies [36].
In classical Hodgkin’s lymphoma, tumor-infiltrating macrophages are linked to a poor prognosis and the presence of EBV [37]. Increasing the number of TAMs is related to a reduction in overall survival, while greater levels of markers are statistically substantially associated with the presence of EBV infection [38].
Hepatitis B virus (HBV)
The most frequent kind of liver cancer is hepatocellular carcinoma (HCC). HBV is a chronic infection that affects over 350million individuals worldwide. At least half of all HCC cases globally are caused by chronic hepatitis B virus (HBV) infection [39]. Here, we highlight evidence of the association between HBV and tumor-promoting myeloid cells.
MDSCs
HCC patients had considerably greater percentages of MDSCs and PMN-MDSCs than chronic hepatitis B patients and healthy controls [40]. Pal et al. have demonstrated that the induction of regulatory T-cells (Tregs) by myeloid-derived suppressor cells in persistent HBV infections featuring high viral surface antigen is long-lasting and persists following antiviral treatment [41]. In the chronic liver failure posed by HBV, the proliferation of myeloid-derived suppressor cells was strongly associated with the severity and course of the disease [42].
Macrophages
Macrophages are monocytic phagocytes with antigen-presentation and cytokine-producing capabilities. The tissue-specific liver macrophages are Kupffer cells dominating other innate immune cells in the organ [43]. From the onset of HBV infection through the beginning and development of HCC, macrophages act as the key mediator of the pathogenic process. Kupffer cells have a role in inflammatory responses and tolerance generation in the early stages of infection [44]. In a specifically modified murine model of HBV infection, liver dysfunction was linked to an enormous frequency of human M2 macrophages [45]. Kupffer cells may impede the progression of HBV-associated HCC by inhibiting T-cell-mediated anti-tumor activity, limiting T-cell activation with PD-L1 expression on monocytes, and causing Tim3+/CD4+ and Tim3+/CD8+ cells to senescence [46, 47].
NK cells
The lymphocytes in the human liver tissue are predominantly natural killer cells (NK cells). Patients with persistent HBV and HCV infection have more NK cells in their liver [44]. A ligand of the NKG2D receptor, MICA, is upregulated in HBV infection, and soluble MICA levels have been linked to modulating responses directed by NK cells, which are essential in developing HCC in HBV-associated HCC patients [44, 48]. Several mechanisms have been shown to selectively impair NK cell function after chronic HBV infection. These include TGF-β and IL-10 stimulation of NK cells and their increased expression of Tim-3 triggered by HBV, hindering their activity [49, 50].
Hepatitis C virus (HCV)
HCV infection affects more than 270million individuals globally. HCV produces a chronic and lifelong infection in most infected individuals. This persistent inflammation in the liver leads to macronodular cirrhosis in 20% of people who contract it. A 4 to 7% yearly risk of progressing to HCC is associated with these individuals [51].
Through the TLR2/PI3K/AKT/STAT3 signaling cascade, HCV induced MDSC-like suppressive monocytes that activated CD4+Foxp3+ Tregs and inhibited the autologous CD4+ T-cell activation [52]. HCV stimulates the accumulation of CD33+ MDSCs, which reduces T-cell responsiveness via the production of ROS [53]. IFN-γ production by natural killer cells is suppressed by MDSCs induced by HCV, which alter cellular metabolism by inhibiting arginase-1 [54]. MDSCs triggered by HCV promote the development of Tregs while inhibiting the activity of effector T-cells [55]. Hepatitis C core protein polarizes granulocytic myeloid-derived suppressor cells via the IL-10/STAT3 signaling [56].
A long non-coding RNA (lncRNA) named HOXA transcript antisense RNA myeloid-specific 1 (HOTAIRM1) targets HOXA1 gene expression to regulate myeloid cell development. HOTAIRM1 enhances MDSCs growth and suppressive activities during HCV infection through the HOXA1-miR124 axis [57]. Exosomes associated with HCV suppress miR-124, which promotes the growth of myeloid-derived suppressor cells [58]. RUNX1 overlapping RNA (RUNXOR) is another lncRNA that targets runt-related transcription factor 1 (RUNX1) and is crucial for myeloid cell development. Exosomes associated with HCV through the STAT3-miR124 axis upregulate RUNXOR and RUNX1, increasing the MDSCs population and suppressive capabilities [59].
Human herpesvirus 8 (HHV-8)
The causative agent of Kaposi’s sarcoma (KS) is Kaposi’s sarcoma herpesvirus (KSHV; also known as human herpesvirus 8 (HHV-8)). KS is the most prevalent neoplasm among untreated HIV patients, although it may also happen in immunosuppressive conditions after organ transplantation [60]. KSHV vFLIP is a latent infection-associated viral oncoprotein. CD11b+Gr1+ cells with suppressor immune phenotype are induced by vFLIP, which remodels myeloid differentiation and causes their proliferation [61]. Based on the evidence, DC exerts decreased antiviral immune responses and altered cytokine production during the HHV-8 infection [62]. It has also been demonstrated that HHV-8 infection is associated with prostate cancer [63]. Moreover, the study on the Iranian population has indicated the high prevalent rate of the HHV-8 genome among patients with cervical cancer [64]. Therefore, it can be perceived that HHV-8 infection may be associated with an increased risk of cervical cancer.
Human papillomavirus (HPV)
Cervical cancer is caused by certain HPV (human papillomavirus) genotypes. Other anogenital cancers and a subset of head and neck cancers seem to be caused by the same genotypes. It is necessary to sustain the malignant development of cervical cancer cells by inducing the expression of particular viral oncoproteins, E6 and E7, which specifically inhibit the tumor suppressors p53 and RB [65]. In malignancies pertaining to HPV, MDSCs are related to both poor clinical outcomes and resistance to treatment. They inhibit the activity of CTLs, downregulate IFN-γ, and increase the frequency of Tregs, all of which contribute to carcinogenesis. Furthermore, compared to normal controls, their levels were elevated, indicating a clear relationship between pathological grade and their levels [66,67,68]. Activating CD8+ effector memory T-cells and controlling MDSCs together allowed protection against cancers caused by the HPV-16 serotype [69].
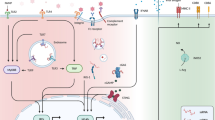
The oncogenic mechanisms of tumor-promoting myeloid cells in reviewed human oncoviral infections have been depicted in Fig.1.

Tumor promoting myeloid cells and their role in pathogenesis of human oncoviruses. Here, five human oncoviruses (Epstein-Barr virus, Hepatitis B and C viruses, Human herpesvirus 8, and human papillomavirus) have been illustrated with their related myeloid cells mostly tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs). These cells along with immune mediators in tumor microenvirnment promote tumor progression, angiogenesis, and migration and suppress anti-tumor effector cells including T and NK cells
Tumor-promoting myeloid cells and potential targets for immunotherapy of human oncoviruses
Due to the immunosuppressive nature of MDSCs and TAMs, these cells have been considered two main potential targets for cancer immunotherapy. Although, different therapeutic approaches to target these immunosuppressive myeloid cells are being investigated. Here, we will provide the strategies for repolarization and revival of tumor-promoting myeloid cells (Table 2).
Blocking recruitment
Blocking the recruitment of MDSCs and TAMs may be a beneficial strategy for reducing tumorigenesis and immunosuppression. CCR2+ TAMs and MDSCs in TME are recruited by the CCL2 chemokine [70,71,72]. Blocking CCL2/CCR2 axis reverses MDSCs infiltration into the tumor, augmenting the effectiveness of the cancer immunotherapy [73]. In breast cancer models, removing CCR2 blockade induces tumor progression, migration, and angiogenesis [74]. Clinical studies are now underway for anti-CCR2 agents, including carlumab (CNTO 888), PF-04136309, MLN1202, BMS-813,160, and CCX872-B [75, 76].
The CXCL12/CXCR4 axis governs TAMs’ migration into hypoxic tumor areas through the endothelial barrier [77]. Targeting the CXCL12/CXCR4 axis in multiple cancer models, including prostate and breast cancer, reduces tumor burden and metastatic susceptibility by preventing TAM infiltration [78, 79].
Depleting macrophage populations in the TME
TAMs are among the most common and important non-neoplastic cell groups in the established TME. The differentiation of macrophages into tumor-suppressive M1 or tumor-promoting M2 types is an important stage in the formation of the TME. Implementing three strategies through this pivotal axis could pave for novel cancer treatment strategies. These strategies could alter M2 TAM survival and apoptotic mechanisms or disrupt their signaling pathways, suppress chemotactic potential toward the tumor, and reprogram M2 TAMs to produce M1 phenotype macrophages [80].
Bisphosphonates elicit myeloid cell cytotoxicity by preferentially targeting phagocytic cells, including TAMs [81]. Zoledronate, a third-generation bisphosphonate, is cytotoxic to TAMs that express matrix metalloproteinase-9 (MMP9) and improves macrophage anti-tumor activity by polarizing monocytes toward pro-inflammatory phenotype [76, 82]. Trabectedin, a drug mainly used for soft tissue malignancies, inhibits TAMs, enhancing anti-cancer adaptive immunity in response to anti-programmed cell death protein 1 (PD-1) treatment [83]. Trabectedin causes mononuclear phagocytes to undergo accelerated apoptosis. In animal tumor models, trabectedin reduced angiogenesis by selectively depleting monocytes/macrophages in the blood, spleens, and tumors [84].
Reprogramming metabolism
Several agents, including growth factors, could modify macrophages’ immune and metabolic responses in their residing microenvironment. This mechanism is reflected in the tricarboxylic acid (TCA) cycle disruption in M1 macrophages with the stimulation of inflammatory mediators resulting in IL-1 and Fatty acid synthesis and switching to pro-inflammatory phenotype [76, 85,86,87,88]. M2 macrophages, on the other hand, have an intact TCA cycle by external anti-inflammatory stimulation, which promotes mitochondrial oxidative phosphorylation (OXPHOS), yielding a higher ATP production [76, 89]. Inhibiting ATP production in M2 macrophages with an ATP synthase or a hexokinase inhibitor decreases anti-inflammatory characteristics and suppresses pro-tumorigenic function [90, 91].
Reprogramming cellular signaling
To induce tumoricidal potential in MDSCs and TAMs, several factors could be used to reprogram their signaling pathways, including colony-stimulating factor 1/colony-stimulating factor 1 receptor (CSF1/CSF1R) blockade, TLR agonists, PI3K inhibitors, CD40 agonists [64], and Class IIa histone deacetylase inhibitors (HDACis) [76, 92, 93]. Promising targets are the macrophage surface receptors that aid antibody-dependent cellular cytotoxicity/phagocytosis (ADCC/ADCP). Macrophages harbor a membrane protein called signal regulatory protein alpha (SIRP-α) binding to CD47 molecules expressed on tumoral cells, which help them evade tumor immunosurveillance [94]. However, anti-SIRPα antibodies cause tumor cell phagocytosis while preserving T-cells [95].
TLRs agonists could induce pro-inflammatory and anti-tumor phenotypes in TAMs. Feng et al. developed a glucomannan polysaccharide with acetyl modification to the degree of 1.8 (acGM-1.8), which stimulates TLR2 signaling and promotes macrophages toward becoming anti-tumor [96]. TLR7/8 agonist-loaded nanoparticles augment cancer immunotherapy via polarizing TAMs [97]. TLR-3 stimulation via modulating IFN-αβ signaling restricts tumor progression by skewing M2 macrophages to the M1 phenotype [98]. TLR 7/8 agonists also stimulate human MDSCs to differentiate toward anti-tumor M1-like macrophages, which may reverse the suppressive action of MDSCs [99].
CSF1/CSF1R blockade in pancreatic cancer models could enhance immune checkpoint T-cell therapy outcomes while reprogramming TAMs [100]. Moreover, blocking the CSF1/CSF1R axis reduces mesothelioma growth and improves anti-PDL1 immunotherapy efficacy [101], and CSF1R inhibition minimizes the development of cervical and mammary tumors in mice by lowering TAMs turnover and increasing the CD8+ T-cell infiltration [102]. Inappropriate response to immunotherapy in indoleamine 2,3-dioxygenase-expressing malignancies may be overcome by targeting MDSCs with CSF1R inhibition [103]. Pro-tumorigenic TAMs are reduced, and pro-tumorigenic PMN-MDSCs are recruited when CSF1R is inhibited [104]. Indeed, CSF1R suppression enabled tumor-infiltrating PMN-MDSCs to be recruited by carcinoma-associated fibroblasts. Thus, CXCR2 inhibitors may augment the anti-cancer effects of CSF1R inhibition by preventing PMN-MDSCs recruitment [104].
TAMs are sensitive to profound and abrupt reprogramming in the presence of a CD40 agonist when CSF-1R signaling is inhibited. Despite the short window of macrophage hyperactivation, simultaneous CSF-1R inhibition plus CD40 stimulation is adequate to establish a pro-inflammatory TME that revives an efficient immune response for T-cell immune checkpoint therapy [105]. Likewise, CD40 agonist, combined with CSF-1R, blockades reconditions TAMs and promotes potent anti-tumor immunity [106]. Activated macrophages with CD40 agonist invaded tumors immediately, were tumoricidal, and aided tumor stroma elimination [107]. In a pancreatic cancer mouse model, dendritic cell vaccination and CD40-agonist combined treatment enable T-cell-dependent anti-tumor immunotherapy [108].
A first in vivo evidence revealed that pharmacological suppression of the PI3K p110δ subunit inhibits the growth of breast cancer by specifically targeting cancer cells and macrophages [109]. Li et al. indicated that TAM accumulation in the glioblastoma microenvironment is suppressed by PI3K inhibition, which results in an extraordinary temozolomide response [110]. A pan-PI3K inhibitor (SF1126) reduced VEGF and other pro-angiogenic factors released by macrophages, blocking tumor-induced angiogenesis [111]. Joshi et al. demonstrated anti-tumor immunity by macrophage Syk-PI3Kγ axis [112]. Additionally, tumor immunosuppression is relieved by SRX3207, a novel dual Syk-PI3K inhibitor [112]. Clotrimazole has anti-cancer characteristics in a mouse melanoma model, functioning as a PI3K inhibitor and causing TAMs to repolarize [113].
HDAC inhibition with trichostatin-A increases anti-PD-L1-mediated tumor suppression and potentiates macrophage anti-tumor activity [114]. TMP195, an HDAC Class IIa inhibitor, may transform tumor-infiltrating monocytes and macrophages into cells able to sustain a robust CD8+ T-cell-mediated anti-tumor immune response in breast cancer and reduce metastasis [115, 116].
Immune checkpoint blockade (ICB)
The PD-1 and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) immune checkpoints are predominantly produced by effector immune cells, including T and NK cells. Targeting these molecules have exciting therapeutic potential by affecting myeloid biology [117, 118]. Because PD-L1 is expressed on MDSCs and TAMs, ICB using anti-PD-L1 may directly impact myeloid cell activities in TME [119]. There is a difference in response to PD-1 and PD-L1 inhibition in myeloid cells, with the latter leading to more potent immune responses by activating inflammasomes and expressing IL-18 [120]. The protective immune response to tumor cells requires inflammasome activation [121].
CD47SIRPα axis has been identified as a critical macrophage immune checkpoint. CD47 is a “don’t eat me” signal that is overexpressed in myeloid malignancies and causes tumors to evade macrophage phagocytosis. CD47 blockade causes leukemic cells to be engulfed and therapeutically eliminated [122]. CD47 blockade combined with trastuzumab eradicates HER2-positive breast cancer cells while also overcoming trastuzumab resistance [123]. Radioresistant breast cancer cells are eliminated when CD47 and HER2 are blocked [124].
Conclusion
According to the clinical and pre-clinical evidence, TAMCs play a dual role in cancer via anti-tumorigenic and pro-tumorigenic effects. TAMCs have pro-tumorigenic and immunosuppressive functions by different mechanisms including TGF-β and IL-10 anti-inflammatory cytokine secretion, ROS production, and mediation of angiogenesis through VEGF and HGF production. Hence, TAMCs could be actively involved in cancer progression, and immune escape results in poor prognosis, adverse clinical outcomes, and a low response rate to cancer treatment. Although diverse cancer-related immunotherapies such as ICBs have been investigated, targeting promoting pathways orchestrated by myeloid cells could shed a light on a new therapeutic approach and may improve cancer immunotherapy. Blocking myeloid cells’ recruitment, macrophage population depletion, reprogramming of metabolism, and cellular signaling might be considered helpful strategies for repolarization and revival of tumor-promoting myeloid cells.
Data availability
Not applicable.
Abbreviations
- mAb:
-
Monoclonal antibody.
- TAM:
-
Tumor-associated macrophage.
- TLR:
-
Toll-like receptor.
- HDAC:
-
Histone deacetylase.
- VEGF:
-
Vascular endothelial growth factor.
- CSF1:
-
Colony-stimulating factor 1.
- CSF1R:
-
Colony-stimulating factor 1 receptor.
- LTA:
-
Lipoteichoic acid.
- BTK:
-
Bruton tyrosine kinase.
- SIRPα:
-
Signal regulatory protein alpha.
References
Mui UN, Haley CT, Tyring SK. Viral Oncology: Molecular Biology and Pathogenesis. J Clin Med. 2017;6(12).
Guven-Maiorov E, Tsai C-J, Nussinov R. Oncoviruses Can Drive Cancer by Rewiring Signaling Pathways Through Interface Mimicry. Frontiers in Oncology. 2019;9.
Tornesello ML, Annunziata C, Tornesello AL, Buonaguro L, Buonaguro FM. Human Oncoviruses and p53 Tumor Suppressor Pathway Deregulation at the Origin of Human Cancers. Cancers (Basel). 2018;10(7).
Dzobo K. The Role of Viruses in Carcinogenesis and Molecular Targeting: From Infection to Being a Component of the Tumor Microenvironment. OMICS. 2021;25(6):358–71.
Goldszmid RS, Dzutsev A, Trinchieri G. Host immune response to infection and cancer: unexpected commonalities. Cell Host Microbe. 2014;15(3):295–305.
Schouppe E, De Baetselier P, Van Ginderachter JA, Sarukhan A. Instruction of myeloid cells by the tumor microenvironment: Open questions on the dynamics and plasticity of different tumor-associated myeloid cell populations. Oncoimmunology. 2012;1(7):1135–45.
Haas L, Obenauf AC. Allies or Enemies-The Multifaceted Role of Myeloid Cells in the Tumor Microenvironment. Front Immunol. 2019;10:2746.
Sica A, Porta C, Morlacchi S, Banfi S, Strauss L, Rimoldi M, et al. Origin and Functions of Tumor-Associated Myeloid Cells (TAMCs). Cancer Microenviron. 2012;5(2):133–49.
Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. 2018;154(1):3–20.
Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-Infiltrating Dendritic Cells in Cancer Pathogenesis. J Immunol. 2015;194(7):2985–91.
Ma Y, Shurin GV, Gutkin DW, Shurin MR. Tumor associated regulatory dendritic cells. Semin Cancer Biol. 2012;22(4):298–306.
Qiu Y, Chen T, Hu R, Zhu R, Li C, Ruan Y, et al. Next frontier in tumor immunotherapy: macrophage-mediated immune evasion. Biomark Res. 2021;9(1):72.
Zhu S, Luo Z, Li X, Han X, Shi S, Zhang T. Tumor-associated macrophages: role in tumorigenesis and immunotherapy implications. J Cancer. 2021;12(1):54–64.
Hourani T, Holden JA, Li W, Lenzo JC, Hadjigol S, O’Brien-Simpson NM. Tumor Associated Macrophages: Origin, Recruitment, Phenotypic Diversity, and Targeting. Frontiers in Oncology. 2021;11.
Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol. 2020;11:583084.
Xia Y, Shen S, Verma IM. NF-κB, an active player in human cancers. Cancer Immunol Res. 2014;2(9):823–30.
Zhang BC, Gao J, Wang J, Rao ZG, Wang BC, Gao JF. Tumor-associated macrophages infiltration is associated with peritumoral lymphangiogenesis and poor prognosis in lung adenocarcinoma. Med Oncol. 2011;28(4):1447–52.
Hu Y, He M-Y, Zhu L-F, Yang C-C, Zhou M-L, Wang Q, et al. Tumor-associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J Experimental Clin Cancer Res. 2016;35(1):12.
Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21(8):485–98.
Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019;120(1):16–25.
Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799–807.
Ohl K, Tenbrock K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front Immunol. 2018;9:2499.
Masucci MT, Minopoli M, Carriero MV. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front Oncol. 2019;9:1146.
Furumaya C, Martinez-Sanz P, Bouti P, Kuijpers TW, Matlung HL. Plasticity in Pro- and Anti-tumor Activity of Neutrophils: Shifting the Balance. Front Immunol. 2020;11:2100.
Tazzyman S, Lewis CE, Murdoch C. Neutrophils: key mediators of tumour angiogenesis. Int J Exp Pathol. 2009;90(3):222–31.
Shen M, Hu P, Donskov F, Wang G, Liu Q, Du J. Tumor-associated neutrophils as a new prognostic factor in cancer: a systematic review and meta-analysis. PLoS ONE. 2014;9(6):e98259.
Shannon-Lowe C, Rickinson AB, Bell AI. Epstein-Barr virus-associated lymphomas. Philos Trans R Soc Lond B Biol Sci. 2017;372(1732).
Collins PJ, Fox CP, George L, Pearce H, Ryan G, De Santo C, et al. Characterizing EBV-associated lymphoproliferative diseases and the role of myeloid-derived suppressor cells. Blood. 2021;137(2):203–15.
Cai TT, Ye SB, Liu YN, He J, Chen QY, Mai HQ, et al. LMP1-mediated glycolysis induces myeloid-derived suppressor cell expansion in nasopharyngeal carcinoma. PLoS Pathog. 2017;13(7):e1006503.
Katahira Y, Higuchi H, Matsushita H, Yahata T, Yamamoto Y, Koike R, et al. Increased Granulopoiesis in the Bone Marrow following Epstein-Barr Virus Infection. Sci Rep. 2019;9(1):13445.
Li X, Li JL, Jiang N, Chen J, Liang ZM, Zhao ZL, et al. Accumulation of LOX-1(+) PMN-MDSCs in nasopharyngeal carcinoma survivors with chronic hepatitis B might permit immune tolerance to epstein-barr virus and relate to tumor recurrence. Aging. 2020;13(1):437–49.
Song Y, Li Q, Liao S, Zhong K, Jin Y, Zeng T. Epstein-Barr virus-encoded miR-BART11 promotes tumor-associated macrophage-induced epithelial-mesenchymal transition via targeting FOXP1 in gastric cancer. Virology. 2020;548:6–16.
Zhang B, Miao T, Shen X, Bao L, Zhang C, Yan C, et al. EB virus-induced ATR activation accelerates nasopharyngeal carcinoma growth via M2-type macrophages polarization. Cell Death Dis. 2020;11(9):742.
Ooft ML, van Ipenburg JA, Sanders ME, Kranendonk M, Hofland I, de Bree R, et al. Prognostic role of tumour-associated macrophages and regulatory T cells in EBV-positive and EBV-negative nasopharyngeal carcinoma. J Clin Pathol. 2018;71(3):267–74.
Sato A, Yamakawa N, Okuyama K, Kotani A, Nakamura N, Ando K. The Critical Interaction Between Epstein-Barr Virus (EBV) Positive B-Cells and Tumor Associated Macrophages (TAMs). Blood. 2014;124(21):2989.
Shimakage M, Sakamoto H. Macrophage involvement in Epstein-Barr virus-related tumors. Exp Ther Med. 2010;1(2):285–91.
Kamper P, Bendix K, Hamilton-Dutoit S, Honoré B, Nyengaard JR, d’Amore F. Tumor-infiltrating macrophages correlate with adverse prognosis and Epstein-Barr virus status in classical Hodgkin’s lymphoma. Haematologica. 2011;96(2):269–76.
Mavili HS, Isisag A, Tan A, Miskioglu M, Baraz LS, Nese N. Relationship of Tumor-Associated Macrophage Population Detected by CD68 PG-M1, CD68 KP1, and CD163 with Latent EBV Infection and Prognosis in Classical Hodgkin Lymphoma. Turk Patoloji Derg. 2021;37(2):130–8.
Xie Y. Hepatitis B, Virus-Associated. Hepatocellular Carcinoma. Adv Exp Med Biol. 2017;1018:11–21.
Li T, Zhang X, Lv Z, Gao L, Yan H. Increased Expression of Myeloid-Derived Suppressor Cells in Patients with HBV-Related Hepatocellular Carcinoma. Biomed Res Int. 2020;2020:6527192.
Pal S, Nandi M, Dey D, Chakraborty BC, Shil A, Ghosh S, et al. Myeloid-derived suppressor cells induce regulatory T cells in chronically HBV infected patients with high levels of hepatitis B surface antigen and persist after antiviral therapy. Aliment Pharmacol Ther. 2019;49(10):1346–59.
Zeng Y, Li Y, Xu Z, Gan W, Lu L, Huang X, et al. Myeloid-derived suppressor cells expansion is closely associated with disease severity and progression in HBV-related acute-on-chronic liver failure. J Med Virol. 2019;91(8):1510–8.
Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14(10):996–1006.
Yang P, Markowitz GJ, Wang XF. The hepatitis B virus-associated tumor microenvironment in hepatocellular carcinoma. Natl Sci Rev. 2014;1(3):396–412.
Bility MT, Cheng L, Zhang Z, Luan Y, Li F, Chi L, et al. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog. 2014;10(3):e1004032.
Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206(6):1327–37.
Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology. 2012;56(4):1342–51.
Tong HV, Toan NL, Song LH, Bock CT, Kremsner PG, Velavan TP. Hepatitis B virus-induced hepatocellular carcinoma: functional roles of MICA variants. J Viral Hepat. 2013;20(10):687–98.
Ju Y, Hou N, Meng J, Wang X, Zhang X, Zhao D, et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J Hepatol. 2010;52(3):322–9.
Peppa D, Micco L, Javaid A, Kennedy PT, Schurich A, Dunn C, et al. Blockade of immunosuppressive cytokines restores NK cell antiviral function in chronic hepatitis B virus infection. PLoS Pathog. 2010;6(12):e1001227.
Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59–73.
Zhai N, Li H, Song H, Yang Y, Cui A, Li T, et al. Hepatitis C Virus Induces MDSCs-Like Monocytes through TLR2/PI3K/AKT/STAT3 Signaling. PLoS ONE. 2017;12(1):e0170516.
Tacke RS, Lee HC, Goh C, Courtney J, Polyak SJ, Rosen HR, et al. Myeloid suppressor cells induced by hepatitis C virus suppress T-cell responses through the production of reactive oxygen species. Hepatology. 2012;55(2):343–53.
Goh CC, Roggerson KM, Lee HC, Golden-Mason L, Rosen HR, Hahn YS. Hepatitis C Virus-Induced Myeloid-Derived Suppressor Cells Suppress NK Cell IFN-γ Production by Altering Cellular Metabolism via Arginase-1. J Immunol. 2016;196(5):2283–92.
Ren JP, Zhao J, Dai J, Griffin JW, Wang L, Wu XY, et al. Hepatitis C virus-induced myeloid-derived suppressor cells regulate T-cell differentiation and function via the signal transducer and activator of transcription 3 pathway. Immunology. 2016;148(4):377–86.
Wang M, Ping Y, Li Z, Li J, Zhang Z, Yue D, et al. Polarization of granulocytic myeloid-derived suppressor cells by hepatitis C core protein is mediated via IL-10/STAT3 signalling. J Viral Hepat. 2019;26(2):246–57.
Thakuri BKC, Zhang J, Zhao J, Nguyen LN, Nguyen LNT, Khanal S, et al. LncRNA HOTAIRM1 promotes MDSC expansion and suppressive functions through the HOXA1-miR124 axis during HCV infection. Sci Rep. 2020;10(1):22033.
Wang L, Cao D, Wang L, Zhao J, Nguyen LN, Dang X, et al. HCV-associated exosomes promote myeloid-derived suppressor cell expansion via inhibiting miR-124 to regulate T follicular cell differentiation and function. Cell Discov. 2018;4:51.
Thakuri BKC, Zhang J, Zhao J, Nguyen LN, Nguyen LNT, Schank M, et al. HCV-Associated Exosomes Upregulate RUNXOR and RUNX1 Expressions to Promote MDSC Expansion and Suppressive Functions through STAT3-miR124 Axis. Cells. 2020;9(12).
Mesri EA, Cesarman E, Boshoff C. Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer. 2010;10(10):707–19.
Ballon G, Akar G, Cesarman E. Systemic expression of Kaposi sarcoma herpesvirus (KSHV) Vflip in endothelial cells leads to a profound proinflammatory phenotype and myeloid lineage remodeling in vivo. PLoS Pathog. 2015;11(1):e1004581.
Campbell DM, Rappocciolo G, Jenkins FJ, Rinaldo CR. Dendritic cells: key players in human herpesvirus 8 infection and pathogenesis. Front Microbiol. 2014;5:452.
Jenkins FJ, Minas TZ, Tang W, Dorsey TH, Ambs S. Human herpesvirus 8 infection is associated with prostate cancer among IFNL4-∆G carriers. Prostate Cancer and Prostatic Diseases. 2022.
Chavoshpour-Mamaghani S, Shoja Z, Mollaei-Kandelous Y, Sharifian K, Jalilvand S. The prevalence of human herpesvirus 8 in normal, premalignant, and malignant cervical samples of Iranian women. Virol J. 2021;18(1):144.
zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2(5):342–50.
Barros MR Jr, de Melo CML, Barros M, de Cássia Pereira de Lima R, de Freitas AC, Venuti A. Activities of stromal and immune cells in HPV-related cancers. J Exp Clin Cancer Res. 2018;37(1):137.
Stone SC, Rossetti RA, Lima AM, Lepique AP. HPV associated tumor cells control tumor microenvironment and leukocytosis in experimental models. Immun Inflamm Dis. 2014;2(2):63–75.
Ma X, Sheng S, Wu J, Jiang Y, Gao X, Cen X, et al. LncRNAs as an intermediate in HPV16 promoting myeloid-derived suppressor cell recruitment of head and neck squamous cell carcinoma. Oncotarget. 2017;8(26):42061–75.
Diniz MO, Sales NS, Silva JR, Ferreira LC. Protection against HPV-16-Associated Tumors Requires the Activation of CD8 + Effector Memory T Cells and the Control of Myeloid-Derived Suppressor Cells. Mol Cancer Ther. 2016;15(8):1920–30.
Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–5.
Ren G, Zhao X, Wang Y, Zhang X, Chen X, Xu C, et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell. 2012;11(6):812–24.
Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, et al. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015;12(2):244–57.
Fridlender ZG, Buchlis G, Kapoor V, Cheng G, Sun J, Singhal S, et al. CCL2 blockade augments cancer immunotherapy. Cancer Res. 2010;70(1):109–18.
Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515(7525):130–3.
Pathria P, Louis TL, Varner JA. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019;40(4):310–27.
Kumar V, Giacomantonio MA, Gujar S. Role of Myeloid Cells in Oncolytic Reovirus-Based Cancer Therapy. Viruses. 2021;13(4).
Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC, et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015;75(17):3479–91.
Scala S. Molecular Pathways: Targeting the CXCR4-CXCL12 Axis–Untapped Potential in the Tumor Microenvironment. Clin Cancer Res. 2015;21(19):4278–85.
Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16(11):2927–31.
Zheng X, Turkowski K, Mora J, Brüne B, Seeger W, Weigert A, et al. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget. 2017;8(29):48436–52.
Rogers TL, Holen I. Tumour macrophages as potential targets of bisphosphonates. J Transl Med. 2011;9:177.
Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114(5):623–33.
Belgiovine C, Frapolli R, Liguori M, Digifico E, Colombo FS, Meroni M, et al. Inhibition of tumor-associated macrophages by trabectedin improves the antitumor adaptive immunity in response to anti-PD-1 therapy. Eur J Immunol. 2021;51(11):2677–86.
Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell. 2013;23(2):249–62.
O’Neill Luke AJ. A Broken Krebs Cycle in Macrophages. Immunity. 2015;42(3):393–4.
Van den Bossche J, O’Neill LA, Menon D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017;38(6):395–406.
O’Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–65.
Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42(3):419–30.
Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Frontiers in Immunology. 2019;10.
Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016;17(3):684–96.
Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity. 2016;45(4):817–30.
Kowal J, Kornete M, Joyce JA. Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy. 2019;11(8):677–89.
Li X, Liu R, Su X, Pan Y, Han X, Shao C, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019;18(1):177.
Barclay AN, Berg TKvd. The Interaction Between Signal Regulatory Protein Alpha (SIRPα) and CD47: Structure, Function, and Therapeutic Target. Annu Rev Immunol. 2014;32(1):25–50.
Andrejeva G, Capoccia BJ, Hiebsch RR, Donio MJ, Darwech IM, Puro RJ, et al. Novel SIRPα Antibodies That Induce Single-Agent Phagocytosis of Tumor Cells while Preserving T Cells. J Immunol. 2021;206(4):712–21.
Feng Y, Mu R, Wang Z, Xing P, Zhang J, Dong L, et al. A toll-like receptor agonist mimicking microbial signal to generate tumor-suppressive macrophages. Nat Commun. 2019;10(1):2272.
Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomedical Eng. 2018;2(8):578–88.
Vidyarthi A, Khan N, Agnihotri T, Negi S, Das DK, Aqdas M, et al. TLR-3 Stimulation Skews M2 Macrophages to M1 Through IFN-αβ Signaling and Restricts Tumor Progression. Front Immunol. 2018;9:1650.
Wang J, Shirota Y, Bayik D, Shirota H, Tross D, Gulley JL, et al. Effect of TLR agonists on the differentiation and function of human monocytic myeloid-derived suppressor cells. J Immunol. 2015;194(9):4215–21.
Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–69.
Magkouta SF, Vaitsi PC, Pappas AG, Iliopoulou M, Kosti CN, Psarra K, et al. CSF1/CSF1R Axis Blockade Limits Mesothelioma and Enhances Efficiency of Anti-PDL1 Immunotherapy. Cancers (Basel). 2021;13(11).
Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology. 2013;2(12):e26968.
Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine. 2016;6:50–8.
Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell. 2017;32(5):654 – 68.e5.
Hoves S, Ooi CH, Wolter C, Sade H, Bissinger S, Schmittnaegel M, et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J Exp Med. 2018;215(3):859–76.
Wiehagen KR, Girgis NM, Yamada DH, Smith AA, Chan SR, Grewal IS, et al. Combination of CD40 Agonism and CSF-1R Blockade Reconditions Tumor-Associated Macrophages and Drives Potent Antitumor Immunity. Cancer Immunol Res. 2017;5(12):1109–21.
Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6.
Lau SP, van Montfoort N, Kinderman P, Lukkes M, Klaase L, van Nimwegen M, et al. Dendritic cell vaccination and CD40-agonist combination therapy licenses T cell-dependent antitumor immunity in a pancreatic carcinoma murine model. J Immunother Cancer. 2020;8(2).
Goulielmaki E, Bermudez-Brito M, Andreou M, Tzenaki N, Tzardi M, de Bree E, et al. Pharmacological inactivation of the PI3K p110δ prevents breast tumour progression by targeting cancer cells and macrophages. Cell Death Dis. 2018;9(6):678.
Li J, Kaneda MM, Ma J, Li M, Shepard RM, Patel K, et al. PI3Kγ inhibition suppresses microglia/TAM accumulation in glioblastoma microenvironment to promote exceptional temozolomide response. Proceedings of the National Academy of Sciences. 2021;118(16):e2009290118.
Joshi S, Singh AR, Zulcic M, Durden DL. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1α and HIF2α stability and tumor growth, angiogenesis, and metastasis. Mol Cancer Res. 2014;12(10):1520–31.
Joshi S, Liu KX, Zulcic M, Singh AR, Skola D, Glass CK, et al. Macrophage Syk-PI3Kγ Inhibits Antitumor Immunity: SRX3207, a Novel Dual Syk-PI3K Inhibitory Chemotype Relieves Tumor Immunosuppression. Mol Cancer Ther. 2020;19(3):755–64.
Ochioni AC, Imbroisi Filho R, Esteves AM, Leandro JGB, Demaria TM, do Nascimento Júnior JX, et al. Clotrimazole presents anticancer properties against a mouse melanoma model acting as a PI3K inhibitor and inducing repolarization of tumor-associated macrophages. Biochim Biophys Acta Mol Basis Dis. 2021;1867(12):166263.
Li X, Su X, Liu R, Pan Y, Fang J, Cao L, et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene. 2021;40(10):1836–50.
Cassetta L, Pollard JW. Repolarizing macrophages improves breast cancer therapy. Cell Res. 2017;27(8):963–4.
Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543(7645):428–32.
Chen J, Sun HW, Yang YY, Chen HT, Yu XJ, Wu WC, et al. Reprogramming immunosuppressive myeloid cells by activated T cells promotes the response to anti-PD-1 therapy in colorectal cancer. Signal Transduct Target Ther. 2021;6(1):4.
Pico de Coaña Y, Masucci G, Hansson J, Kiessling R. Myeloid-derived suppressor cells and their role in CTLA-4 blockade therapy. Cancer Immunol Immunother. 2014;63(9):977–83.
Liu M, Zhou J, Liu X, Feng Y, Yang W, Wu F, et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut. 2020;69(2):365–79.
Bar N, Costa F, Das R, Duffy A, Samur M, McCachren S, et al. Differential effects of PD-L1 versus PD-1 blockade on myeloid inflammation in human cancer. JCI Insight. 2020;5(12).
Gasparoto TH, de Oliveira CE, de Freitas LT, Pinheiro CR, Hori JI, Garlet GP, et al. Inflammasome activation is critical to the protective immune response during chemically induced squamous cell carcinoma. PLoS ONE. 2014;9(9):e107170.
Chao MP, Takimoto CH, Feng DD, McKenna K, Gip P, Liu J, et al. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front Oncol. 2019;9:1380.
Upton R, Banuelos A, Feng D, Biswas T, Kao K, McKenna K, et al. Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proceedings of the National Academy of Sciences. 2021;118(29):e2026849118.
Candas-Green D, Xie B, Huang J, Fan M, Wang A, Menaa C, et al. Dual blockade of CD47 and HER2 eliminates radioresistant breast cancer cells. Nat Commun. 2020;11(1):4591.
Liao YX, Fu ZZ, Zhou CH, Shan LC, Wang ZY, Yin F, et al. AMD3100 reduces CXCR4-mediated survival and metastasis of osteosarcoma by inhibiting JNK and Akt, but not p38 or Erk1/2, pathways in in vitro and mouse experiments. Oncol Rep. 2015;34(1):33–42.
Evans CA, Liu T, Lescarbeau A, Nair SJ, Grenier L, Pradeilles JA, et al. Discovery of a Selective Phosphoinositide-3-Kinase (PI3K)-γ Inhibitor (IPI-549) as an Immuno-Oncology Clinical Candidate. ACS Med Chem Lett. 2016;7(9):862–7.
Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10:31.
Zhu S, Jung J, Victor E, Arceo J, Gokhale S, Xie P. Clinical Trials of the BTK Inhibitors Ibrutinib and Acalabrutinib in Human Diseases Beyond B Cell Malignancies. Front Oncol. 2021;11:737943.
Yang L, Zhang Y. Tumor-associated macrophages, potential targets for cancer treatment. Biomark Res. 2017;5:25.
Brana I, Calles A, LoRusso PM, Yee LK, Puchalski TA, Seetharam S, et al. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol. 2015;10(1):111–23.
Loberg RD, Ying C, Craig M, Yan L, Snyder LA, Pienta KJ. CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia. 2007;9(7):556–62.
Zollo M, Di Dato V, Spano D, De Martino D, Liguori L, Marino N, et al. Targeting monocyte chemotactic protein-1 synthesis with bindarit induces tumor regression in prostate and breast cancer animal models. Clin Exp Metastasis. 2012;29(6):585–601.
Pradel LP, Ooi CH, Romagnoli S, Cannarile MA, Sade H, Rüttinger D, et al. Macrophage Susceptibility to Emactuzumab (RG7155) Treatment. Mol Cancer Ther. 2016;15(12):3077–86.
Gambardella V, Castillo J, Tarazona N, Gimeno-Valiente F, Martínez-Ciarpaglini C, Cabeza-Segura M, et al. The role of tumor-associated macrophages in gastric cancer development and their potential as a therapeutic target. Cancer Treat Rev. 2020;86:102015.
Manji GA, Van Tine BA, Lee SM, Raufi AG, Pellicciotta I, Hirbe AC, et al. A Phase I Study of the Combination of Pexidartinib and Sirolimus to Target Tumor-Associated Macrophages in Unresectable Sarcoma and Malignant Peripheral Nerve Sheath Tumors. Clin Cancer Res. 2021;27(20):5519–27.
Harb WA, Johnson ML, Goldman JW, Weise AM, Call JA, Dudek AZ, et al. A phase 1b/2 study of ARRY-382, an oral inhibitor of colony stimulating factor 1 receptor (CSF1R), in combination with pembrolizumab (Pembro) for the treatment of patients (Pts) with advanced solid tumors. J Clin Oncol. 2017;35(15_suppl):TPS3110-TPS.
Lu X, Meng T. Depletion of tumor-associated macrophages enhances the anti-tumor effect of docetaxel in a murine epithelial ovarian cancer. Immunobiology. 2019;224(3):355–61.
Papadopoulos KP, Gluck L, Martin LP, Olszanski AJ, Tolcher AW, Ngarmchamnanrith G, et al. First-in-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients with Advanced Solid Tumors. Clin Cancer Res. 2017;23(19):5703–10.
Laoui D, Van Overmeire E, De Baetselier P, Van Ginderachter JA, Raes G. Functional Relationship between Tumor-Associated Macrophages and Macrophage Colony-Stimulating Factor as Contributors to Cancer Progression. Front Immunol. 2014;5:489.
Dowlati A, Harvey RD, Carvajal RD, Hamid O, Klempner SJ, Kauh JSW, et al. LY3022855, an anti-colony stimulating factor-1 receptor (CSF-1R) monoclonal antibody, in patients with advanced solid tumors refractory to standard therapy: phase 1 dose-escalation trial. Invest New Drugs. 2021;39(4):1057–71.
Vonderheide RH. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med. 2020;71:47–58.
Byrne KT, Betts CB, Mick R, Sivagnanam S, Bajor DL, Laheru DA, et al. Neoadjuvant Selicrelumab, an Agonist CD40 Antibody, Induces Changes in the Tumor Microenvironment in Patients with Resectable Pancreatic Cancer. Clin Cancer Res. 2021;27(16):4574–86.
O’Hara MH, O’Reilly EM, Varadhachary G, Wolff RA, Wainberg ZA, Ko AH, et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol. 2021;22(1):118–31.
Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25(7):876–83.
Cen X, Zhu G, Yang J, Yang J, Guo J, Jin J, et al. TLR1/2 Specific Small-Molecule Agonist Suppresses Leukemia Cancer Cell Growth by Stimulating Cytotoxic T Lymphocytes. Adv Sci (Weinh). 2019;6(10):1802042.
Megías J, Martínez A, San-Miguel T, Gil-Benso R, Muñoz-Hidalgo L, Albert-Bellver D, et al. Pam(3)CSK(4), a TLR2 ligand, induces differentiation of glioblastoma stem cells and confers susceptibility to temozolomide. Invest New Drugs. 2020;38(2):299–310.
Long EM, Millen B, Kubes P, Robbins SM. Lipoteichoic acid induces unique inflammatory responses when compared to other toll-like receptor 2 ligands. PLoS ONE. 2009;4(5):e5601.
Sultan H, Wu J, Fesenkova VI, Fan AE, Addis D, Salazar AM, et al. Poly-IC enhances the effectiveness of cancer immunotherapy by promoting T cell tumor infiltration. J Immunother Cancer. 2020;8(2).
Gao H-X, Bhattacharya S, Matheny CJ, Yanamandra N, Zhang S-Y, Emerich H, et al. Synergy of TLR4 agonist GSK1795091, an innate immune activator, with agonistic antibody against co-stimulatory immune checkpoint molecule OX40 in cancer immunotherapy. J Clin Oncol. 2018;36(15_suppl):12055-.
Nilsen NJ, Deininger S, Nonstad U, Skjeldal F, Husebye H, Rodionov D, et al. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J Leukoc Biol. 2008;84(1):280–91.
Diab A, Marcondes M, Kotzin B, Tagliaferri MA, Hoch U, Li Y, et al. Phase Ib: Preliminary clinical activity and immune activation for NKTR-262 [TLR 7/8 agonist] plus NKTR-214 [CD122-biased agonist] in patients (pts) with locally advanced or metastatic solid tumors (REVEAL Phase Ib/II Trial). J Clin Oncol. 2019;37(8_suppl):26-.
Chi H, Li C, Zhao FS, Zhang L, Ng TB, Jin G, et al. Anti-tumor Activity of Toll-Like Receptor 7 Agonists. Front Pharmacol. 2017;8:304.
Zanker DJ, Spurling AJ, Brockwell NK, Owen KL, Zakhour JM, Robinson T, et al. Intratumoral administration of the Toll-like receptor 7/8 agonist 3M-052 enhances interferon-driven tumor immunogenicity and suppresses metastatic spread in preclinical triple-negative breast cancer. Clin Transl Immunology. 2020;9(9):e1177.
Wang D, Jiang W, Zhu F, Mao X, Agrawal S. Modulation of the tumor microenvironment by intratumoral administration of IMO-2125, a novel TLR9 agonist, for cancer immunotherapy. Int J Oncol. 2018;53(3):1193–203.
Sabree SA, Voigt AP, Blackwell SE, Vishwakarma A, Chimenti MS, Salem AK, et al. Direct and indirect immune effects of CMP-001, a virus-like particle containing a TLR9 agonist. J Immunother Cancer. 2021;9(6).
Wang S, Campos J, Gallotta M, Gong M, Crain C, Naik E, et al. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8 + T cells. Proc Natl Acad Sci U S A. 2016;113(46):E7240-e9.
Mathias MD, Sockolosky JT, Chang AY, Tan KS, Liu C, Garcia KC, et al. CD47 blockade enhances therapeutic activity of TCR mimic antibodies to ultra-low density cancer epitopes. Leukemia. 2017;31(10):2254–7.
Petrova PS, Viller NN, Wong M, Pang X, Lin GH, Dodge K, et al. TTI-621 (SIRPαFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin Cancer Res. 2017;23(4):1068–79.
Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D, et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J Clin Oncol. 2019;37(12):946–53.
La Fleur L, Boura VF, Alexeyenko A, Berglund A, Pontén V, Mattsson JSM, et al. Expression of scavenger receptor MARCO defines a targetable tumor-associated macrophage subset in non-small cell lung cancer. Int J Cancer. 2018;143(7):1741–52.
Min AKT, Mimura K, Nakajima S, Okayama H, Saito K, Sakamoto W, et al. Therapeutic potential of anti-VEGF receptor 2 therapy targeting for M2-tumor-associated macrophages in colorectal cancer. Cancer Immunol Immunother. 2021;70(2):289–98.
Zhang Q, Wang J, Yadav DK, Bai X, Liang T. Glucose Metabolism: The Metabolic Signature of Tumor Associated Macrophage. Front Immunol. 2021;12:702580.
Lee C, Jeong H, Bae Y, Shin K, Kang S, Kim H, et al. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J Immunother Cancer. 2019;7(1):147.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Conception and manuscript design: R.J. Collection of data: A.A. P.F. S.P. A.R.D. and R.J. Manuscript writing: A.A. P.F. S.P. A.R.D. and R.J. Designing of the figure: A.R.D. Made important revisions and confirmed final revision: R .J. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Aghamajidi, A., Farhangnia, P., Pashangzadeh, S. et al. Tumor-promoting myeloid cells in the pathogenesis of human oncoviruses: potential targets for immunotherapy. Cancer Cell Int 22, 327 (2022). https://doi.org/10.1186/s12935-022-02727-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-022-02727-3