
Abstract
Background
Antenatal infection/inflammation is associated with disturbances in neuronal connectivity, impaired cortical growth and poor neurodevelopmental outcomes. The pathophysiological substrate that underpins these changes is poorly understood. We tested the hypothesis that progressive inflammation in late gestation fetal sheep would alter cortical neuronal microstructure and neural function assessed using electroencephalogram band power analysis.
Methods
Fetal sheep (0.85 of gestation) were surgically instrumented for continuous electroencephalogram (EEG) recording and randomly assigned to repeated saline (control; n = 9) or LPS (0 h = 300 ng, 24 h = 600 ng, 48 h = 1200 ng; n = 8) infusions to induce inflammation. Sheep were euthanised 4 days after the first LPS infusion for assessment of inflammatory gene expression, histopathology and neuronal dendritic morphology in the somatosensory cortex.
Results
LPS infusions increased delta power between 8 and 50 h, with reduced beta power from 18 to 96 h (P < 0.05 vs. control). Basal dendritic length, numbers of dendritic terminals, dendritic arborisation and numbers of dendritic spines were reduced in LPS-exposed fetuses (P < 0.05 vs. control) within the somatosensory cortex. Numbers of microglia and interleukin (IL)-1β immunoreactivity were increased in LPS-exposed fetuses compared with controls (P < 0.05). There were no differences in total numbers of cortical NeuN + neurons or cortical area between the groups.
Conclusions
Exposure to antenatal infection/inflammation was associated with impaired dendritic arborisation, spine number and loss of high-frequency EEG activity, despite normal numbers of neurons, that may contribute to disturbed cortical development and connectivity.
Similar content being viewed by others

Introduction
Exposure to antenatal infection/inflammation is a leading cause of brain injury and neurodevelopmental impairment [1]. For example, exposure to infection/inflammation increases the risk of cerebral palsy by 2- to 12-fold in near-term/term infants [2, 3]. Improving our understanding of the pathogenesis of brain injury is essential for better injury detection and development of interventions to improve outcomes, and reduce the socioeconomic burdens on the affected individuals, their families and society [4].
Perinatal infection/inflammation is common in low, middle and high-income countries [5], and is associated with grey matter abnormalities. For example, neuroimaging studies have shown that chorioamnionitis is independently associated with reduced sulcal depth and cortical volume in the temporal lobe, without overt grey matter injury [6, 7].
In rodents, chorioamnionitis induced with a single intrauterine injection of lipopolysaccharide (LPS) was associated with reduced dendritic arborisation in cortical neuronal cultures [8]. In pregnant rat dams, twelve hourly intraperitoneal (i.p.) injections of inactivated group B Streptococcus (109 CFU) between G19 and G22 (term) led to cortical thinning and neuromotor impairments in the offspring at postnatal day 40 [9]. Similarly, mild-to-moderate inflammation in neonatal rats using daily i.p. LPS (0.3 mg/kg) injections from P1–3 reduced dendritic arborisation and spine formation in cortical pyramidal neurons without overt neuronal loss [10].
While these preclinical data provide a compelling link between acute inflammation and impaired cortical development, the most common clinical fetal and neonatal scenario involves a progressive inflammatory response without severe fetal compromise [11]. Consistent with this, in fetal sheep live bacterial inoculation is associated with progressive systemic inflammation [12]. Similarly, recent clinical evidence strongly suggests that antenatal inflammation is a progressive process that persists into the newborn period [13] and supports the concept that sustained inflammation is associated with long-term impairments in brain development [14,15,16,17].
It is important to find ways to rapidly and non-invasively identify such progressive inflammation-induced injury at the bedside. Electroencephalography (EEG) is widely used to identify functional impairments in cases of preterm and term encephalopathy [18, 19]. However, the relationship between EEG and abnormal development of the neuronal microstructure is poorly understood. Thus, in the present study we tested the hypothesis that progressive antenatal inflammation induced by repeated, increasing-dose infusions of Gramnegative LPS would be associated with cortical inflammation and altered neuronal microstructural development. Furthermore, we hypothesised that pathological changes associated with altered cortical neuronal microstructural development could be detected using EEG assessment of neural function.
Materials and methods
All procedures were approved by the Hudson Institute of Medical Research Animal Ethics committee and were conducted in accordance with the National Health and Medical Research Council Code of Practice for the Care and Use of Animals for Scientific Purposes (Eighth Edition). The experiments are reported in accordance with the ARRIVE guidelines for reporting animal research [20]. In this study, we compared two groups of interest: (i) vehicle controls and (ii) antenatal inflammation. The key outcome measures were: (i) cortical inflammation, (ii) neuronal microstructural development, assessed using dendritic length, numbers of dendritic terminals, neuronal arborisation and numbers of dendritic spines and (iii) EEG measures of neuronal function. Seventeen pregnant Border-Leicester ewes bearing singleton or twin fetuses underwent aseptic surgery at either 124 or 125 days of gestation. Food but not water was withdrawn approximately 18 h before surgery. Anaesthesia was induced by i.v injection of sodium thiopentone (20 mL) and maintained using 2–3% isoflurane in oxygen (Bomac Animal Health, New South Wales, Australia). Ewes received prophylactic antibiotics (ampicillin: 1 g i.v; Austrapen, Lennon Healthcare, St. Leonards, NSW, Australia, and engemycin: 500 mg i.v; Schering-Plough, Upper Hutt, New Zealand) immediately before surgery. Isoflurane levels, heart rate, oxygen saturation, and respiratory rate were continuously monitored throughout surgery by trained anaesthetic staff.
Fetal instrumentation
A midline maternal laparotomy was performed, the fetus was exposed and partially removed from the uterus for implantation of polyvinyl catheters into the right brachiocephalic artery and amniotic cavity. In twin pregnancies, only one twin was instrumented. Two pairs of electroencephalograph (EEG) electrodes (AS633-7SSF; Cooner Wire, Chatsworth, CA, USA) were placed through burr holes onto the dura over the parasagittal parietal (somatosensory) cortex (10 and 20 mm anterior to bregma, and 10 mm lateral) and secured using surgical bone wax and cyanoacrylate glue. A catheter was inserted into the left fetal axillary vein for administration of post-operative antibiotics and lipopolysaccharide (LPS) or vehicle (saline). The fetus was returned to the uterus in its original orientation and all fetal leads were exteriorised through the maternal flank. A catheter was inserted into the maternal jugular vein for administration of post-operative antibiotics and euthanasia at the end of the experimental period. At the completion of surgery, ewes received fentanyl for 3 days via a transdermal patch placed on the left hind leg (75 μg/h; Janssen Cilag, North Ryde, NSW, USA).
Ewes were housed together in separate pens in a temperature controlled (20 ± 2 °C and relative humidity of 50 ± 10%) room with a 12-h light–dark cycle with access to food and water ad libitum. Four to five days of post-operative recovery were allowed before experiments commenced. Ewes and fetuses received daily i.v. infusions of ampicillin (800 mg, maternal i.v. and 200 mg, fetal i.v.) and engemycin (500 mg, maternal i.v.) for three consecutive days after surgery. Catheters were maintained patent with a continuous infusion of heparinised saline (25 IU/mL) at a rate of (0.2 mL/h).
Experimental recordings
Fetal EEG was continuously recorded from 24 h prior to the first saline or LPS infusion (129 days of gestation) until the end of the experiment (134 days of gestation). The analogue fetal EEG signal was bandpass-filtered with a cut-off frequency set at 1 and 22 Hz and digitised at a sampling frequency of 400 Hz. EEG power was derived from the analogue signal, whilst spectral edge was calculated as the frequency below which 90% of the intensity was present. Relative (%) spectral power in the Δ (0–3.9 Hz), θ (4–7.9 Hz), ɑ (8–12.9 Hz), and β (13–22 Hz) frequency bands were quantified. This involved calculating power spectra, by fast Fourier transform, of the EEG on sequential epochs using a 10-s Hanning window to minimise spectral leakage, as previously described [21, 22].
Experimental protocol
Experiments started at 129 days of gestation (term is ~ 147 days). This study examined 0.85 gestation fetal sheep, at an age when brain development is broadly equivalent to that of a near term/term human infant [23].
Fetuses were randomly allocated to two groups: vehicle (saline, n = 9 [6 males, 3 females]) or LPS (Escherichia coli, O55:B5, MilliporeSigma, MO, USA; n = 8 [6 males, 2 females]). Fetuses received 300 ng, 600 ng, and 1200 ng infusions of LPS diluted in 2 mL of saline i.v. (infusion rate: 1 mL/min) at 0, 24 and 48 h, respectively, along with a 3 mL vehicle (saline) infusion at a rate of 0.75 mL/h starting 1 h after the saline infusion, as previously described [24]. Control and LPS groups received the 3 mL vehicle infusion. Inflammation was confirmed based on increased plasma cytokine concentrations relative to baseline after LPS infusion. Serial cytokine measurements have been previously reported in Kelly et al. [24]. This model is relevant to the fetal inflammatory response syndrome caused by chorioamnionitis and reproduces the acute inflammatory exacerbations associated with adverse neurodevelopment [25, 27, 27]. Controls received an equivalent volume of saline at the same infusion rate. Randomisation was stratified by cohort to control for the time of year and twin pregnancy. Fetal preductal arterial blood samples were collected every morning (0900 h) starting from 30 min before the start of the experiment until the day of post-mortem for pH, blood gases, and glucose and lactate concentrations (ABL 90 Flex Plus analyser; Radiometer, Brønshøj, Denmark). Four days after the start of infusions, sheep were euthanised by intravenous injection of pentobarbitone sodium (100 mg/kg, Lethabarb, Virbac, NSW, Australia).
Brain collection and processing
At post-mortem the right hemisphere was immersion-fixed with 10% phosphate-buffered formalin for 3 days before processing and embedding using a standard paraffin tissue preparation. Using a brain mould, the right hemisphere was cut with a blocking blade into 5-mm-thick coronal blocks. Blocks from the forebrain, approximately 23 mm anterior to stereotaxic zero, with a clearly visible lateral gyrus containing the somatosensory cortex were sectioned into 8-μm-thick coronal sections using a microtome (Leica Microsystems, Victoria, Australia).
Region matched tissue sections from the left hemisphere were rinsed in distilled water and immersion-fixed using a commercially available FD Rapid Golgi Stain Kit (FD Neurotechnologies Inc., MD, USA). Tissue containing a clearly visible lateral gyrus containing the somatosensory cortex was frozen and sectioned with a Leica VT1200S vibratome at 100 μm. The sections were mounted onto coverslips, processed for Golgi visualisation, dehydrated in a graded series of alcohol solutions and cover slipped.
Gene expression analysis
Remaining grey matter tissue from the lateral gyrus, adjacent to the section collected for Golgi staining, was dissected, snap-frozen in liquid nitrogen and stored at − 80 °C for mRNA analysis of inflammatory genes. The tissue was homogenised and total mRNA was isolated using an RNeasy Midi Kit (QIAGEN, Venlo, Netherlands) and reverse transcribed into single stranded cDNA (SuperScript III First-Strand Synthesis System, Invitrogen, MA, USA). Relative mRNA expression levels of interleukin (IL)1A, IL1B, and IL6 were measured by qRT-PCR using an Applied Biosystems Quantstudio 6 Real-Time PCR system. Relative mRNA levels of the genes of interest were normalised to the 18S RNA for each sample by subtracting the cycle threshold (Ct) value for 18S from the Ct value for the gene of interest (ΔCt). mRNA levels of genes of interest were normalised using the formula 2 − ΔCt and the results expressed as a fold change from control. A threshold value (Ct) for each sample was measured in triplicate and a control sample containing no cDNA template was included in each run. Details of the primers used are presented in Table 1.
Immunohistochemistry
Slides were baked at 60 °C for 1 h then dewaxed in xylene, rehydrated in increasing concentrations of ethanol and washed in 0.1 mol/L phosphate buffered saline (PBS). Antigen retrieval was performed in citrate buffer (pH 6) using a microwave for 15 min. Endogenous peroxide quenching was performed by incubating slides in 0.1% H2O2 in methanol. Non-specific antigen blocking was performed using 3% normal goat serum. Sections were labelled with 1:250 rabbit anti-IL-1β (cat#: NB600-633, Novus, CO, USA), 1:200 rabbit anti-ionised calcium binding adaptor molecule 1 (Iba-1, cat#: ab153696, Abcam, Cambridge, UK) 1:200 rabbit anti-glial fibrillary acidic protein (GFAP; cat#: ab68428, Abcam) 1:350 rabbit anti-neuronal nuclei (NeuN, cat#: ab177487, Abcam), and 1:800 rabbit anti-Caspase3 (cat#: AF835, R&D Systems, MS, USA), overnight at 4 °C. Sections were incubated in biotin conjugated IgG goat anti-rabbit (1:200; Dako, Victoria, Australia) for 3 h at room temperature before being incubated in avidin–biotin complex (MilliporeSigma) for 45 min at room temperature. Sections were reacted with 3,3’-diaminobenzidine tetrahydrochloride (MilliporeSigma). The reaction was stopped in PBS and slides were then dehydrated in xylene followed by increasing concentrations of ethanol, mounted in dibutyl phthalate polystyrene xylene and cover slipped.
ApopTag was used to detect single and double-stranded breaks in DNA associated with apoptosis [28]. Staining was carried out according to manufacturer’s instructions (MilliporeSigma, s7100, ApopTag Peroxidase in Situ Apoptosis Detection Kit). In brief, tissue was dewaxed in xylene, rehydrated in increasing concentrations of ethanol, and washed in PBS. The tissue was then pre-treated with proteinase K for 15 min, washed in PBS, and background peroxidase activity quenched in 3.0% hydrogen peroxide for 5 min. The equilibration buffer was added for 10 s, before the TdT enzyme was added and left for 1 h at 37 °C. The reaction was stopped in stop buffer for 10 min, then washed before adding the anti-digoxigenin conjugate for 30 min at room temperature. Finally, peroxidase substrate was added for 6 min before the tissue was counterstained in 50% haematoxylin (cat#MH-1NPR, Trajan Scientific, VIC, Australia), dehydrated in xylene followed by increasing concentrations of ethanol, mounted in dibutyl phthalate polystyrene xylene and cover slipped.
Immunohistochemistry analysis
Cortical areas were quantified using QuPath imaging software (version 0.2.3) [29]. Microglia (Iba-1 + cells), neuronal nuclei (NeuN + cells), and caspase 3 + cells were visualised using light microscopy (Olympus, Tokyo, Japan) at 40 × magnification using CellSens imaging software (version 2.3; Olympus). Caspase 3 + cells displaying both immunostaining and apoptotic bodies were counted. NeuN + cells were counted only if they were morphologically normal, while cells displaying condensed or fragmented nuclei were not counted [30]. IL-1β-stained sections were scored at 20 × magnification using an immunoreactivity scoring system adapted from Girard et al. [31]. Scoring was based on the intensity of staining whereby 1 = light, 2 = moderate, 3 = moderate to intense and 4 = intense, as previously reported [32]. The area fraction of GFAP (astrocyte) staining was quantified in ImageJ software (v2.0, LOCI, University of Wisconsin) using a standard intensity threshold. ApopTag + cells were quantified using QuPath imaging software. Total numbers of positive cells were counted within the lateral gyrus from the parietal lobe between cortical layers 3 and 5. For all other immunohistochemical analyses, positive cells or immunoreactivity were quantified for each field of view (1 field from the base of the gyrus and 1 field from the top of the gyrus; Fig. 1) from two sections per subject using ImageJ. For each field of view, average scores from two slides from the right hemisphere were calculated. The size of the field of view was the same for all assessments (20 x magnification = 0.489 mm2 and 40 x magnification = 0.130 mm2). All imaging and cell counts were performed by an assessor who was blinded to the treatment group by independent coding of slides and data files.

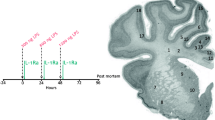
Schematic outlining the study design. The study consisted of two groups: control (vehicle, n = 9) and LPS (n = 8). The solid lines show the timing of the lipopolysaccharide (LPS)/vehicle infusions which were given over 2 min at increasing doses (300 ng, 600 ng, and 1200 ng). Controls received an equivalent volume of vehicle (saline) during the infusion period. Continuous electroencephalogram (EEG) recordings were performed throughout the experiential period. At 96 h, brains were collected for Golgi staining to examine neuronal arborisation and numbers of dendritic spines, immunohistochemistry to assess neuroinflammation, neuronal numbers and cortical area and mRNA assessment of proinflammatory proteins. Boxes indicate regions of interest used for immunohistochemical and Golgi analysis. Image source:[86]
Golgi analysis
Coded region-matched Golgi-stained tissue sections (10 serial sections per subject) were used to assess basal dendrites from pyramidal neurons in layers 3 and 5 of the lateral cortex as described previously [33, 34]. The Golgi staining tended to produce less complete filling of the apical dendrites relative to the basal dendrites. Thus, apical dendrites were not analysed. Basal dendrites were visualised using an Olympus BX61 stereology microscope (Olympus) equipped with an Olympus DP73 colour camera (× 0.5 lens) at 60 × magnification and CellSens imaging software (version 2.3; Olympus). From each subject, basal dendrites from a total of 20 pyramidal neurons selected from 10 serial sections of the lateral gyrus met the inclusion criteria for imaging. A total of 10 neurons were selected from the base of the gyrus and 10 were selected from the top of the gyrus (Fig. 1). We found that 20 was the maximum number of neurons per subject that could be selected based on the pre-defined selection criteria. Neurons were selected based on morphological criteria [34]: triangular shaped soma and apical dendrites perpendicular to the pial surface, complete Golgi impregnation of the cell that permitted visualisation of the entire dendritic arbour and spines, neuronal soma and processes not obscured by other neurons, glia or blood vessels, and neurons exhibiting a complete basilar dendritic tree without truncated or cut processes. Pyramidal neuronal subtypes were not distinguished. A preliminary analysis of n = 4 subjects per group showed no significant differences between neurons selected from the base of the gyrus and neurons selected from the top of the gyrus for dendritic length (control: gyrus base = 2874 ± 495, gyrus top = 3261 ± 517; LPS: gyrus base = 1663 ± 137, gyrus top = 1945 ± 173) and numbers of terminals (control: gyrus base = 42 ± 2, gyrus top = 48 ± 3; LPS: gyrus base = 30 ± 3, gyrus top = 32 ± 2). Nevertheless, equal numbers of neurons from the gyrus base (n = 10) and top (n = 10) were analysed from each subject to ensure our data were not confounded by potential regional differences in neuronal development. Images were cropped and separated into individual channels using ImageJ. The images were then imported into Imaris (version 9.2.1, Bitplane, Oxford Instruments Company, Abington, UK). Measures of dendritic complexity including summated dendritic length, numbers of dendritic terminals and Sholl analysis (numbers of dendrite intersections per Sholl ring) were assessed using the Imaris filament tracer tool. Sholl intersections were analysed using 5 μm interval concentric rings centred on the soma. Numbers of dendritic spines were quantified using the Imaris filament tracer and dendritic spine classification (filopodia, long thin, stubby, mushroom) was performed using the MATLAB spine classification extension (MATLAB, R2019a, Mathworks Inc., CA, USA).
Data analysis and statistics
Offline analysis of physiological data was performed using LabChart Pro software (v8.1.3; ADInstruments, Sydney, NSW, Australia). EEG data were processed as hourly averages and presented from 24 h before the first saline or LPS infusion until the end of the experiment. Due to small differences in baseline spectral edge frequency between the group, spectral edge frequency was normalised by subtracting the baseline values (average of 24 h before the first saline/LPS infusion) from the absolute value. EEG data during the baseline, 24 h period after LPS/saline infusion, and recovery (from 24 h after the final LPS infusion until the end of the study, i.e. 72–96 h) periods were analysed separately. Sleep stage cycling was assessed using the raw EEG spectral edge frequency trace during the last 5 h of the experimental period (91–96 h). Sleep stage cycling was defined as a repetitive alternating pattern of high and low-frequency activity, with each phase lasting approximately 20 min, as previously described [35]. Data were tested for normality using the Shapiro–Wilk test. Histological and PCR data were analysed using an unpaired t-test. Mann–Whitney U-tests were used for testing non-parametric data. For EEG data and Sholl analysis of dendritic morphology, when statistical significance was found between groups, group and time (EEG) or group and radius (Sholl analysis) post hoc comparisons were made using the Fisher’s least significant difference test [36]. Linear and non-linear regression were used to assess the relationship between EEG spectral edge frequency and band power with neuronal microstructure. Post hoc power analysis for summated dendritic length showed 85% power to detect a minimum difference of 970 µm. Statistical significance was accepted when P < 0.05. Data are presented as scatter plots with mean ± standard error of the mean (SEM).
Results
Baseline period
Before LPS exposure, fetal arterial blood gases, pH, glucose, lactate and EEG power and frequency did not differ between groups and were within the normal range for our laboratory. Blood gas data and EEG power have been previously published in Kelly et al. [24].
Confirmation of systemic inflammation
All LPS-exposed fetuses had increased plasma cytokine levels relative to baseline after LPS infusions [24], confirming the induction of a systemic inflammatory response.
Spectral edge frequency (SEF)
After the first LPS infusion, SEF was lower in the LPS group at 7 h (P < 0.05 vs. controls, Fig. 2A). After the second LPS infusion, SEF was reduced in the LPS group from 28–31 h (i.e., 4–7 h after the second infusion; P < 0.05 vs. controls). After the third LPS infusion, SEF was reduced in the LPS group from 50–52, 57–58 and at 65 h (i.e., at 2–4 h, 9–10 h and 17 h after the third infusion, P < 0.05 vs. controls). Qualitative assessment of EEG spectral edge frequency during the last 5 h of the recording period (91–96 h) showed sleep stage cycling was present in all control and LPS-exposed fetuses.

Neurophysiological changes over time. From the top down, the figure shows spectral edge frequency (A), %delta activity (B), %theta activity (C), %alpha activity (D), and %beta activity (E) in the control (black, n = 9) and LPS (red n = 8) groups. Vertical lines indicate the timings of LPS administration. Data are hourly means ± standard error (SE). *P < 0.05 vs. control
Spectral band power analysis
After the first LPS infusion, delta band power increased in the LPS group from 7–15 h (P < 0.05 vs. controls, Fig. 2B). After the third LPS infusion, %delta band power was higher in the LPS group from 48–50 h (i.e., 0–2 h after the third infusion, P < 0.05 vs controls, Fig. 2B). There were no differences in %theta and %alpha band powers after LPS infusions between groups (Fig. 2C, D).
After the first LPS infusion, %beta band power decreased in the LPS group from 7–9 h and 16–18 h and at 22 h (P < 0.05 vs. controls, Fig. 2E). After the second LPS infusion, %beta band power was lower in the LPS group from 31–33 h and 38–40 h and at 47 h (i.e., at 7–9 h, 14–16 h, and 23 h after the second LPS infusion; P < 0.05 vs. controls, Fig. 2E). After the third LPS infusion, %beta band power was lower at 48 h and between 55–66 h, 73–86 h, and 92–94 h (i.e., at 0 h, 7–18 h, 25–38 h, and 44–46 h after the third LPS infusion; P < 0.05 vs. controls, Fig. 2E).
Gene expression analysis
There were no significant changes in mRNA expression of IL1A, IL1B (P = 0.06) and IL6 (P = 0.09) in the somatosensory cortex in the LPS group compared with controls (Fig. 3).

Interleukin (IL)1A, IL1B, and IL6 mRNA levels in the somatosensory cortex in tissue sections adjacent to samples processed for Golgi staining from controls (white circles, n = 7, two subjects had undetectable values), and LPS (black circles; IL1B, n = 7 [one subject had undetectable values], IL1A, n = 8, and IL6, n = 8) groups. Data are means ± SE and are expressed as the fold change from the mean control values
Neuronal basal dendrite morphology
The summated basal dendritic length of cortical pyramidal neurons was significantly reduced in the LPS group (by ~ 36%) compared to control (control: 2716 ± 314 µm vs. LPS: 1738 ± 92 µm; P < 0.05, Figs. 4A, 6A). The number of basal dendritic terminals was significantly reduced in the LPS group (by ~ 31%) compared with controls (control: 46 ± 3 vs. LPS: 32 ± 3, P < 0.01, Figs. 4B, 6B). Sholl analysis of pyramidal neuron complexity showed reduced dendritic arborisation in the LPS group at 40–230 μm away from the soma (P < 0.05 vs. controls; Fig. 4C).

Summated dendritic length (μm) (A), number of dendritic terminals (B) and Sholl analysis (C) showing the number of dendritic intersections (dendritic arborisation) indicated by the number of intersections at 5 μm intervals away from the soma in the control (white circles, n = 7; two subjects had limited Golgi penetration) and LPS (black circles, n = 7; 1 subject had limited Golgi penetration) groups. Data are means ± SE, *P < 0.05 vs control
Neuronal basal dendritic spine number and morphology
The total number of basal dendritic spines was reduced in the LPS group compared with controls (P < 0.05, Figs. 5E, 6C). Spine morphology classification showed reduced numbers of long thin spines in the LPS group compared to control (P < 0.05; Fig. 5B). There were no differences in numbers of filopodia, stubby or mushroom spines between the groups (Fig. 5A, C, D, respectively).

Numbers of filopodia, long thin, stubby, mushroom and total dendritic spines in control (white circles, n = 7; two subjects had limited Golgi penetration) and LPS (black circles, n = 7; 1 subject had limited Golgi penetration) groups. Data are means ± SE, *P < 0.05 vs control

Representative Golgi stained (A) and traced (B) images of basal dendrites and dendritic spines (C) from pyramidal neurons in the somatosensory cortex from control and LPS-exposed subjects. Scale bar panel A = 100 μm, panel B = 50 μm and panel C = 15 μm
Histopathology
Immunoreactivity of IL-1β was increased in the somatosensory cortex of the LPS group (P < 0.05 vs. controls; Figs. 7A, 8A). Numbers of Iba-1 + microglia were significantly increased in the LPS group (P < 0.01 vs. controls, Figs. 7B, 8B). There was no difference in area fraction of GFAP + astrocyte staining between the groups (Figs. 7C, 8C). Numbers of caspase3 + cells were increased in the LPS group compared with controls (P < 0.05; Figs. 7D, 8E). There were no differences in the total numbers of ApopTag + cells between the groups (Figs. 7E, 8F). Finally, there were no differences in either numbers of NeuN + neurons (Figs. 7F, 8D) or cortical area (control: 2.7 × 107 ± 2.4 × 106 µm2 vs LPS: 2.6 × 107 ± 3.4 × 106 µm2) in the lateral gyrus between the groups.

Interleukin 1(IL)-1β immunoreactivity scores (A), numbers of ionised calcium binding adaptor molecule (IBA-1) + microglia (B), glial fibrillary acidic protein (GFAP +) area fraction staining (C), numbers of caspase 3 + cells (D), numbers of ApopTag (TUNEL) + cells (E) numbers of NeuN + neurons (F), in the lateral gyrus (LG) in control (open circles, n = 9) and LPS groups (black circles, n = 8) groups. Data are means ± SE, *P < 0.05 vs control

Representative photomicrographs showing immunohistochemical staining of IL-1β (A), IBA-1 (B), GFAP (C), NeuN (D), caspase3 (E) and ApopTag (TUNEL, F) in the lateral gyrus. Arrowheads point to Caspase 3 + cells and arrows point to ApopTag (TUNEL +) cells. Scale bar = 100 μm
Correlative analysis
Mean %beta band power during the final 12 h of the experiment was positively correlated with dendritic length (linear regression: R2 = 0.30, p = 0.0417) and neuronal arborisation (linear regression: R2 = 0.30, P = 0.0423). There were no significant correlations between beta band power and numbers of dendritic terminals and numbers of dendritic spines. There were no significant correlations between spectral edge frequency, or the %delta, %theta and %alpha spectral bands and markers of neuronal microstructure.
Discussion
This study demonstrates that antenatal inflammation reduced the length, number and complexity (arborisation) of basal dendrites, and reduced the numbers of dendritic spines on pyramidal neurons within the somatosensory cortex in late gestation fetal sheep. The reduction in dendritic length and complexity was associated with an increased number of microglia and IL-1β-positive staining but was not associated with changes in the number of cortical neurons or cortical area. Functionally, the inflammation-induced reduction in dendritic length and complexity were associated with transient increases in delta (slow wave) activity, reduced beta (fast wave) activity and an overall reduction in the spectral edge frequency of the EEG. Given the association between exposure to perinatal inflammation and reductions in cortical growth and connectivity [6, 7, 37], the present study provide critical new insight into the deficits in neuronal structure and function arising from antenatal inflammation.
Clinically, Gram-negative infections, including E. coli, continue to be among the most common pathogens linked to perinatal infection/inflammation and increased risk of perinatal brain injury [38, 39]. We sought to reproduce features of Gram-negative infection/inflammation using repeated increasing doses of LPS infusions to promote a chronic progressive fetal inflammatory response that is commonly associated with adverse neurodevelopmental outcomes [24, 40]. By contrast, most of the previous preclinical studies have focused on the pathophysiological consequences of single or repeated bolus doses of LPS or other infectious/inflammatory stimuli [41].
This study showed that inflammation induced by increasing doses of LPS infusions was associated with increased numbers of microglia and greater immunoreactivity of IL-1β in the somatosensory cortex but no significant increases in cortical IL1B (p = 0.06), IL6 (p = 0.09) or IL1A mRNA expression. Consistent with this observation, human post-mortem studies have reported increased numbers of microglia and IL-1β immunoreactivity in areas of white and grey matter inflammation and injury [42,43,44]. Indeed, cerebral recognition of pathogen-associated molecular patterns such as LPS by innate immune receptors, including toll like receptor 4, on microglia and other immune cells leads to glial cell activation and nuclear factor kappa B induced transcription of bioactive IL-1β [45]. Furthermore, circulating cytokines, including IL-1β, can penetrate the blood brain barrier [46,47,48] to recruit and activate microglia within the central nervous system [45]. Thus, the concomitant increase in numbers of microglia and IL-1β immunoreactivity observed in this study strongly supports sustained inflammation in the somatosensory cortex 4 days after beginning intravenous LPS infusions.
In this experimental model of antenatal inflammation, we have previously reported progressive increases in concentrations of systemic pro- and anti-inflammatory cytokines (IL-1β, tumour necrosis factor [TNF], IL-6 and IL-10), in addition to diffuse white matter gliosis and reduced numbers of precursor oligodendrocytes [24]. Clinically, in large prospective studies of preterm infants, increased concentrations of these inflammatory proteins in cord blood and postnatal blood samples have been associated with perinatal brain injury and impaired neurodevelopment in childhood [49]. The present study shows that exposure to inflammation did not affect overall numbers of neurons (NeuN +) or neuronal density in the areas of the somatosensory cortex evaluated in this study, suggesting a lack of overt cortical injury. This is further confirmed by the similar numbers of TUNEL + cells between groups, suggesting no effect of LPS-exposure on acute cell death in the somatosensory cortex. This observation is consistent with neonatal experimental and clinical studies showing limited or no neuronal cell death in cases of perinatal encephalopathy, including after systemic inflammation [10, 49, 50]. By contrast, we observed increased numbers of caspase 3 + cells in LPS-exposed fetuses compared to controls. This finding of increased numbers of caspase-3-positive cells without increased cell death has been reported in the adult and perinatal brain [51,52,53,54] and is likely to be linked to other roles played by caspases, which include immune/microglial activation and cell differentiation [54,55,56].
We observed a reduction in neuronal dendritic complexity in LPS-exposed fetuses as shown by reduced dendritic length, numbers of dendritic terminals, dendritic arborisation, and numbers of dendritic spines. In humans, the marked cortical expansion that occurs during the last trimester primarily reflects the prolific increase in neuronal dendritic growth and complexity during this stage of development [57, 58]. Our observations suggest that at this period in late gestation, neuronal development within the somatosensory cortex in the developing fetus is highly vulnerable to inflammation-induced impairments of dendritic arborisation and spine formation. These data are consistent with previous studies that reported reduced neuronal arborisation in the frontal cortex of fetal sheep after acute cerebral ischaemia at mid-gestation, and reduced dendritic number and spine density in the retrosplenial cortex of newborn rabbits (P1) after a single bolus of intra-amniotic LPS (20 µg/kg) [59]. Similarly, in separate rodent studies examining the long-term effects of prenatal and early postnatal LPS-induced inflammation, reduced dendritic arborisation was seen in the motor cortex and medial prefrontal cortex on postnatal days 21 and 60 [10, 60]. Furthermore, these data support a link between diffuse white matter injury, which we have previously reported in the same experimental paradigm [24], and impaired neuronal development. For example, human case series have shown reduced dendritic length in cases of both diffuse and necrotic white matter injury [61, 62]. Similarly, moderate LPS-induced inflammation in neonatal rodents (from P1-P3) was associated with reduced dendritic arborisation in the motor cortex, diffuse white matter injury, and impaired myelination and motor function on postnatal day 21 [10].
The somatosensory cortex has been shown to synapse with cervical excitatory neurons and modulate locomotion independently of the motor cortex [63]. Consistent with this observation, we have previously reported reduced fetal movements in the same fetal sheep paradigm, as shown by reduced nuchal electromyographic activity from 3 days after starting LPS infusions until the time of post-mortem [24]. Although we did not evaluate neuronal complexity within the motor cortex in this study, our data raise the possibility that impaired neuronal development in the somatosensory cortex may contribute to the inflammation-induced reduction in fetal body movements observed in this preclinical model of antenatal infection/inflammation. Taken together, these data support a close link between impaired neuronal development in the somatosensory cortex and inhibition of motor function.
Although the precise mechanism/s underpinning the inflammation-induced impairment in neuronal development are yet to be identified, it is likely to include a direct effect of inflammation on the central nervous system. For example, in vitro studies have shown that cortical neurons exposed to inflammatory cytokines, including IL-1β, IL-6, TNF, and interferon gamma, show reduced dendritic branching and synapse formation [64, 65]. This is supported by our findings of increased numbers of cortical microglia, which are known to secrete proinflammatory cytokines, along with increased IL-1β immunoreactivity observed in the LPS-exposed fetuses. Microglial processes have also been shown to interact with synapses to eliminate spines, suggesting a direct effect of microglial activation on spine density [66, 67]. Furthermore, reduced circulating concentrations of neural growth factors, including nerve growth factor and brain derived neurotrophic factor, have been reported in human and animal studies of perinatal infection/inflammation [59, 68, 69].
Consistent with the inflammation-induced reduction in neuronal complexity in the present study, neuronal activity in LPS-exposed fetuses was impaired, as shown by an overall reduction in the spectral edge frequency of EEG activity along with an increase in the proportion of EEG activity in the delta band and a reduced proportion of activity in the beta band. Collectively, these data indicate loss of high-frequency activity after LPS-exposure with a shift to lower frequency activity. The inflammation-induced change in EEG spectra could reflect alterations to fetal behaviour/sleep stages. The fetus is never awake, but rather cycles between low voltage (high frequency) and high voltage (low frequency) sleep [70]. Qualitative analysis of EEG frequency signals over the last 5 h of the experimental period showed sleep stage cycling was present in all control and LPS-exposed fetuses. Consistent with these data, pre- and post-natal immune challenges in mice have shown inflammation-induced increases in slow wave sleep in association with a shift in spectral band power (increased low-frequency and reduced high-frequency activity) that was consistent with our analysis [71]. Although sleep stage cycling was observed in all fetuses in the present study at the end of the recording period, immediately prior to post-mortem, it is possible that the inflammation-induced changes in EEG spectra are associated with subtle changes in the proportions of high and low-frequency sleep.
We have previously reported that there were no differences in myelin density or numbers of mature myelinating oligodendrocytes at this timepoint in this cohort of LPS-exposed fetuses [24]. This suggests that the changes in EEG frequency in the present study are not related to altered myelination. Alternatively, the inflammation-induced reduction in high-frequency activity may reflect inhibition of synaptic activity. This could be due to the reduced neuronal arborisation and numbers of dendritic spines on cortical neurons directly underlying the EEG electrodes (i.e., a direct functional consequence of inflammation-induced changes in neuronal pathology). Alternatively, elevated central levels of IL-1β have been shown to induce NMDA-mediated suppression of synaptic function [72]. Consistent with this, TNF inhibition using the soluble TNF receptor antagonist, etanercept, reduced the magnitude of EEG suppression in fetal sheep exposed to LPS [73], possibly due to reduced NMDA receptor activation [74]. Furthermore, in vivo and in vitro studies have shown that both LPS- and IL-1β-induced central inflammation actively mediate EEG suppression through the release of inhibitory neuromodulators, such as allopregnanolone and adenosine [75, 76]. Taken together, the present data suggest that inflammation-induced suppression of EEG activity is mediated by a combination of activation of anti-excitotoxic mediators as well as reduced complexity of the neuronal microstructure.
Clinical studies have shown that reduced EEG frequency strongly predicts subsequent brain injury and neurodevelopmental impairment in preterm and term infants. For example, in a cohort study, reduced EEG frequency was associated with the severity of neonatal white matter injury [77]. Similarly, depression of the EEG background pattern was associated with both motor and cognitive impairment in preterm and term infants with evidence of central inflammation [78, 79]. Furthermore, increased latency of somatosensory evoked potentials was reported in children with bilateral spastic cerebral palsy. Notably, the latency of somatosensory evoked potentials correlated with a history of exposure to perinatal infection/inflammation [80]. Functional MRI studies have shown reduced cortical functional connectivity in fetuses exposed to inflammation before birth [37]. Similarly, reduced cortical functional connectivity was observed in preterm infants without evidence of overt cortical injury [81]. Subsequent investigation of infants with moderate to severe white matter injury, but without overt cortical injury, showed a reduction in cortical functional connectivity that correlated with the severity of white matter injury [82]. Our data suggest that in the absence of overt neuronal injury or white matter loss, reduced neuronal complexity and synaptic density may contribute to reduced functional connectivity within and between major grey matter structures after exposure to perinatal inflammation.
This study was not designed to test the effect of sex and is not large enough to determine whether there were sex-specific effects of antenatal inflammation on neuronal microstructure or EEG activity. Previous studies in perinatal rodents reported sex-dependant effects in central and peripheral immune activation, as well as neuronal development [83, 84]. Further studies evaluating the impact of sex on inflammation-induced changes to neuronal structure and function in large animals are needed.
In conclusion, this study demonstrates that an inflammation-induced reduction in high-frequency spectral band power was associated with reduced cortical neuronal arborisation and dendritic spine density. Collectively, these data support the concept that inflammation-induced impairments in neuronal maturation and function, rather than overt neuronal loss, make a key contribution to disturbed cortical development and connectivity, and subsequent impaired neurodevelopmental outcomes, in infants exposed to perinatal inflammation. We propose that early EEG monitoring combined with neuroimaging modalities that enable more sensitive assessment of brain microstructure [10, 85] and therapeutics designed to mitigate systemic and central inflammation, could provide an effective approach for early detection and therapeutic intervention.
Availability of data and materials
The datasets used during the current study are available from the corresponding author upon reasonable request.
References
Martinello K, Hart AR, Yap S, Mitra S, Robertson NJ. Management and investigation of neonatal encephalopathy: 2017 update. Arch Dis Child Fetal Neonatal Ed. 2017;102:F346-f358.
Eunson P. The long-term health, social, and financial burden of hypoxic–ischaemic encephalopathy. Dev Med Child Neurol. 2015;57:48–50.
Economic costs associated with mental retardation. cerebral palsy, hearing loss, and vision impairment—United States, 2003. MMWR Morb Mortal Wkly Rep. 2004;53:57–9.
Shih STF, Tonmukayakul U, Imms C, Reddihough D, Graham HK, Cox L, Carter R. Economic evaluation and cost of interventions for cerebral palsy: a systematic review. Dev Med Child Neurol. 2018;60:543–58.
Becroft DM, Thompson JM, Mitchell EA. Placental chorioamnionitis at term: epidemiology and follow-up in childhood. Pediatr Dev Pathol. 2010;13:282–90.
Jain VG, Kline JE, He L, Kline-Fath BM, Altaye M, Muglia LJ, DeFranco EA, Ambalavanan N, Parikh NA. Acute histologic chorioamnionitis independently and directly increases the risk for brain abnormalities seen on magnetic resonance imaging in very preterm infants. Am J Obstet Gynecol. 2022;227:623.e621-623.e613.
Hatfield T, Wing DA, Buss C, Head K, Muftuler LT, Davis EP. Magnetic resonance imaging demonstrates long-term changes in brain structure in children born preterm and exposed to chorioamnionitis. Am J Obstet Gynecol. 2011;205(384):e381-388.
Burd I, Brown A, Gonzalez JM, Chai J, Elovitz MA. A mouse model of term chorioamnionitis: unraveling causes of adverse neurological outcomes. Reprod Sci. 2011;18:900–7.
Allard MJ, Brochu ME, Bergeron JD, Segura M, Sébire G. Causal role of group B Streptococcus-induced acute chorioamnionitis in intrauterine growth retardation and cerebral palsy-like impairments. J Dev Orig Health Dis. 2019;10:595–602.
Prasad JD, van de Looij Y, Gunn KC, Ranchhod SM, White PB, Berry MJ, Bennet L, Sizonenko SV, Gunn AJ, Dean JM. Long-term coordinated microstructural disruptions of the developing neocortex and subcortical white matter after early postnatal systemic inflammation. Brain Behav Immun. 2021;94:338–56.
Sweeney EL, Dando SJ, Kallapur SG, Knox CL. The human ureaplasma species as causative agents of chorioamnionitis. Clin Microbiol Rev. 2017;30:349–79.
Moss TJ, Nitsos I, Ikegami M, Jobe AH, Newnham JP. Experimental intrauterine ureaplasma infection in sheep. Am J Obstet Gynecol. 2005;192:1179–86.
Oh JW, Park CW, Moon KC, Park JS, Jun JK. The relationship among the progression of inflammation in umbilical cord, fetal inflammatory response, early-onset neonatal sepsis, and chorioamnionitis. PLoS ONE. 2019;14: e0225328.
Spann MN, Monk C, Scheinost D, Peterson BS. Maternal immune activation during the third trimester is associated with neonatal functional connectivity of the salience network and fetal to toddler behavior. J Neurosci. 2018;38:2877–86.
Brown AS, Sourander A, Hinkka-Yli-Salomäki S, McKeague IW, Sundvall J, Surcel HM. Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry. 2014;19:259–64.
Jiang NM, Tofail F, Ma JZ, Haque R, Kirkpatrick B, Nelson CA, Petri WA. Early life inflammation and neurodevelopmental outcome in Bangladeshi infants growing up in adversity. Am J Trop Med Hyg. 2017;97:974–9.
Jiang NM, Tofail F, Moonah SN, Scharf RJ, Taniuchi M, Ma JZ, Hamadani JD, Gurley ES, Houpt ER, Azziz-Baumgartner E, et al. Febrile illness and pro-inflammatory cytokines are associated with lower neurodevelopmental scores in Bangladeshi infants living in poverty. BMC Pediatr. 2014;14:50.
Tsuchida TN. EEG background patterns and prognostication of neonatal encephalopathy in the era of hypothermia. J Clin Neurophysiol. 2013;30:122–5.
Al Naqeeb N, Edwards AD, Cowan FM, Azzopardi D. Assessment of neonatal encephalopathy by amplitude-integrated electroencephalography. Pediatrics. 1999;103:1263–71.
Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. J Physiol. 2020;598:3793–801.
Keogh MJ, Bennet L, Drury PP, Booth LC, Mathai S, Naylor AS, Fraser M, Gunn AJ. Subclinical exposure to low-dose endotoxin impairs EEG maturation in preterm fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2012;303:R270-278.
Galinsky R, Dhillon SK, Lear CA, Yamaguchi K, Wassink G, Gunn AJ, Bennet L. Magnesium sulfate and sex differences in cardiovascular and neural adaptations during normoxia and asphyxia in preterm fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2018;315:R205-r217.
McIntosh TK, Vink R, Yamakami I, Faden AI. Magnesium protects against neurological deficit after brain injury. Brain Res. 1989;482:252–60.
Kelly SB, Stojanovska V, Zahra VA, Moxham A, Miller SL, Moss TJM, Hooper SB, Nold MF, Nold-Petry CA, Dean JM, et al. Interleukin-1 blockade attenuates white matter inflammation and oligodendrocyte loss after progressive systemic lipopolysaccharide exposure in near-term fetal sheep. J Neuroinflamm. 2021;18:189.
Gotsch F, Romero R, Kusanovic JP, Mazaki-Tovi S, Pineles BL, Erez O, Espinoza J, Hassan SS. The fetal inflammatory response syndrome. Clin Obstet Gynecol. 2007;50:652–83.
Grether JK, Nelson KB. Maternal infection and cerebral palsy in infants of normal birth weight. [Erratum appears in JAMA 1998;279(2):118]. JAMA. 1997;278:207–11.
Yanowitz TD, Jordan JA, Gilmour CH, Towbin R, Bowen A, Roberts JM, Brozanski BS. Hemodynamic disturbances in premature infants born after chorioamnionitis: association with cord blood cytokine concentrations. Pediatr Res. 2002;51:310–6.
Mohan C, Long K, Mutneja M, Ma J. Detection of end-stage apoptosis by ApopTag® TUNEL technique. Methods Mol Biol. 2015;1219:43–56.
Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7:16878.
Pozo Devoto VM, Chavez JC, Fiszer de Plazas S. Acute hypoxia and programmed cell death in developing CNS: differential vulnerability of chick optic tectum layers. Neuroscience. 2006;142:645–53.
Girard S, Kadhim H, Larouche A, Roy M, Gobeil F, Sébire G. Pro-inflammatory disequilibrium of the IL-1 beta/IL-1ra ratio in an experimental model of perinatal brain damages induced by lipopolysaccharide and hypoxia-ischemia. Cytokine. 2008;43:54–62.
Kelly SB, Stojanovska V, Zahra VA, Moxham A, Miller SL, Moss TJM, Hooper SB, Nold MF, Nold-Petry CA, Dean JM, et al. Interleukin-1 blockade attenuates white matter inflammation and oligodendrocyte loss after progressive systemic lipopolysaccharide exposure in near-term fetal sheep. J Neuroinflamm. 2021; 18.
Dudink I, White TA, Ardalan M, Mallard C, Ballerin G, Creed SJ, Pham Y, Sutherland AE, Castillo-Melendez M, Allison BJ, Miller SL. An optimized and detailed step-by-step protocol for the analysis of neuronal morphology in golgi-stained fetal sheep brain. Dev Neurosci. 2022;44:344–62.
Dean JM, McClendon E, Hansen K, Azimi-Zonooz A, Chen K, Riddle A, Gong X, Sharifnia E, Hagen M, Ahmad T, et al. Prenatal cerebral ischemia disrupts MRI-defined cortical microstructure through disturbances in neuronal arborization. Sci Transl Med. 2013;5:168ra167.
Galinsky R, Dhillon SK, Kelly SB, Wassink G, Davidson JO, Lear CA, van den Heuij LG, Bennet L, Gunn AJ. Magnesium sulphate reduces tertiary gliosis but does not improve EEG recovery or white or grey matter cell survival after asphyxia in preterm fetal sheep. J Physiol. 2023. https://doi.org/10.1113/JP284381.
Levin JR, Serlin RC, Seaman MA. A controlled, powerful multiple-comparison strategy for several situations. Psychol Bull. 1994;115:153–9.
Thomason ME, Scheinost D, Manning JH, Grove LE, Hect J, Marshall N, Hernandez-Andrade E, Berman S, Pappas A, Yeo L, et al. Weak functional connectivity in the human fetal brain prior to preterm birth. Sci Rep. 2017;7:39286.
Stoll BJ, Puopolo KM, Hansen NI, Sánchez PJ, Bell EF, Carlo WA, Cotten CM, D’Angio CT, Kazzi SNJ, Poindexter BB, et al. Early-onset neonatal sepsis 2015 to 2017, the rise of Escherichia coli, and the need for novel prevention strategies. JAMA Pediatr. 2020;174: e200593.
Flannery DD, Edwards EM, Puopolo KM, Horbar JD. Early-onset sepsis among very preterm infants. Pediatrics. 2021; 148.
Galinsky R, Polglase GR, Hooper SB, Black MJ, Moss TJ. The consequences of chorioamnionitis: preterm birth and effects on development. J Pregnancy. 2013;2013: 412831.
Dean JM, Shi Z, Fleiss B, Gunn KC, Groenendaal F, van Bel F, Derrick M, Juul SE, Tan S, Gressens P, et al. A critical review of models of perinatal infection. Dev Neurosci. 2015;37:289–304.
Girard S, Sébire G, Kadhim H. Proinflammatory orientation of the interleukin 1 system and downstream induction of matrix metalloproteinase 9 in the pathophysiology of human perinatal white matter damage. J Neuropathol Exp Neurol. 2010;69:1116–29.
Kadhim H, Tabarki B, De Prez C, Sebire G. Cytokine immunoreactivity in cortical and subcortical neurons in periventricular leukomalacia: are cytokines implicated in neuronal dysfunction in cerebral palsy? Acta Neuropathol. 2003;105:209–16.
Kadhim H, Tabarki B, Verellen G, De Prez C, Rona AM, Sebire G. Inflammatory cytokines in the pathogenesis of periventricular leukomalacia. Neurology. 2001;56:1278–84.
Kelly SB, Green E, Hunt RW, Nold-Petry CA, Gunn AJ, Nold MF, Galinsky R. Interleukin-1: an important target for perinatal neuroprotection? Neural Regen Res. 2023;18:47–50.
Sadowska GB, Chen X, Zhang J, Lim YP, Cummings EE, Makeyev O, Besio WG, Gaitanis J, Padbury JF, Banks WA, Stonestreet BS. Interleukin-1β transfer across the blood-brain barrier in the ovine fetus. J Cereb Blood Flow Metab. 2015;35:1388–95.
Threlkeld SW, Lynch JL, Lynch KM, Sadowska GB, Banks WA, Stonestreet BS. Ovine proinflammatory cytokines cross the murine blood-brain barrier by a common saturable transport mechanism. NeuroImmunoModulation. 2010;17:405–10.
Skinner RA, Gibson RM, Rothwell NJ, Pinteaux E, Penny JI. Transport of interleukin-1 across cerebromicrovascular endothelial cells. Br J Pharmacol. 2009;156:1115–23.
Galinsky R, Lear CA, Dean JM, Wassink G, Dhillon SK, Fraser M, Davidson JO, Bennet L, Gunn AJ. Complex interactions between hypoxia-ischemia and inflammation in preterm brain injury. Dev Med Child Neurol. 2018;60:126–33.
Rathbone R, Counsell SJ, Kapellou O, Dyet L, Kennea N, Hajnal J, Allsop JM, Cowan F, Edwards AD. Perinatal cortical growth and childhood neurocognitive abilities. Neurology. 2011;77:1510–7.
Martinello KA, Meehan C, Avdic-Belltheus A, Lingam I, Ragab S, Hristova M, Tann CJ, Peebles D, Hagberg H, Wolfs T, et al. Acute LPS sensitization and continuous infusion exacerbates hypoxic brain injury in a piglet model of neonatal encephalopathy. Sci Rep. 2019;9:10184.
Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, Cano J, Brundin P, Englund E, Venero JL, Joseph B. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472:319–24.
Villapol S, Bonnin P, Fau S, Baud O, Renolleau S, Charriaut-Marlangue C. Unilateral blood flow decrease induces bilateral and symmetric responses in the immature brain. Am J Pathol. 2009;175:2111–20.
Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol. 2011;69:743–58.
Abraham MC, Shaham S. Death without caspases, caspases without death. Trends Cell Biol. 2004;14:184–93.
Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol. 2020;38:567–95.
Mrzljak L, Uylings HB, Kostovic I, van Eden CG. Prenatal development of neurons in the human prefrontal cortex. II. A quantitative Golgi study. J Comp Neurol. 1992;316:485–96.
Lu D, He L, Xiang W, Ai WM, Cao Y, Wang XS, Pan A, Luo XG, Li Z, Yan XX. Somal and dendritic development of human CA3 pyramidal neurons from midgestation to middle childhood: a quantitative Golgi study. Anat Rec (Hoboken). 2013;296:123–32.
Balakrishnan B, Dai H, Janisse J, Romero R, Kannan S. Maternal endotoxin exposure results in abnormal neuronal architecture in the newborn rabbit. Dev Neurosci. 2013;35:396–405.
Baharnoori M, Brake WG, Srivastava LK. Prenatal immune challenge induces developmental changes in the morphology of pyramidal neurons of the prefrontal cortex and hippocampus in rats. Schizophr Res. 2009;107:99–109.
Stolp HB, Fleiss B, Arai Y, Supramaniam V, Vontell R, Birtles S, Yates AG, Baburamani AA, Thornton C, Rutherford M, et al. Interneuron development is disrupted in preterm brains with diffuse white matter injury: observations in mouse and human. Front Physiol. 2019;10:955.
Takashima S, Becker LE, Chan FW. Retardation of neuronal maturation in premature infants compared with term infants of the same postconceptional age. Pediatrics. 1982;69:33–9.
Karadimas SK, Satkunendrarajah K, Laliberte AM, Ringuette D, Weisspapir I, Li L, Gosgnach S, Fehlings MG. Sensory cortical control of movement. Nat Neurosci. 2020;23:75–84.
Gilmore JH, Jarskog LF, Vadlamudi S, Lauder J. Prenatal infection and risk for schizophrenia: IL-I beta, IL-6, and TNF alpha inhibit cortical neuron dendrite development. Neuropsychopharmacology. 2004;29:1221–9.
Neumann H, Schweigreiter R, Yamashita T, Rosenkranz K, Wekerle H, Barde YA. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a rho-dependent mechanism. J Neurosci. 2002;22:854–62.
Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29:3974–80.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–8.
Skogstrand K, Hagen CM, Borbye-Lorenzen N, Christiansen M, Bybjerg-Grauholm J, Bækvad-Hansen M, Werge T, Børglum A, Mors O, Nordentoft M, et al. Reduced neonatal brain-derived neurotrophic factor is associated with autism spectrum disorders. Transl Psychiatry. 2019;9:252.
Holm M, Morken TS, Fichorova RN, VanderVeen DK, Allred EN, Dammann O, Leviton A. Systemic inflammation-associated proteins and retinopathy of prematurity in infants born before the 28th week of gestation. Invest Ophthalmol Vis Sci. 2017;58:6419–28.
Mellor DJ, Diesch TJ, Gunn AJ, Bennet L. The importance of “awareness” for understanding fetal pain. Brain Res Brain Res Rev. 2005;49:455–71.
Missig G, Mokler EL, Robbins JO, Alexander AJ, McDougle CJ, Carlezon WA Jr. Perinatal immune activation produces persistent sleep alterations and epileptiform activity in male mice. Neuropsychopharmacology. 2018;43:482–91.
Coogan A, O’Connor JJ. Inhibition of NMDA receptor-mediated synaptic transmission in the rat dentate gyrus in vitro by IL-1 beta. NeuroReport. 1997;8:2107–10.
Galinsky R, Dhillon SK, Dean JM, Davidson JO, Lear CA, Wassink G, Nott F, Kelly SB, Fraser M, Yuill C, et al. Tumor necrosis factor inhibition attenuates white matter gliosis after systemic inflammation in preterm fetal sheep. J Neuroinflammation. 2020;17:92.
Jara JH, Singh BB, Floden AM, Combs CK. Tumor necrosis factor alpha stimulates NMDA receptor activity in mouse cortical neurons resulting in ERK-dependent death. J Neurochem. 2007;100:1407–20.
Billiards SS, Walker DW, Canny BJ, Hirst JJ. Endotoxin increases sleep and brain allopregnanolone concentrations in newborn lambs. Pediatr Res. 2002;52:892–9.
Luk WP, Zhang Y, White TD, Lue FA, Wu C, Jiang CG, Zhang L, Moldofsky H. Adenosine: a mediator of interleukin-1beta-induced hippocampal synaptic inhibition. J Neurosci. 1999;19:4238–44.
Inder TE, Buckland L, Williams CE, Spencer C, Gunning MI, Darlow BA, Volpe JJ, Gluckman PD. Lowered electroencephalographic spectral edge frequency predicts the presence of cerebral white matter injury in premature infants. Pediatrics. 2003;111:27–33.
Hayakawa F, Okumura A, Kato T, Kuno K, Watanabe K. Dysmature EEG pattern in EEGs of preterm infants with cognitive impairment: maturation arrest caused by prolonged mild CNS depression. Brain Dev. 1997;19:122–5.
Sävman K, Blennow M, Gustafson K, Tarkowski E, Hagberg H. Cytokine response in cerebrospinal fluid after birth asphyxia. Pediatr Res. 1998;43:746–51.
Teflioudi EP, Zafeiriou DI, Vargiami E, Kontopoulos E, Tsikoulas I. Somatosensory evoked potentials in children with bilateral spastic cerebral palsy. Pediatr Neurol. 2011;44:177–82.
Smyser CD, Inder TE, Shimony JS, Hill JE, Degnan AJ, Snyder AZ, Neil JJ. Longitudinal analysis of neural network development in preterm infants. Cereb Cortex. 2010;20:2852–62.
Smyser CD, Snyder AZ, Shimony JS, Blazey TM, Inder TE, Neil JJ. Effects of white matter injury on resting state fMRI measures in prematurely born infants. PLoS ONE. 2013;8: e68098.
Smith PLP, Mottahedin A, Svedin P, Mohn CJ, Hagberg H, Ek J, Mallard C. Peripheral myeloid cells contribute to brain injury in male neonatal mice. J Neuroinflamm. 2018;15:301.
Ardalan M, Chumak T, Vexler Z, Mallard C. Sex-dependent effects of perinatal inflammation on the brain: implication for neuro-psychiatric disorders. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20092270.
Galinsky R, van de Looij Y, Mitchell N, Dean JM, Dhillon SK, Yamaguchi K, Lear CA, Wassink G, Davidson JO, Nott F, et al. Magnetic resonance imaging correlates of white matter gliosis and injury in preterm fetal sheep exposed to progressive systemic inflammation. Int J Mol Sci. 2020;21:8891.
Johnson JI, Sudheimer KD, Davis KK, Kerndt GM, Winn BM. The sheep brain atlas. https://msu.edu/~brains/brains/sheep/index.html. Accessed 14 May 2023.
Funding
This study was supported by project grants from the National Health and Medical Research Council of Australia (R.G.; 1090890 and 1164954), the Cerebral Palsy Alliance, Harold and Cora Brennen Benevolent Trust, Health Research Council of New Zealand (17/601, 22/559) and the Victorian Government’s Operational Infrastructure Support Program.
Author information
Authors and Affiliations
Contributions
SBK, AJG, JMD and RG conceptualised and designed the study. SBK, VZ, AM, ID, SLM, SBH, JMD, LB, AJG, GRP, RG undertook experiments, formal analysis and interpretation of the data. SBK and RG undertook the Golgi analysis, immunohistochemistry, cell quantification, analysis and preparation of figures. RG provided overall oversight of the research. All authors critically reviewed the manuscript and approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures were approved by the Hudson Institute of Medical Research Animal Ethics committee and were conducted in accordance with the National Health and Medical Research Council Code of Practice for the Care and Use of Animals for Scientific Purposes (Eighth Edition).
Consent for publications
Not applicable.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kelly, S.B., Dean, J.M., Zahra, V.A. et al. Progressive inflammation reduces high-frequency EEG activity and cortical dendritic arborisation in late gestation fetal sheep. J Neuroinflammation 20, 124 (2023). https://doi.org/10.1186/s12974-023-02805-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-023-02805-x