
Abstract
Mature B cell neoplasms, previously indolent non-Hodgkin lymphomas (iNHLs), are a heterogeneous group of malignancies sharing similar disease courses and treatment paradigms. Most patients with iNHL have an excellent prognosis, and in many, treatment can be deferred for years. However, some patients will have an accelerated course and may experience transformation into aggressive lymphomas. In this review, we focus on management concepts shared across iNHLs, as well as histology-specific strategies. We address open questions in the field, including the influence of genomics and molecular pathway alterations on treatment decisions. In addition, we review the management of uncommon clinical entities including nodular lymphocyte-predominant Hodgkin lymphoma, hairy cell leukemia, splenic lymphoma and primary lymphoma of extranodal sites. Finally, we include a perspective on novel targeted therapies, antibodies, antibody–drug conjugates, bispecific T cell engagers and chimeric antigen receptor T cell therapy.
Similar content being viewed by others


Introduction
Mature B cell neoplasms, commonly known as indolent non-Hodgkin lymphomas (iNHLs), are a heterogeneous group of malignancies sharing similar disease courses and treatment paradigms. While these lymphomas are generally considered incurable, most patients can expect a lifespan similar to that of the age-matched population, with the exception of those who are young at diagnosis and those who, following initial systemic treatment, progress rapidly or experience transformation into aggressive lymphomas (8–10% risk at 5 years) [1,2,3,4,5,6,7,8,9]. A major premise in managing iNHL is that earlier treatment does not improve survival, and many patients can be observed for years before treatment is indicated [10,11,12].
Providing an exhaustive review of each iNHL is beyond the scope of this paper, and excellent disease-specific reviews have been previously published [13,14,15,16,17,18,19]. In this review, we will focus on general concepts in the treatment of iNHLs, with specific attention to unique histologies and scenarios in which management should depart from common paradigms.
Localized disease
As lymphocytes populate nearly every organ system, the occurrence of an isolated nodal or extranodal site of disease is intriguing. It raises the question of whether localized presentation is a matter of chance (i.e., incidental early identification of a process bound to disseminate with time) or represents an inherent biological property “directing” lymphoma to a distinct region. The latter hypothesis is supported by similarly low rates of progression in rigorously staged patients with localized follicular lymphoma (FL) treated with observation compared with radiotherapy (RT) [20]. Further, several studies have suggested that localized FL is often characterized by a unique genetic profile, lower rates of BCL2/IGH translocation and a higher dependence on local microenvironmental features [21,22,23,24]. Similar observations apply to unique iNHL histologies involving extranodal sites (see Unique anatomical presentations below) [25,26,27].
Radiotherapy
Localized disease, present in 15–30% of patients, is one of the few situations in which iNHL is considered curable. Following RT, over 90% of patients will achieve a complete response (CR) and very few recurrences occur within the irradiated field [28, 29]. Approximately 50% will remain free from progression at 10 years with few relapses occurring beyond that time (i.e., suggesting cure) [30,31,32,33,34]. Current-day response rates may be considerably higher due to improved staging with positron emission tomography (PET), molecular testing of bone marrow (BM) and modern improvements in RT delivery with reduced toxicity [31]. Nonetheless, fewer than 30% of eligible patients are treated with RT despite its curative potential, in part due to skepticism about RT safety [35,36,37]. Contemporary treatment paradigms utilize involved-site radiotherapy (ISRT), which focuses treatment on radiographically apparent areas of disease. Compared to historical paradigms of total lymphoid irradiation or involved-field RT (IFRT), ISRT has significantly reduced the exposed anatomic areas [38, 39]. Depending on the irradiated site, less than 3% of patients will develop severe acute toxicities and less than 1.5% will develop late toxicities. The most common side effects are local skin erythema and mucositis, which are usually manageable with supportive care strategies and resolve within a few weeks (Table 1). Concerns about secondary malignancies in irradiated patients have not been supported in the population of patients with FL and marginal zone lymphoma (MZL) treated with RT [40,41,42,43]. Another possible explanation for the low utilization rate of RT is a sentiment among some clinicians that active intervention is not justified considering the excellent outcomes seen with observation (with up to 20% experience spontaneous remission) [44]. However, a SEER analysis of 6568 patients with localized grade 1–2 FL found that upfront RT was associated with improved disease-specific survival (DSS) and overall survival (OS) compared to patients who did not receive RT (10 y and 20 y DSS 79% and 63% for RT vs. 66% and 51% for no RT) [45]. In contrast, several reports suggested similar OS for upfront RT compared with observation; most notably, a multicenter retrospective review of 256 patients with FL rigorously staged with PET and a BM biopsy and treated with RT (n = 171) or watchful waiting (WW) (n = 85) demonstrated no difference in the time to first chemotherapy (TTC) treatment (4-year TTC 75–80%) [20, 37, 46, 47].
A more challenging question is whether patients with a localized major site of disease and minimal systemic involvement (for example, minute BM involvement) benefit from RT, which would no longer offer curative intent. Overall, approximately 40–70% of patients with FL, 4–10% of those with mucosa-associated lymphoid tissue (MALT) lymphoma, 30–60% of those with other MZL, and 50–90% of those with mantle cell lymphoma (MCL) will have morphologic BM involvement [48,49,50,51,52,53,54,55,56,57]. Molecular involvement of BM by flow cytometry or PCR, regardless of the presence or absence of morphologic involvement, can be seen in 40–65% of patients with FL at diagnosis, including in some with stage I/II disease. Overall, these patients have a much shorter PFS after RT (~ 5-year PFS of 50% vs. 95%; n = 67) and after systemic chemoimmunotherapy (CIT) (3-year PFS 40% vs. 85%; n = 53), comparable to that of patients with morphological BM involvement; however this difference is not significant among patients with limited stage disease [58,59,60]. Similarly, some patients with FL will have evidence of minimal residual disease (MRD), defined as detectable circulating BCL2/IGH + cells, at diagnosis despite negative molecular BM assessment [58]. Although half of these patients will become MRD-negative after RT, this is not associated with a decreased chance of relapse, and MRD-driven consolidation with rituximab after IFRT can improve PFS for these patients [58].
Overall, we favor ISRT for localized disease based on its low toxicity and probable PFS benefit. In the modern era, we treat most localized cases with definitive intent with 24 Gy in 12 fractions based on the noninferior outcomes from the BNLI RT dose de-escalation study [29].
Outcomes of patients who progress after primary RT are favorable, with a 3-year freedom from progression (FFP) of 57% [61]. In certain cases in which RT toxicity may be higher or a long course of RT cumbersome (e.g., orbital MALT, salivary gland MALT), we offer patients the option of a stepwise approach starting with very low dose RT regimen of 2 Gy × 2 based on promising data from a single institutional series (see RT for symptomatic advanced-stage disease) [62, 63]. Though several authors have advocated a considerable PFS advantage for CIT with or without RT over RT alone, particularly for localized bulky disease (defined in this context as > 5 cm), this is not our practice [64, 65]. At our institution, we rigorously stage patients with PET, incorporating this imaging modality into our RT simulation scan. We treat proximal stage II disease in a single field using modern RT approaches (i.e., intensity modulated RT, IMRT). If a patient does have more distant stage II disease, we will often treat two or more sites using separate isocenters. Given our recommended doses of typically less than 30 Gy, we rarely encounter dose-limiting toxicity. Although some will be considered for consolidative systemic therapies after RT, many patients will be cured with RT alone. Similarly, we rarely add RT or CIT consolidation for fully resected disease [47]. We do not routinely offer proton RT, which theoretically has improved conformality with lower RT exposure to adjacent tissue, as efficacy data are limited, there can be some uncertainty of dose range near critical normal structures and toxicity of ISRT using IMRT is not a major concern [66].
Systemic disease
Indications for treatment
Multiple studies have demonstrated that early treatment of asymptomatic patients with low-burden iNHL does not prolong OS or reduce the rate of histologic transformation (HT) compared to expectant management [11, 12, 67, 68]. Therefore, most clinicians observe patients until conventionally defined treatment criteria are met. Typically, treatment is indicated for patients with symptomatic or high-burden disease and those with end-organ compromise, based on early studies in FL by the Groupe d’Etude des Lymphomes Folliculaires (GELF) and British National Lymphoma Investigation (BNLI). Symptomatic/high burden nodal disease is defined as any mass > 7 cm; involvement of ≥ 3 nodal sites each ≥ 3 cm; splenomegaly below the umbilical line; pleural or peritoneal effusion; cytopenias; and/or > 5.0 k/mcl circulating blood lymphoma cells [11]. Some also consider macroscopic involvement of cortical bone, kidneys and liver, and rapid progression over the preceding 3 months to be indicators of more aggressive disease, justifying the initiation of therapy [12, 69]. While these criteria are based on data from the pre-rituximab era, they seem to hold true in current-day practice [11, 12, 70]. They have also been extrapolated to the management of patients with low-risk MCL, nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL), Waldenstrom macroglobulinemia (WM) and MZL with the exception of presence of circulating lymphoma cells in the peripheral blood in the leukemic iNHLs [71,72,73,74]. Reassuringly, none of the aforementioned studies demonstrated a significant increase in the risk of HT in patients initially observed compared to those who were treated early.
Lack of survival advantage, however, does not necessarily imply lack of benefit. In defined groups of patients, early anti-CD20 monotherapy can be relevant. In some elderly or frail patients with progressive, low-burden disease, early treatment with single-agent rituximab may prolong the chemotherapy-free interval. Rituximab can also have a role in the treatment of patients whose quality of life (QoL) is heavily affected by an expectant approach and/or for whom the lymphadenopathy is cosmetically burdensome [75]. In these patients, rituximab monotherapy is associated with a 71% overall response rate (ORR), 12% complete response (CR) rate and median time to treatment failure (TTF) of 4 years, which may be prolonged by adding maintenance therapy or retreating at progression [76].
Concerns associated with the early use of rituximab include the slightly increased risk of infection (1% for rituximab induction vs. 4% for rituximab maintenance) and future resistance to anti-CD20 agents [75]. In the RESORT trial, which evaluated frontline treatment with rituximab monotherapy followed by rituximab maintenance (RM), the additional exposure to rituximab was not associated with later resistance; however, this was not a research question addressed by the trial [76, 77]. Further data may be provided by a phase III trial comparing WW with rituximab as first-line treatment in patients with advanced-stage FL with low tumor burden (FLORA study, JCOG1411) [78].
In our practice, we do not recommend treatment initiation for asymptomatic patients with advanced, low-burden iNHL because of the lack of survival benefit, because 20–30% of patients will not meet treatment criteria for at least 10 years, and the median time to treatment in newly diagnosed patients is 3 years [12]. Although some clinicians believe that early treatment may instill a sense of security in patients, thus improving QoL, in our experience, patients’ QoL is generally preserved with careful, regular follow-up, judicious use of imaging, transparent communication and proactive management of anxiety and/or depression. In the rare instances in which we consider early treatment with rituximab monotherapy for FL, we generally administer 4 weekly doses without RM, though may consider a second round of rituximab at weeks 15–18, particularly in patients with a partial response (PR) [79]. In those with MZL, LPL/WM and other rare iNHL, such as NLPHL, we adopt a similar approach, but consider RM following induction [71,72,73,74, 76, 77].
Risk prediction and prognostic genomic biomarkers
Several studies have sought to identify clinical and molecular markers of worse prognosis in iNHL, mostly borrowing from FL. The original Follicular Lymphoma International Prognostic Index (FLIPI) was developed based on a cohort of 4167 patients followed in the pre-rituximab era and subsequently validated with current treatment approaches [80,81,82]. It is based on measures of disease burden (stage, > 4 LN regions), measures of disease activity or end-organ damage (hemoglobin < 12, LDH > upper limit of normal), and patient factors (age > 60). FLIPI2, an updated version developed in the rituximab era, uses BM involvement and largest mass > 6 cm in lieu of stage and nodal regions, and beta-2-microglobulin (β2M) instead of LDH [60]. Both studies used these factors to generate three risk groups: low risk (0–1 risk factors), intermediate risk (2 risk factors), and high risk (3–5 risk factors) with respective 5 y OS by FLIPI of 91% versus 78% versus 51%, and 10 y OS of 71% versus 51% versus 36% [80]. The PRIMA-PI uses only two risk factors (β2M > 3 mg/L and BM involvement) to derive similar risk groups [82]. Poor performance status, which is not part of FLIPI, FLIPI2, or PRIMA-PI, should be considered a negative prognostic feature and was not included due to few patients represented in the cohorts [83].
Finally, m7-FLIPI adds mutational data of seven key genes, four with strong prognostic power (EZH2 and ARID1A—‘good’; EP300, FOXO1—‘poor’) and three that are borderline (MEF2B—‘good’; CREBBP, CARD11—‘poor’) [83]. The m7-FLIPI applies only to patients treated with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) or R-CVP (rituximab, cyclophosphamide, vincristine, prednisone), refining the identification of high-risk patients by reallocating about a third of high-risk per FLIPI alone to the low-risk group. Per m7-FLIPI, patients with high-risk FLIPI are high risk only if there is a ‘poor’ mutation or poor performance status, while patients with EZH2 mutations (~ 20–25% of FLs) are nearly uniformly assigned low risk [83]. The strength of this score is in its negative predictive value, whereby 90% and 80% of low-risk patients can expect to be progression free at 1 and 2 years, respectively, while about 50% of high-risk patients will be refractory to treatment or experience progression of disease (POD) during that time. Importantly, the inclusion of mutations that may be associated with specific clinical features (high turnover and elevated LDH) in the prognostic score may underestimate the prognostic effect of the underlying genomic features. Further, the m7-FLIPI was developed on a small cohort and its prognostic value may not be reproducible in patients treated with diverse regimens (e.g., EZH2 mutations seem to confer better prognosis with R-CHOP but not bendamustine rituximab—BR) [84, 85].
Prognostic scores in other iNHL are not as stringently validated but tend to follow the same logic of incorporating measures of disease burden, disease activity and patient age and performance status. For splenic MZL (SMZL), scores include hemoglobin, platelets, LDH and presence of extra-hilar lymphadenopathy; for MALT lymphoma, age, stage and LDH; and for WM, scores also incorporate IgM levels and β2M [86,87,88,89]. In practice, these risk scores cannot be used to guide earlier treatment and are not accurate enough to provide patient counseling [90]. We use these scores to ensure a diligent workup of high-risk patients to rule out an underlying aggressive lymphoma.
Frontline treatment
In patients meeting criteria for treatment, regimens are based on a backbone of an anti-CD20 agent (rituximab, R; ofatumumab; obinutuzumab, G). In the case of FL, anti-CD20 therapy is combined with either chemotherapy (BR/G or R/G-CHOP) or R-lenalidomide. Regarding the initial choice of anti-CD20 agent, the GALLIUM study compared R-chemotherapy to G-chemotherapy in 1202 patients with FL and demonstrated longer PFS with obinutuzumab (3 y PFS 80% vs. 73%), though this advantage was driven by the bendamustine subgroup and by patients younger than age 60 [91, 92].
As for choice of chemotherapy backbone, bendamustine-based treatment resulted in a 5-year PFS of 65%, compared with 56% for CHOP-based therapy (Table 2) [93,94,95,96]. Initial findings from the STiL trial suggested superiority of BR over R-CHOP, particularly in MCL, FL, and WM, but not in MZL [96]. However, these findings were not reproduced in the larger GALLIUM trial, and STiL has been criticized for the crossover design potentially leading to underestimation of the activity of R-CHOP [93]. Nonetheless, it is clear that bendamustine-based treatment is not inferior to CHOP [91, 95]. One concern with bendamustine is the increased though statistically insignificant risk of nonrelapse mortality demonstrated in the long-term follow-up of the BRIGHT and GALLIUM studies (5% vs. 2% GALLIUM and 11% vs. 7% BRIGHT) [91, 95]. Importantly, this statistically insignificant risk was not driven by infection and a significant proportion of deaths in the bendamustine groups were noted several years from treatment. Thus, it is hard to interpret these observations, particularly in the setting of lower rates of progression with bendamustine-based treatment leading to no difference in overall survival. One scenario in which oncologists often favor R/G-CHOP is in suspected yet unproven HT, in those with high LDH or high FDG avidity on diagnostic PET. However, in a recent report from the GALLIUM study, there was no association between treatment arm and subsequent transformation in patients with high standardized uptake value (SUV) at baseline (assessed for SUVmax > 10 and for SUVmax > 20) [97].
The addition of maintenance anti-CD20 once every 2 months for 2 years is associated with a considerable increase in PFS after frontline and second-line treatment with CHOP-based therapy. The PRIMA study, which evaluated RM in 1018 patients who attained an initial PR or better with R-chemotherapy (mostly R-CHOP), demonstrated that RM was associated with a 6.5-year increase in PFS (median PFS 10.5 vs. 4.1y; 10y PFS 51% vs. 35%) [94]. This benefit was independent of the depth of response (CR vs. PR). However, even without RM, the median time to next chemotherapy was over 9 years, and there was no difference in OS (10y OS ~ 80% in both arms). There was also no difference in the rate of transformation between the arms (8–9%), and although the rate of early progression (24 m from starting chemotherapy) was lower in the RM arm (~ 12% vs. 25%), the limited efficacy of RM in more aggressive disease explains the equivalent OS [98]. Serious infectious complications or lack of response to subsequent treatments after prolonged rituximab exposure was not of significant concern after initial R-CHOP [94]. However, the addition of maintenance after bendamustine-based regimens is controversial and may be associated with an increased risk of severe, sometimes fatal, late infections, particularly in older patients (12–17% vs. 4–6%, fatal in 4–6% vs. 2%) [91, 99]. The role of RM in non-FL histologies is poorly defined with nonrandomized observational studies suggesting a potential benefit in WM and MZL (WM median PFS 56 m with RM vs. 29 m without), however, with increased rates of infection (43% vs. 25%) [100,101,102,103].
In FL, lenalidomide has been used in lieu of chemotherapy. The RELEVANCE trial compared R-lenalidomide (R2) to R-chemotherapy (72% R-CHOP) in 1030 patients with FL (50% high-risk FLIPI; 13% grade 3A; 40% bulky > 7 cm disease). It demonstrated similar response rates and PFS between regimens (R2: ORR 86%, CR 48%, 3y PFS of 77%; R-chemo: ORR 92%, CR 53%, 3y PFS 78%) which may be an underestimation as 20% of patients were not evaluable [104]. Preliminary results from a phase II study suggest outcomes are similar when using obinutuzumab in place of rituximab (n = 90; CR by PET 92%; 12% discontinuation rate) [105]. The main drawback of lenalidomide-based treatment is its length of 18–24 months and the prevalent yet manageable low-grade rash, diarrhea and constipation (each in about a third of the patients—for a review of management of these side-effects see [106]. Compared to R-CHOP, there is no hair loss and there are lower rates of febrile neutropenia (2% vs. 7%) [104].
For iNHL other than FL (e.g., MZL, MALT, NLPHL), initial treatment with rituximab monotherapy results in high response rates and long PFS (ORR 73–78%, CR by CT 42–55%, PFS 5–6 years), and incorporation of chemotherapy may be reserved for patients with bulkier disease or in whom a rapid response is warranted [107,108,109]. The role of lenalidomide in MZL is not clear with a subanalysis from the AUGMENT trial in 63 relapsed/refractory (R/R) cases showing no advantage over rituximab monotherapy (though limited by sample size and imbalance in baseline prognostic factors) [110]. For other lymphomas, including WM or SLL, initial treatment with ibrutinib or venetoclax (in SLL) may be preferred, at a cost of indefinite treatment as opposed to short-term chemotherapy [111, 112].
Finally, patients with hairy cell leukemia (HCL) attain exceptionally good responses with minimal toxicity using a combination of rituximab and cladribine with near-uniform CR and long-term (7–10 years) PFS and MRD negativity can be achieved with minimal toxicities using a combination of cladribine with concurrent rituximab, which is our practice [113, 114]. Durable remissions can also be attained with cladribine or pentostatin monotherapy (CR rate > 75%, PR rate > 5%, 4-year OS > 95%) which is the standard of care in many centers [115,116,117,118]. Of note, treatment with purine analogs in this setting may be associated with early profound cytopenias and blood counts should be evaluated regularly during treatment. The role of the BRAF-inhibitor vemurafenib is being explored in the upfront setting for HCL and has shown promising response rates [119].
In summary, frontline R/G-CHOP, B-R/G and R2 have comparable response rates in FL and treatment selection should be based on patient-specific factors. We prefer frontline B-R/G for its favorable toxicity profile and short treatment duration but avoid adding maintenance in this setting. In patients who are chemotherapy averse or in whom side effects of cytotoxic chemotherapy would be problematic, we offer R2. We consider obinutuzumab in place of rituximab for younger patients with FL (age < 70) particularly in the context of a bendamustine-based treatment [92]. Though still in its infancy, genomic biomarkers may contribute to management decisions. In FL, a recent report from the GALLIUM study suggested superiority of R-CHOP over BR in EZH2-mutated cases (20–25% of the population; HR 0.25) [84, 120]. For patients with TP53, NOTCH1 and SF3B1 mutations, targeted agents or clinical trials should be considered [121, 122].
RT for symptomatic advanced-stage disease
In symptomatic advanced-stage disease, the role of RT is primarily palliative. We rely on a program utilizing very low doses of RT (VLDRT), typically 2 Gy × 2, as compared to the standard full-dose regimens of 24–30 Gy [17, 123]. VLDRT has proven highly effective in controlling lymphomatous lesions irrespective of overall disease stage, histology or number of prior lines of systemic therapy [124]. VLDRT is associated with an ORR of > 80% with an anticipated 5-year local PFS of ~ 75% (though inferior to 24 Gy ORR > 90% and 5yPFS 91%) [125, 126].
We reserve VLDRT, rather than ISRT, for patients who require RT to multiple disease sites, have detectable BM involvement, and/or are frail. We reassess patients 10 weeks after VLDRT and consider additional 4 Gy or escalation to full dose RT depending on the residual disease.
Imaging and biomarkers for diagnosis and response assessment
Although PET is known to be sensitive and specific for detection of residual disease in aggressive lymphomas, the use of PET in iNHL is controversial due to high variability in FDG-uptake [127, 128]. Baseline PET is important in identifying cases of transformation [129]. Following CIT, response assessment by PET is superior to that by CT, as it identifies more than twice as many patients with a CR (Deauville ≤ 3 consistent with CR in 76% of patients). This is associated with a 30mPFS of 87% compared to 55% for patients with Deauville > 3 [130, 131].
More recently, circulating tumor DNA (ctDNA) has gained attention as a new method of MRD assessment [132]. The advent of high-throughput sequencing (HTS)-based approaches has made it possible to detect small amounts of ctDNA that is continually shed into the bloodstream [133]. Preliminary data in DLBCL, HL and FL suggest that ctDNA has promise as a marker of MRD and predictor of recurrence [134,135,136,137]. In an ongoing study, we aim to prospectively analyze ctDNA levels in patients with newly diagnosed FL to benchmark levels in correlation to treatment response with the goal of utilizing this as an early endpoint in future clinical trials (NCT04468841).
Unique anatomical presentations
While the majority of iNHLs present with predominantly nodal and BM involvement, there are several unique clinical presentations of organ-specific extranodal lymphomas that warrant distinct treatment strategies.
Splenic lymphoma and splenectomy
One unique clinical presentation is that of iNHL with circulating peripheral blood lymphoma cells with or without splenomegaly, but with minimal to no lymphadenopathy (LAD). This presentation is characteristic of SMZL, but can also be seen in a subset of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), MCL, HCL and WM [138, 139]. All tend to be characterized by an indolent course and may be associated with shared molecular pathway abnormalities [140]. Treatment paradigms vary between institutions, but in our experience these patients may be managed expectantly in a similar fashion to those with SMZL. For SMZL, some cases are hepatitis C virus (HCV) associated and regression of the MZL can be achieved with antiviral therapy; for this reason, we test patients with newly diagnosed SMZL for HCV prior to proceeding with treatment [141, 142]. Our preference for frontline treatment is rituximab monotherapy, which is associated with a response rate of 95%, 5y PFS 73% (vs. 58% for splenectomy) and 10y PFS of 60%, outcomes comparable to those of rituximab plus chemotherapy [143,144,145]. We plan for the possibility of splenectomy at diagnosis and if feasible defer any treatment until pre-splenectomy vaccinations for encapsulated bacteria are completed. In patients who fail rituximab, we use BR (ORR 91%, CR rate 73%, 3y PFS 90%) with close observation and referral for splenectomy in those who fail to achieve a good response or progress early [146]. In the latter case, we aim to attain some control of the size of the spleen (preferably < 20 cm craniocaudal axis) to allow for a laparoscopic procedure [147, 148]. The risk of perioperative mortality seems to be lower than anticipated in the case of iNHL, less than 2% in a cohort of nearly 2000 patients [149]. Post-splenectomy infections are noted in approximately 3%, with overwhelming infections in 1.4% of patients, mostly within the first 3 years following surgery [150, 151]. Postsurgical splenic and portal vein thromboses remain relatively common complications of splenectomy, with an average incidence of 2–3% [152]. An alternative to surgery is low-dose splenic radiation (4–8 Gy), which is not anticipated to result in significant adverse events (AEs), particularly if the adjacent kidney is not included in the radiated field [153, 154]. Data about the feasibility of this approach come from a limited historical case series, and a clinical trial in patients who have failed rituximab is about to be open [155].
Gastrointestinal (GI)
The GI tract is the most common site of origin of extranodal lymphomas, and secondary GI involvement can occur in approximately 10% of patients [156]. For gastric MALT lymphoma associated with active H. pylori infection, eradication of the bacteria can lead to an estimated 10-year OS 95% and EFS 86% (CR rate 50–90%), while in the remainder, gastric radiation is associated with up to 100% CR rate, and 10-year relapse-free survival 77% [157,158,159]. Of note, 15–30% of patients present with an 11;18 translocation involving the MALT1 gene on chromosome 18 (API2-MALT1) and have a lower response rate to antibiotics and in some cases resistance to rituximab monotherapy [160,161,162]. The role of other translocations involving the MALT1 gene (e.g., 14; 18) is less clear, though these entities seem molecularly similar. iNHL involving other sites in the GI tract are mostly MALT lymphomas and FL that in most cases are characterized by an exceedingly indolent course [163,164,165]. Unique in this regard is the rare entity of immunoproliferative small intestinal disease (IPSID, also known as alpha heavy chain disease or Mediterranean lymphoma) which is a variant of MALT which secretes alpha heavy chains. This subtype typically presents in younger male patients, is associated with Mediterranean decent and often manifests with abdominal pain, chronic diarrhea, malabsorption, and severe weight loss [166].
Lungs
Primary malignant lymphomas of the lung are very rare. These are mostly extranodal MZLs of bronchial-associated lymphoid tissue (BALT), though primary FL or MCL of the lungs has been reported anecdotally or by extension from widespread disease [167]. Low frequencies of persistent infection with Chlamydia pneumoniae, C. trachomatis, and C. psittaci have been identified in pulmonary MALT lymphomas, though BALT has not been directly connected to a single pathogenic agent [168]. Although some oncologists may feel compelled to treat BALT early, we recently demonstrated that BALT lymphoma is an indolent disease that can be managed by local excision or expectantly in most patients, without systemic therapy for many years [169].
Central nervous system (CNS)
CNS involvement by iNHL occurs in less than 3% of patients [170]. Although it has been most well described in LPL/WM as Bing-Neel syndrome, it can also occur in SLL, MCL and FL [171, 172]. Treatment, which involves systemic and intrathecal chemotherapy, should be considered carefully due to low response rates and high toxicity [173]. Recent data have shown significant response rates to ibrutinib therapy which has been given at a dose of 420 mg as well as 560 mg with or without rituximab, which is our preference in WM, MCL and SLL [174, 175]. Though further investigation is required, focal RT and whole brain RT (WBRT) have been demonstrated safe and effective in some cases [176].
Skin
The two main subtypes of cutaneous iNHL are primary cutaneous follicle center lymphoma (PCFCL) and primary cutaneous marginal zone lymphoma (PCMZL). There are two known subtypes of PCMZL, one which demonstrates diffuse proliferation of neoplastic B cells expressing IgM and CXCR3 (clinically similar to MALT lymphoma) and the other which expresses class-switched immunoglobulins and shows a predominance of T cell infiltration [177]. PCFCL is comprised of large cleaved cells (which may be confused with DLBCL) that are usually localized on the head, scalp or trunk, and are mostly BCL2-negative without t(14;18) translocation [178]. Although the underlying etiology of cutaneous lymphomas is unclear, Borrelia burgdorferi infection (Lyme disease) has been significantly associated with primary cutaneous lymphomas in endemic geographic regions, though it remains unclear whether treatment of B. burgdorferi infection leads to regression of the lymphoma [179, 180]. Both PCMZL and PCFCL are indolent diseases which can be managed expectantly, with localized RT or with rituximab monotherapy [181].
Ocular
Extranodal MZL accounts for greater than 50% of ocular adnexal lymphomas (OAL), which are rare and most often localized [182, 183]. Similar to the potential infectious drivers seen in other MZLs, a subset of OAMZL patients will have an underlying C. psittaci infection [184]. Although a phase II study including 27 patients with newly diagnosed or relapsed OAMZL treated with doxycycline showed an ORR of 48% and a 2-year failure-free survival of 66%, this is not standard practice [185]. The majority of cases reported in the literature are treated with either standard-dose (24–30.6 Gy) or high-dose (> 30.6 Gy) IMRT with overall response rate greater than 90% and 5-year local control rates approaching 100% and overall survival greater than 90% [183].
Relapsed and refractory disease
Once relapse is suspected, it is important to rule out HT to a more aggressive lymphoma particularly if progression occurs within the first 12–24 months after prior therapy (POD12/24). POD12 and POD24 have received much traction in recent years as an indicator of more aggressive disease requiring distinct treatment approaches [186]. However, it is important to note that the poor prognosis of POD12/24 is mainly driven by a subset of patients who experience HT (estimated at 30–40% of those with POD24 treated with R-CHOP and 75% of those with POD24 treated with BR) [9, 187,188,189]. These rates may be lower with the incorporation of PET in the diagnostic workup of iNHL, which allows for early recognition of HT prior to initiation of frontline therapy. [129, 187] Long-term outcomes of patients who experience early progression with an indolent histology are considerably superior, and it is not clear that they should be managed differently than patients with a later progression [129, 187, 190]. After HT has been excluded, patients may be observed for a period of time if they are asymptomatic without signs of end-organ compromise. If low disease volume or comorbidities are present, retreatment with a course of single agent rituximab can be considered, resulting in a median time to treatment failure of approximately 4 years [76, 79, 191]. Substituting rituximab with obinutuzumab in this setting does not translate into an improvement in PFS [192]. Ultimately, in patients meeting criteria for treatment (as with frontline disease), our preference is to choose a noncross-reacting frontline regimen, typically associated with response rates of 85% for R-CHOP, 82% for BR, and 78% for R2 (Table 3) [110, 193]. In patients with multiple relapses, we opt for enrollment on a clinical trial or use one of the targeted therapies discussed below.
Targeted therapies
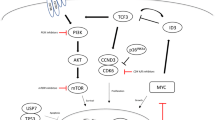
PI3K3-inhibitors
Many iNHLs depend on the phosphatidylinositol 3-kinase delta (PI3Kδ) pathway, and there are now three PI3K-inhibitors (PI3Ki) approved for the treatment of R/R FL after two prior lines of therapy (idelalisib, copanlisib, and duvelisib—overall N ~ 450, ORR of 50–60%, CR 5–15%, median PFS 10–12 m in a heavily pretreated population) [95, 194,195,196]. Responses to PI3Ki should be seen within the first few months of treatment (median ~ 3 m) and are observed at similar rates among patients who had prior POD24 and among varied subtypes of R/R iNHL [197].
Idelalisib, a PI3Kδ-inhibitor, was the first drug to be studied (N = 125) and demonstrated an ORR of 57% (6% CR), median PFS 11 m and OS 20 m. Duvelisib, an oral inhibitor of the PI3K-δ and -γ isoforms, received approval based on an open-label, global phase II trial (DYNAMO) (N = 129), which demonstrated ORR 47%, median PFS 10 m [195]. Unfortunately, toxicity with both agents is nearly universal leading to drug discontinuation in approximately 25%, dose reduction in 20–40%, and treatment interruptions or delays in 65–75% of patients [195, 196]. Further, there are black-box warnings for severe transaminitis (~ 15%), diarrhea (~ 15%), pneumonitis (~ 5%) and bowel perforation (~ 0.5%) (idelalisib). Other common grade 3 AEs are neutropenia (27%) and increased risk of pneumocystis jiroveci pneumonia (PJP) and CMV reactivation (both rare), which require PJP prophylaxis and regular CMV RNA monitoring. For diarrhea, there are two peaks, early (easily manageable) and late ~ 6 m (which resembles inflammatory colitis). Colitis and transaminitis appear to occur more frequently in patients with fewer prior lines of therapy [198,199,200]. Transaminitis tends to occur early within the first 3 months (recommendation to monitor laboratories every 1–2 weeks for the first 6m).
Copanlisib, a PI3K α,δ-inhibitor, was evaluated in a phase II study (CHRONOS-1) (N = 142) with ORR 59% (CR 15%), median PFS 11 m. Toxicity was again nearly universal leading to drug discontinuation in 25%, dose reduction in 37%, and treatment interruptions or delays in 74% of patients, though most commonly hyperglycemia (50%) or diarrhea (35%). It does not require PJP or CMV prophylaxis and has no black-box warnings [201].
Of note, preliminary data from ME-401 (a novel PI3Ki) suggest that intermittent treatment for 7 days out of a 28-day cycle may offset the risk of severe AEs while maintaining high response rates [202]. Meanwhile, preliminary data for parsaclisib (highly selective PI3Kδ-inhibitor) demonstrate objective response rates of ~ 70% for relapsed or refractory iNHLs [203]. Umbralisib is another PI3Ki under investigation which confers fewer episodes of autoimmune-like toxicities.
BTK-inhibition
Ibrutinib, acalabrutinib and zanabrutinib bind covalently at the cysteine 481 site on the BTK receptor and have activity in SLL, WM, and MZL. All rarely lead to a CR, but most patients have stable disease or better and benefit from a 2-year PFS of 80% in WM and a median PFS of 14 months in MZL [111, 204]. In WM, patients with MYD88 mutations and CXCR4 wild type have improved response to ibrutinib, while those with both MYD88 and CXCR4 mutations have an inferior response to treatment which can be offset with the addition of rituximab and patients with wild-type MYD88 and CXCR4 mutations are unlikely to respond [111]. In MZL, the frequency of MYD88 mutation is less than 5% while ORR to ibrutinib 48%, suggesting that responses are independent of MYD88 mutation status [204, 205]. Notwithstanding, in a recent analysis in R/R MZL, patients with TNFAIP3 mutations had better responses to ibrutinib and those with MYD88L265P mutations had longer PFS [206]. Conversely, patients with mutations in KMT2D and CARD11 derived less benefit from treatment. CARD11 mutations have also been demonstrated in association with primary resistance to ibrutinib in several other studies, while development of mutations at the C481 site of BTK and in PLC-gamma is associated with acquired resistance to this class of drugs [207, 208]. To overcome these mechanisms, there are novel agents currently in clinical trials which target BTK in a noncovalent manner, have additional cellular targets (e.g., SRC kinases) or have targets downstream from MYD88 such as IRAK-4 and MALT1 (NCT03740529, NCT03893682, NCT03328078, NCT03900598, NCT03162536).
BCL-2 inhibition
BCL-2 is an anti-apoptotic protein implicated in the pathobiology of many iNHLs, most known in the context of the BCL2:IGH (t14;18) translocation in FL. Venetoclax, an oral BCL2 inhibitor, has considerable activity in CLL and MCL, but surprisingly disappointing activity in FL (ORR 38%, CR 14% PFS 13 m) [209]. Evaluation in other iNHL has been limited, though the drug is expected to have activity in MZL and WM [210]. Importantly, the drug has a manageable toxicity profile [209, 211]. To promote activity in FL, several trials are evaluating combination therapy with PI3K inhibition and with epigenetic targeting with EZH2 inhibitors.
EZH2 inhibition (FL)
Next-generation deep sequencing has uncovered mutations in chromatin-modifying genes (CMG), including KMT2D, CREBBP, and EZH2 in the majority of FL patients, which provide attractive therapeutic targets [212]. Tazemetostat, a drug targeting EZH2, has become one of the most promising novel therapeutics in FL with an ORR 77% in EZH2mut and 34% in EZH2wt, median PFS of 14 months and 11 months, respectively, and a manageable toxicity profile [213]. The drug has been recently approved by the FDA in patients with relapsed FL who have failed at least two lines of therapy. Several ongoing trials are evaluating EZH2i in combination with other targeted agents (NCT04224493, NCT02601950).
Antibodies, antibody–drug conjugates (ADC) and bispecific T cell engagers (BiTE)
Several novel antibodies, ADCs and BiTEs are in advanced stages of clinical trials and have been recently recommended by the NCCN guidelines. These include the anti-CD79b ADC polatuzumab vedotin, the CD3-CD20 BiTE mosunetuzumab and the anti-CD47 antibody magrolimab (Hu5F9-G4). Polatuzumab is NCCN recommended in FL and MCL (but not MZL) for use after ≥ 2 prior lines of therapy with rituximab (ORR 70%, CR 45%, median PFS 15 m; N = 20) or in combination with BR (CR by PET 70%, median PFS 17 m; N = 39) [214,215,216]. Of note, there was no difference between BR-polatuzumab and BR alone possibly owing to a high discontinuation rate for peripheral neuropathy (grade 1–2 observed in 40%). The drug is also associated with approximately a 40% incidence of grade 1–2 diarrhea.
Mosunetuzumab was evaluated in FL (n = 69) with an ORR of 64% and CR of 44% [217]. The main concern with this agent is cytokine release syndrome (CRS), which is seen in approximately 25% of patients and is usually self-limited and responsive to standard of care management, including tocilizumab. Similar CD3-CD20 BiTEs are being investigated as monotherapy and in combination with other agents (e.g., lenalidomide, polatuzumab, and the anti-PD-L1 antibody atezolizumab) (NCT04246086, NCT03671018, NCT02500407). Magrolimab (anti-CD47 antibody) in combination with rituximab is associated with an ORR of 61% and CR 24% and a median time to response of 2 months with a favorable toxicity profile [218].
Finally, ibritumomab tiuxetan (Zevalin) is an anti-CD20 antibody linked to radioisotopes, which is FDA approved in FL and is not widely used due to logistical difficulties and concern for cytopenias and secondary myelodysplastic syndrome. While the overall PFS is only 9 months, PFS is nearly 4 years in the 35% of patients who achieve a CR [219]. The treatment is administered as a single infusion in the outpatient setting after two doses of rituximab. We reserve this treatment for patients who have exhausted other options.
Indications for hematopoietic cell transplantation (HCT) and chimeric antigen receptor (CAR) T cells
The role of hematopoietic cell transplantation (HCT) in iNHL is controversial given the advent of novel agents and steady reductions over time in nonrelapse mortality (NRM) after HCT [220]. Consolidative autologous HCT (AHCT) has been most studied in patients with iNHL with POD24, where several reports have demonstrated a median PFS of 3–5 years, which is longer than the expected PFS with CIT alone and may potentially confer superior OS in high-risk patients who undergo AHCT within one year of initial treatment failure [221,222,223]. However, these data are limited by their retrospective nature, and AHCT has not been compared directly to novel treatments such as R2 [110, 224, 225]. With NRM rates less than 4% even in appropriately selected elderly patients, we offer AHCT consolidation to patients with refractory or early relapsed, high-risk FL who are in chemosensitive remission after salvage therapy, allowing us to reserve novel therapies for subsequent lines of treatment [221, 226].
The use and appropriate timing of allogeneic (allo)-HCT remains a complex treatment decision in patients with R/R iNHL, given concerns about high rates of NRM and graft-versus-host disease (GVHD) [227, 228]. In a large retrospective registry cohort of heavily pretreated patients with a history of POD24, allo-HCT was associated with impressive 5-year OS of 75% and 50% for matched related and unrelated donors, respectively, despite a median of three prior lines of therapy and 40% of patients transplanted with refractory disease [222]. However, there were considerable rates of NRM at 5 years (33% and 17% for matched related and unrelated donors, respectively), mainly secondary to GVHD [222]. It is plausible that with better patient selection and more modern HCT techniques, such as the use of reduced intensity (RIC) or non-myeloablative (NMA) conditioning, NRM would be considerably lower [220, 229, 230]. Haploidentical allo-HCT with posttransplantation cyclophosphamide (PT-Cy) has largely removed the barrier of donor availability with comparable efficacy and markedly lower rates of chronic GVHD compared to standard allo-HCT (12% vs. 49%, respectively) [231, 232]. We consider RIC or NMA allo-HCT, preferably on a clinical trial, for medically appropriate patients who relapse after AHCT and/or those with multiple relapses and short remission durations who demonstrate treatment sensitivity [233].
Decisions regarding HCT will become even more challenging with the forthcoming FDA approval of CD19-directed chimeric antigen receptor (CAR) T cells in iNHL. CAR T is associated with CR rates up to 88% with sustained remission in up to 89% of FL patients with an initial response, over median follow-up of 28.6 months [234,235,236]. Recently presented data from the phase 2 ZUMA-5 trial which evaluated commercially available axicabtagene ciloleucel in patients with FL and MZL demonstrated ORR 94% (95% in FL, 86% in MZL), with CR rate of 79% [237]. While high-grade CRS and neurotoxicity remain concerns, occurring at rates of 13% and 28%, respectively, our growing experience utilizing appropriately-timed anti-cytokine blockade and corticosteroids should help mitigate severe toxicities [234, 237, 238]. Future studies will evaluate CAR T cells for other iNHL histologies [239].
Conclusion
The treatment of iNHLs is one of the most fascinating and rapidly developing areas of oncology, in which clinicians need to balance potential toxicities with the benefits of treatment. An intimate familiarity with disease course and an understanding of molecular pathways are required to tailor therapy for each patient. The ideal treatment plan is one that will remain effective for many years while preserving the patients’ quality of life and longevity.
Availability of data and materials
Not applicable.
Abbreviations
- ADC:
-
Antibody–drug conjugate
- AHCT:
-
Autologous hematopoietic cell transplantation
- allo-HCT:
-
Allogeneic hematopoietic cell transplantation
- β2M:
-
Beta-2-microglobulin
- BALT:
-
Bronchial-associated lymphoid tissue
- BiTE:
-
Bispecific T cell engagers
- BM:
-
Bone marrow
- BNLI:
-
British National Lymphoma Investigation
- BR:
-
Bendamustine–rituximab
- CAR-T:
-
Chimeric antigen receptor T cell
- CIT:
-
Chemoimmunotherapy
- CNS:
-
Central nervous system
- CR:
-
Complete response
- ctDNA:
-
Circulating tumor DNA
- FFP:
-
Freedom from progression
- FL:
-
Follicular lymphoma
- FLIPI:
-
Follicular Lymphoma International Prognostic Index
- G:
-
Obinutuzumab
- GELF:
-
Groupe d’Etude des Lymphomes Folliculaires
- GI:
-
Gastrointestinal
- GVHD:
-
Graft-versus-host-disease
- HCL:
-
Hairy cell leukemia
- HCT:
-
Hematopoietic cell transplantation
- HCV:
-
Hepatitis C virus
- HTS:
-
High-throughput sequencing
- IFRT:
-
Involved-field radiotherapy
- ISRT:
-
Involved-site radiotherapy
- iNHL:
-
Indolent non-Hodgkin lymphoma
- LAD:
-
Lymphadenopathy
- MALT:
-
Mucosa-associated lymphoid tissue
- MCL:
-
Mantle cell lymphoma
- MRD:
-
Minimal residual disease
- MZL:
-
Marginal zone lymphoma
- NLPHL:
-
Nodular lymphocyte-predominant Hodgkin lymphoma
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PCFCL:
-
Primary cutaneous follicle center lymphoma
- PCMZL:
-
Primary cutaneous marginal zone lymphoma
- PET:
-
Positron emission tomography
- PI3Ki:
-
PI3K-inhibitor
- PJP:
-
Pneumocystis jiroveci pneumonia
- POD:
-
Progression of disease
- POD12/24:
-
Progression of disease in 12/24 months
- PR:
-
Partial response
- QoL:
-
Quality of life
- R:
-
Rituximab
- R2 :
-
Rituximab, lenalidomide
- R/R:
-
Relapsed/refractory
- R-CHOP:
-
Rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone
- R-CVP:
-
Rituximab, cyclophosphamide, vincristine, prednisone
- RM:
-
Rituximab maintenance
- RT:
-
Radiotherapy
- SMZL:
-
Splenic marginal zone lymphoma
- SUV:
-
Standard uptake value
- TTC:
-
Time to first chemotherapy
- TTF:
-
Time to treatment failure
- WBRT:
-
Whole brain radiotherapy
- WM:
-
Waldenstrom macroglobulinemia
- WW:
-
Watchful waiting
References
Magnano L, et al. Life expectancy of follicular lymphoma patients in complete response at 30 months is similar to that of the Spanish general population. Br J Haematol. 2019;185:480–91.
Tarella C, et al. Life expectancy in follicular lymphoma is mainly determined by response to first LINE treatment: a long-term survey on 597 patients. Blood. 2015;126:3989–3989.
Sarkozy C, et al. Cause of death in follicular lymphoma in the first decade of the rituximab era: a pooled analysis of French and US cohorts. J Clin Oncol. 2019;37:144–52.
van de Schans SAM, van Steenbergen LN, Coebergh JWW, Janssen-Heijnen MLG, van Spronsen DJ. Actual prognosis during follow-up of survivors of B-cell non-Hodgkin lymphoma in the Netherlands. Haematologica. 2014;99:339–45.
Howlader N, et al. Cancer statistics review, 1975–2017—SEER statistics. SEER cancer statistics review, 1975–2017, National Cancer Institute, Bethesda, MD. https://seer.cancer.gov/csr/1975_2017/ (2019).
Lobetti-Bodoni C, et al. The importance of age in prognosis of follicular lymphoma: clinical features and life expectancy of patients younger than 40 years. Blood. 2011;118:1593–1593.
Batlevi CL, et al. Follicular lymphoma in the modern era: survival, treatment outcomes, and identification of high-risk subgroups. Blood Cancer J. 2020a;10:74.
Link BK, et al. Rates and outcomes of follicular lymphoma transformation in the immunochemotherapy era: A report from the university of Iowa/mayo clinic specialized program of research excellence molecular epidemiology resource. J Clin Oncol. 2013;31:3272–8.
Sortais C, et al. Progression of disease within 2 years (POD24) is a clinically relevant endpoint to identify high-risk follicular lymphoma patients in real life. Ann Hematol. 2020;99:1595–604.
Advani R, Rosenberg SA, Horning SJ. Stage I and II follicular non-Hodgkin’s lymphoma: long-term follow-up of no initial therapy. J Clin Oncol Off J Am Soc Clin Oncol. 2004;22:1454–9.
Brice P, et al. Comparison in low-tumor-burden follicular lymphomas between an initial no-treatment policy, prednimustine, or interferon alfa: a randomized study from the Groupe d’Etude des Lymphomes Folliculaires. Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol. 1997;15:1110–7.
Ardeshna KM, et al. Long-term effect of a watch and wait policy versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma: a randomised controlled trial. Lancet. 2003;362:516–22.
Casulo C. How I manage patients with follicular lymphoma. Br J Haematol. 2019;186:513–23.
Yang JC, Yahalom J. Early-stage follicular lymphoma: what is the preferred treatment strategy? J Clin Oncol. 2018;36:2904–6.
Matasar MJ, et al. Follicular lymphoma: recent and emerging therapies, treatment strategies, and remaining unmet needs. Oncologist. 2019;24:e1236–50.
Diamond B, Kumar A. Mantle cell lymphoma: current and emerging treatment strategies and unanswered questions. Hematol Oncol Clin North Am. 2019;33:613–26.
Yahalom J. Radiotherapy of follicular lymphoma: updated role and new rules. Curr Treat Options Oncol. 2014;15:262–8.
Ayyappan S, William BM. Marginal zone lymphoma: clinicopathologic variations and approaches to therapy. Curr Oncol Rep. 2018;20:33.
Treon SP, et al. Genomic landscape of Waldenström macroglobulinemia and its impact on treatment strategies. J Clin Oncol. 2020;38:1198–208.
Tobin JWD, et al. Outcomes of stage I/II follicular lymphoma in the PET era: an international study from the Australian Lymphoma Alliance. Blood Adv. 2019;3:2804–11.
Katzenberger T, et al. A distinctive subtype of t(14;18)-negative nodal follicular non-Hodgkin lymphoma characterized by a predominantly diffuse growth pattern and deletions in the chromosomal region 1p36. Blood. 2009;113:1053–61.
Staiger AM, et al. Localized- and advanced-stage follicular lymphomas differ in their gene expression profiles. Blood. 2020;135:181–90.
Leich E, et al. Follicular lymphomas with and without translocation t(14;18) differ in gene expression profiles and genetic alterations. Blood. 2009;114:826–34.
Leich E, et al. Similar clinical features in follicular lymphomas with and without breaks in the BCL2 locus. Leukemia. 2016;30:854–60.
Soubeyran P, et al. Is there any place for a wait-and-see policy in stage I0 follicular lymphoma? A study of 43 consecutive patients in a single center. Ann Oncol. 1996;7:713–8.
Schmatz AI, et al. Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol. 2011;29:1445–51.
Beaton C, Davies M, Beynon J. The management of primary small bowel and colon lymphoma-a review. Int J Colorectal Dis. 2012;27:555–63.
Eichenauer DA, et al. Long-term course of patients with stage IA nodular lymphocyte-predominant Hodgkin lymphoma: a report from the German Hodgkin study group. J Clin Oncol. 2015;33:2857–62.
Lowry L, et al. Reduced dose radiotherapy for local control in non-Hodgkin lymphoma: a randomised phase III trial. Radiother Oncol. 2011;100:86–92.
Wilder RB, et al. Long-term results with radiotherapy for stage I-II follicular lymphomas. Int J Radiat Oncol. 2001;51:1219–27.
Brady JL, et al. Definitive radiotherapy for localized follicular lymphoma staged by 18F-FDG PET-CT: a collaborative study by ILROG. Blood. 2019a;133:237–45.
Lo A, et al. Long-term outcomes for patients with limited-stage follicular lymphoma: update of a population-based study. Blood. 2020;136:1006–10.
Teckie S, et al. Long-term outcome of 487 patients with early-stage extra-nodal marginal zone lymphoma. Ann Oncol. 2017;28:1064–9.
Barzenje DA, et al. Long-term outcome for patients with early stage marginal zone lymphoma and mantle cell lymphoma. Leuk Lymphoma. 2017;58:623–32.
Friedberg JW, et al. Follicular lymphoma in the united states: first report of the national lymphocare study. J Clin Oncol. 2009;27:1202–8.
Ruella M, et al. Addition of rituximab to involved-field radiation therapy prolongs progression-free survival in stage I–II follicular lymphoma: results of a multicenter study. Int J Radiat Oncol Biol Phys. 2016;94:783–91.
Friedberg JW, et al. Effectiveness of first-line management strategies for stage I follicular lymphoma: analysis of the National Lymphocare Study. J Clin Oncol. 2012;30:3368–75.
Illidge T, et al. Modern radiation therapy for nodal non-hodgkin lymphoma—target definition and dose guidelines from the international lymphoma radiation oncology group. Int J Radiat Oncol. 2014;89:49–58.
Yahalom J, et al. Modern radiation therapy for extranodal lymphomas: field and dose guidelines from the International Lymphoma Radiation Oncology Group. Int J Radiat Oncol Biol Phys. 2015;92:11–31.
Tsang RW, Gospodarowicz MK. Low-grade non-hodgkin lymphomas. Semin Radiat Oncol. 2007;17:198–205.
Guadagnolo BA, et al. Long-term outcome and mortality trends in early-stage, Grade 1–2 follicular lymphoma treated with radiation therapy. Int J Radiat Oncol. 2006;64:928–34.
Au WY, et al. Incidence of second neoplasms in patients with MALT lymphoma: no increase in risk above the background population. Ann Oncol Off J Eur Soc Med Oncol. 1999;10:317–21.
Goda JS, et al. Long-term outcome in localized extranodal mucosa-associated lymphoid tissue lymphomas treated with radiotherapy. Cancer. 2010;116:3815–24.
Krikorian JG, Portlock CS, Cooney P, Rosenberg SA. Spontaneous regression of non-Hodgkin’s lymphoma: a report of nine cases. Cancer. 1980;46:2093–9.
Pugh TJ, Ballonoff A, Newman F, Rabinovitch R. Improved survival in patients with early stage low-grade follicular lymphoma treated with radiation. Cancer. 2010;116:3843–51.
Michallet A-SAS, et al. Early stage follicular lymphoma: what is the clinical impact of the first-line treatment strategy? J Hematol Oncol. 2013;6:45.
Barzenje DA, et al. Radiotherapy compared to other strategies in the treatment of stage I/II follicular lymphoma: a study of 404 patients with a median follow-up of 15 years. PLoS ONE. 2015;10:e0131158.
Anderson T, et al. Malignant lymphoma. 1. The histology and staging of 473 patients at the National Cancer Institute. Cancer. 1982;50:2699–707.
Choi SI, et al. Prevalence and implications of bone marrow involvement in patients with gastric mucosa-associated lymphoid tissue lymphoma. Gut Liver. 2018;12:278–87.
Brandwein-Gensler MS, Mahadevia P, Gnepp DR. Chapter 13—hematopoietic lesions—diagnostic surgical pathology of the head and neck. 2nd ed. Philadelphia: W.B. Saunders; 2009. p. 933–74.
Arcaini L, et al. Primary nodal marginal zone B-cell lymphoma: clinical features and prognostic assessment of a rare disease. Br J Haematol. 2007;136:301–4.
Carbone A, et al. Monocytoid B-cell lymphoma with bone marrow and peripheral blood involvement at presentation. Am J Clin Pathol. 1989;92:228–36.
Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. J Clin Oncol. 1998;16:2780–95.
Cohen PL, Kurtin PJ, Donovan KA, Hanson CA. Bone marrow and peripheral blood involvement in mantle cell lymphoma. Br J Haematol. 1998;101:302–10.
Pittaluga S, et al. Prognostic significance of bone marrow trephine and peripheral blood smears in 55 patients with mantle cell lymphoma. Leuk Lymphoma. 1996;21:115–25.
Vasef MA, Medeiros LJ, Koo C, McCourty A, Brynes RK. Cyclin D1 immunohistochemical staining is useful in distinguishing mantle cell lymphoma from other low-grade B-cell neoplasms in bone marrow. Am J Clin Pathol. 1997;108:302–7.
Wasman J, Rosenthal NS, Farhi DC. Mantle cell lymphoma. Morphologic findings in bone marrow involvement. Am J Clin Pathol. 1996;106:196–200.
Pulsoni A, et al. Minimal residual disease monitoring in early stage follicular lymphoma can predict prognosis and drive treatment with rituximab after radiotherapy. Br J Haematol. 2020;188:249–58.
Kato D, et al. The clinical impact of minimal bone marrow involvement on the outcome of patients with follicular lymphoma. Blood. 2016;128:2966–2966.
Federico M, et al. Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol. 2009;27:4555–62.
Binkley MS, et al. Salvage treatment and survival for relapsed follicular lymphoma following primary radiation therapy: a collaborative study on behalf of ILROG. Int J Radiat Oncol Biol Phys. 2019;104:522–9.
Gunther JR, et al. Radiation therapy for salivary gland MALT lymphoma: ultra-low dose treatment achieves encouraging early outcomes and spares salivary function. Leuk Lymphoma. 2020;61:171–5.
Pinnix CC, et al. Ultra-low-dose radiotherapy for definitive management of ocular adnexal B-cell lymphoma. Head Neck. 2017;39:1095–100.
MacManus M, et al. Randomized trial of systemic therapy after involved-field radiotherapy in patients with early-stage follicular lymphoma: TROG 99.03. J Clin Oncol. 2018;36:2918–25.
Seymour JF, et al. Long-term follow-up of a prospective study of combined modality therapy for stage I–II indolent non-Hodgkin’s lymphoma. J Clin Oncol. 2003;21:2115–22.
Brady JL, et al. Definitive radiotherapy for localized follicular lymphoma staged by18F-FDG PET-CT: a collaborative study by ILROG. Blood. 2019b;133:237–45.
Horning SJ, Rosenberg SA. The natural history of initially untreated low-grade non-Hodgkin’s lymphomas. N Engl J Med. 1984;311:1471–5.
Young RC, et al. The treatment of indolent lymphomas: watchful waiting v aggressive combined modality treatment. Semin Hematol. 1988;25:11–6.
Morabito F, et al. Prospective study of indolent non-follicular non-Hodgkin’s lymphoma: validation of Gruppo Italiano Per Lo Studio Dei Linfomi (GISL) prognostic criteria for watch and wait policy. Leuk Lymphoma. 2002;43:1933–8.
Barta SK, et al. Randomized phase 3 study in low-grade lymphoma comparing maintenance anti-CD20 antibody with observation after induction therapy: a trial of the ECOG-ACRIN cancer research group (E1496). Cancer. 2016;122:2996–3004.
Borchmann S, et al. Active surveillance for nodular lymphocyte-predominant Hodgkin lymphoma. Blood. 2019;133:2121–9.
Kumar A, et al. Clinical presentation determines selection of patients for initial observation in mantle cell lymphoma. Haematologica. 2019;104:e163–6.
Kyle RA, et al. Prognostic markers and criteria to initiate therapy in Waldenstrom’s macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. In: Seminars in oncology. vol. 30. W.B. Saunders; 2003. p. 116–20.
Cheah CY, Opat S, Trotman J, Marlton P. Front-line management of indolent non-Hodgkin lymphoma in Australia. Part 2: mantle cell lymphoma and marginal zone lymphoma. Intern Med J. 2019;49:1070–80.
Ardeshna KM, et al. Rituximab versus a watch-and-wait approach in patients with advanced-stage, asymptomatic, non-bulky follicular lymphoma: An open-label randomised phase 3 trial. Lancet Oncol. 2014;15:424–35.
Kahl BS, et al. Rituximab extended schedule or re-treatment trial for low-tumor burden follicular lymphoma: eastern cooperative oncology group protocol E4402. J Clin Oncol. 2014;32:3096–102.
Williams ME, et al. Rituximab extended schedule or retreatment trial for low tumour burden non-follicular indolent B-cell non-Hodgkin lymphomas: Eastern Cooperative Oncology Group Protocol E4402. Br J Haematol. 2016;173:867–75.
Miyamoto K, et al. Phase III study of watchful waiting vs. rituximab as first-line treatment in advanced stage follicular lymphoma with low tumour burden (JCOG1411, FLORA study). Jpn J Clin Oncol. 2018;48:777–80.
Martinelli G, et al. Long-term follow-up of patients with follicular lymphoma receiving single-agent rituximab at two different schedules in trial SAKK 35/98. J Clin Oncol. 2010;28:4480–4.
Buske C, et al. The Follicular Lymphoma International Prognostic Index (FLIPI) separates high-risk from intermediate- or low-risk patients with advanced-stage follicular lymphoma treated front-line with rituximab and the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) with respect to treatment outcome. Blood. 2006;108:1504–8.
Nooka AK, et al. Examination of the follicular lymphoma international prognostic index (FLIPI) in the National LymphoCare study (NLCS): a prospective US patient cohort treated predominantly in community practices. Ann Oncol Off J Eur Soc Med Oncol. 2013;24:441–8.
Bachy E, et al. A simplified scoring system in de novo follicular lymphoma treated initially with immunochemotherapy. Blood. 2018;132:49–58.
Pastore A, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16:1111–22.
Jurinovic V, et al. Evaluation of the m7-FLIPI in patients with follicular lymphoma treated within the gallium trial: EZH2 mutation status may be a predictive marker for differential efficacy of chemotherapy. Blood. 2019;134:122–122.
Lockmer S, et al. M7-FLIPI is not prognostic in follicular lymphoma patients with first-line rituximab chemo-free therapy. Br J Haematol. 2020;188:259–67.
Montalbán C, et al. Risk stratification for Splenic Marginal Zone Lymphoma based on haemoglobin concentration, platelet count, high lactate dehydrogenase level and extrahilar lymphadenopathy: development and validation on 593 cases. Br J Haematol. 2012;159:164–71.
Arcaini L, et al. Splenic marginal zone lymphoma: A prognostic model for clinical use. Blood. 2006;107:4643–9.
Thieblemont C, et al. A MALT lymphoma prognostic index. Blood. 2017;130:1409–17.
Morel P, et al. International prognostic scoring system for Waldenström macroglobulinemia. Blood. 2009;113:4163–70.
Solal-Céligny P, et al. Follicular lymphoma international prognostic index. Blood. 2004;104:1258–65.
Marcus R, et al. Obinutuzumab for the first-line treatment of follicular lymphoma. N Engl J Med. 2017;377:1331–44.
Hiddemann W, et al. Immunochemotherapy with obinutuzumab or rituximab for previously untreated follicular lymphoma in the GALLIUM study: influence of chemotherapy on efficacy and safety. J Clin Oncol. 2018;36:2395–404.
Salles G, et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet. 2011;377:42–51.
Bachy E, et al. Sustained progression-free survival benefit of rituximab maintenance in patients with follicular lymphoma: long-term results of the PRIMA study. J Clin Oncol. 2019;37:2815–24.
Flinn IW, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood. 2014a;123:2944–52.
Rummel MJ, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381:1203–10.
Mir F, et al. Baseline SUVmax did not predict histological transformation in the phase 3 GALLIUM study in follicular lymphoma. Blood. 2020;135:1214–8.
Lugtenburg EJ, et al. Rituximab maintenance for patients with diffuse large B-cell lymphoma in first complete remission: Results from a randomized HOVON-Nordic Lymphoma Group phase III study. J Clin Oncol. 2019;37:7507–7507.
Hill BT, et al. Maintenance rituximab or observation after frontline treatment with bendamustine-rituximab for follicular lymphoma. Br J Haematol. 2019;184:524–35.
Treon SP, et al. Extended rituximab therapy in Waldenström’s macroglobulinemia. Ann Oncol Off J Eur Soc Med Oncol. 2005;16:132–8.
Treon SP, et al. Maintenance Rituximab is associated with improved clinical outcome in rituximab naïve patients with Waldenstrom Macroglobulinaemia who respond to a rituximab-containing regimen. Br J Haematol. 2011;154:357–62.
Dimopoulos MA, et al. Extended rituximab therapy for previously untreated patients with Waldenström’s macroglobulinemia. Clin Lymphoma. 2002;3:163–6.
Hochster H, et al. Maintenance rituximab after cyclophosphamide, vincristine, and prednisone prolongs progression-free survival in advanced indolent lymphoma: Results of the randomized phase III ECOG1496 study. J Clin Oncol. 2009;27:1607–14.
Morschhauser F, et al. Rituximab plus lenalidomide in advanced untreated follicular lymphoma. N Engl J Med. 2018;379:934–47.
Nastoupil LJ, et al. Results of a phase II study of obinutuzumab in combination with lenalidomide in previously untreated, high tumor burden follicular lymphoma (FL). Blood. 2019;134:125–125.
Cheson BD, Morschhauser F, Martin P. Management of adverse events from the combination of rituximab and lenalidomide in the treatment of patients with follicular and low-grade non-Hodgkin lymphoma. Clin Lymphoma Myeloma Leuk. 2020;20:563–71.
Advani RH, et al. Mature results of a phase II study of rituximab therapy for nodular lymphocyte-predominant Hodgkin lymphoma. J Clin Oncol. 2014;32:912–8.
Conconi A, et al. Clinical activity of rituximab in extranodal marginal zone B-cell lymphoma of MALT type. Blood. 2003;102:2741–5.
Zucca E, et al. Final results of the IELSG-19 randomized trial of mucosa-associated lymphoid tissue lymphoma: Improved event-free and progression-free survival with rituximab plus chlorambucil versus either chlorambucil or rituximab monotherapy. J Clin Oncol. 2017;35:1905–12.
Leonard JP, et al. AUGMENT: a phase III study of lenalidomide plus rituximab versus placebo plus rituximab in relapsed or refractory indolent lymphoma. J Clin Oncol. 2019;37:1188–99.
Dimopoulos MA, et al. Phase 3 trial of ibrutinib plus rituximab in Waldenström’s macroglobulinemia. N Engl J Med. 2018;378:2399–410.
Fischer K, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380:2225–36.
Chihara D, et al. Long-term durable remission by cladribine followed by rituximab in patients with hairy cell leukaemia: update of a phase II trial. Br J Haematol. 2016;174:760–6.
Chihara D, et al. Randomized phase II study of first-line cladribine with concurrent or delayed rituximab in patients with hairy cell leukemia. J Clin Oncol. 2020;38:1527–8.
Else M, et al. Long-term follow-up of 233 patients with hairy cell leukaemia, treated initially with pentostatin or cladribine, at a median of 16 years from diagnosis. Br J Haematol. 2009;145:733–40.
Else M, et al. Long remissions in hairy cell leukemia with purine analogs: a report of 219 patients with a median follow-up of 12.5 years. Cancer. 2005;104:2442–8.
Goodman GR, Burian C, Koziol JA, Saven A. Extended follow-up of patients with hairy cell leukemia after treatment with cladribine. J Clin Oncol. 2003;21:591–6.
Chadha P, et al. Treatment of hairy cell leukemia with 2-chlorodeoxyadenosine (2-CdA): long-term follow-up of the Northwestern University experience. Blood. 2005;106:241–6.
Park JH, et al. First line chemo-free therapy with the BRAF inhibitor vemurafenib combined with obinutuzumab is effective in patients with Hcl. Blood. 2019;134:3998.
Vindi J, et al. A clinicogenetic risk model (m7-FLIPI) prospectively identifies one-half of patients with early disease progression of follicular lymphoma after first-line immunochemotherapy. Blood. 2015;126:333–333.
Eskelund CW, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017;130:1903–10.
Stilgenbauer S, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123:3247–54.
Chau K, et al. Outcomes of 210 patients with follicular and marginal zone lymphomas treated with 4 Gy of radiation therapy. Blood. 2019;134:2827.
Russo AL, et al. Low-dose involved-field radiation in the treatment of non-hodgkin lymphoma: predictors of response and treatment failure. Int J Radiat Oncol Biol Phys. 2013;86:121–7.
Hoskin PJ, et al. 4 Gy versus 24 Gy radiotherapy for patients with indolent lymphoma (FORT): a randomised phase 3 non-inferiority trial. Lancet Oncol. 2014;15:457–63.
Hoskin P, et al. Long term follow-up of FoRT: a phase 3 multi-center prospective randomized trial of radiation therapy for follicular and marginal zone lymphoma. Hematol Oncol. 2019;37:219–20.
Brepoels L, et al. Aggressive and indolent non-Hodgkin’s lymphoma: response assessment by Integrated International Workshop Criteria. Leuk Lymphoma. 2007;48:1522–30.
Albano D, et al. Prognostic role of baseline 18 F-FDG PET/CT parameters in MALT lymphoma. Hematol Oncol. 2019;37:39–46.
Batlevi CL, et al. Positron-emission tomography–based staging reduces the prognostic impact of early disease progression in patients with follicular lymphoma. Eur J Cancer. 2020b;126:78–90.
Trotman J, et al. Prognostic value of PET-CT after fi rst-line therapy in patients with follicular lymphoma: A pooled analysis of central scan review in three multicentre studies. Lancet Haematol. 2014;1:e17–27.
Trotman J, et al. Prognostic value of end-of-induction PET response after first-line immunochemotherapy for follicular lymphoma (GALLIUM): secondary analysis of a randomised, phase 3 trial. Lancet Oncol. 2018;19:1530–42.
Scherer F, Kurtz DM, Diehn M, Alizadeh AA. High-throughput sequencing for noninvasive disease detection in hematologic malignancies. Blood. 2017;130:440–52.
Pantel K, Alix-Panabières C. Liquid biopsy and minimal residual disease—latest advances and implications for cure. Nat Rev Clin Oncol. 2019;16:409–24.
Kurtz DM, et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood. 2015;125:3679–87.
Roschewski M, et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: a correlative biomarker study. Lancet Oncol. 2015;16:541–9.
Spina V, et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood. 2018;131:2413–25.
Sarkozy C, et al. The prognostic value of clonal heterogeneity and quantitative assessment of plasma circulating clonal IG-VDJ sequences at diagnosis in patients with follicular lymphoma. Oncotarget. 2017;8:8765–74.
Royo C, et al. Non-nodal type of mantle cell lymphoma is a specific biological and clinical subgroup of the disease. Leukemia. 2012;26:1895–8.
Angelopoulou MK, et al. The splenic form of mantle cell lymphoma. Eur J Haematol. 2002;68:12–21.
Pangalis GA, Angelopoulou MK, Vassilakopoulos TP, Siakantaris MP, Kittas C. B-chronic lymphocytic leukemia, small lymphocytic lymphoma, and lymphoplasmacytic lymphoma, including Waldenström’s macroglobulinemia: a clinical, morphologic, and biologic spectrum of similar disorders. Semin Hematol. 1999;36:104–14.
Armand M, Besson C, Hermine O, Davi F. Hepatitis C virus—associated marginal zone lymphoma. Best Pract Res Clin Haematol. 2017;30:41–9.
Fetica B, et al. High prevalence of viral hepatitis in a series of splenic marginal zone lymphomas from Romania. Blood Cancer J. 2016;6:e498.
Kalpadakis C, et al. Treatment of splenic marginal zone lymphoma with rituximab monotherapy: progress report and comparison with splenectomy. Oncologist. 2013;18:190–7.
Kalpadakis C, et al. Rituximab monotherapy in splenic marginal zone lymphoma: prolonged responses and potential benefit from maintenance. Blood. 2018;132:666–70.
Tsimberidou AM, et al. Outcomes in patients with splenic marginal zone lymphoma and marginal zone lymphoma treated with rituximab with or without chemotherapy or chemotherapy alone. Cancer. 2006;107:125–35.
Iannitto E, et al. Efficacy of bendamustine and rituximab in splenic marginal zone lymphoma: results from the phase II BRISMA/IELSG36 study. Br J Haematol. 2018;183:755–65.
Targarona EM, Cerdán G, Gracia E, Rodríguez M, Trias M. Results of laparoscopic splenectomy for treatment of malignant conditions. HPB (Oxford). 2001;3:251–5.
Targarona EM, et al. Splenomegaly should not be considered a contraindication for laparoscopic splenectomy. Ann Surg. 1998;228:35–9.
Fallah J, Olszewski AJ. Diagnostic and therapeutic splenectomy for splenic lymphomas: analysis of the National Cancer Data Base. Hematology. 2019;24:378–86.
Bisharat N, Omari H, Lavi I, Raz R. Risk of infection and death among post-splenectomy patients. J Infect. 2001;43:182–6.
Kyaw MH, et al. Evaluation of severe infection and survival after splenectomy. Am J Med. 2006;119:276.e1-276.e7.
Bagrodia N, et al. Morbidity and mortality following elective splenectomy for benign and malignant hematologic conditions analysis of the american college of surgeons national surgical quality improvement program data. JAMA Surg. 2014;149:1022–9.
Zaorsky NG, et al. Splenic irradiation for splenomegaly: a systematic review. Cancer Treat Rev. 2017;53:47–52.
Trip AK, et al. Radiation-induced dose-dependent changes of the spleen following postoperative chemoradiotherapy for gastric cancer. Radiother Oncol. 2015;116:239–44.
El Weshi A, et al. Low and medium dose spleen radiation therapy are able to induce long-term responses in splenic lymphoma with villous lymphocytes. Br J Haematol. 1998;103:1212–3.
Aisenberg AC. Coherent view of non-Hodgkin’s lymphoma. J Clin Oncol. 1995;13:2656–75.
Nakamura S, et al. Long-term clinical outcome of gastric MALT lymphoma after eradication of Helicobacter pylori: a multicentre cohort follow-up study of 420 patients in Japan. Gut. 2012;61:507–13.
Ohkubo Y, et al. Radiotherapy for localized gastric mucosa-associated lymphoid tissue lymphoma: long-term outcomes over 10 years. J Radiat Res. 2017;58:537.
Zullo A, et al. Effects of helicobacter pylori eradication on early stage gastric mucosa-associated lymphoid tissue lymphoma. Clin Gastroenterol Hepatol. 2010;8:105–10.
Liu H, et al. Resistance of t(11;18) positive gastric mucosa-associated lymphoid tissue lymphoma to Helicobacter pylori eradication therapy. Lancet. 2001;357:39–40.
Lévy M, et al. Rituximab and chlorambucil versus rituximab alone in gastric mucosa-associated lymphoid tissue lymphoma according to t(11;18) status: a monocentric non-randomized observational study. Leuk Lymphoma. 2013;54:940–4.
Ye H, et al. MALT lymphoma with t(14;18)(q32;q21)/IGH-MALT1 is characterized by strong cytoplasmic MALT1 and BCL10 expression. J Pathol. 2005;205:293–301.
Koch P, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the german multicenter study GIT NHL 01/92. J Clin Oncol. 2001;19:3861–73.
Papaxoinis G, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: a clinicopathologic study of 128 cases in Greece. A Hellenic Cooperative Oncology Group study (HeCOG). Leuk Lymphoma. 2006;47:2140–6.
Marks E, Shi Y. Duodenal-type follicular lymphoma a clinicopathologic review. Arch Pathol Lab Med. 2018;142:542–7.
Salem P, et al. Primary small intestinal lymphoma in adults. A comparative study of IPSID versus non-IPSID in the middle east. Cancer. 1987;59:1670–6.
Li G, Hansmann M-L, Zwingers T, Lennert K. Primary lymphomas of the lung: morphological, immunohistochemical and clinical features. Histopathology. 1990;16:519–31.
Chanudet E, et al. Chlamydiae and Mycoplasma infections in pulmonary MALT lymphoma. Br J Cancer. 2007;97:949–51.
Leyfman Y, et al. Expectant management of extranodal marginal zone lymphoma of bronchial-associated lymphoid tissue (BALT). Blood. 2019;134:2826–2826.
Hollender A, et al. Central nervous system involvement following diagnosis of non-Hodgkin’s lymphoma: a risk model. Ann Oncol Off J Eur Soc Med Oncol. 2002;13:1099–107.
Simon L, et al. Bing-Neel syndrome, a rare complication of waldenström macroglobulinemia: analysis of 44 cases and review of the literature. A study on behalf of the French Innovative Leukemia Organization (FILO). Haematologica. 2015;100:1587–94.
Castillo JJ, et al. Central nervous system involvement by Waldenström macroglobulinaemia (Bing-Neel syndrome): a multi-institutional retrospective study. Br J Haematol. 2016;172:709–15.
Castillo JJ, Treon SP. How we manage Bing-Neel syndrome. Br J Haematol. 2019;187:277–85.
Cabannes-Hamy A, et al. Efficacy of ibrutinib in the treatment of Bing-Neel syndrome. Am J Hematol. 2016;91:E17–9.
Bernard S, et al. Activity of ibrutinib in mantle cell lymphoma patients with central nervous system relapse. Blood. 2015;126:1695–8.
de la Fuente MI, et al. Marginal zone dural lymphoma: the Memorial Sloan Kettering Cancer Center and University of Miami experiences. Leuk Lymphoma. 2017;58:882–8.
Edinger JT, Kant JA, Swerdlow SH. Cutaneous marginal zone lymphomas have distinctive features and include 2 subsets. Am J Surg Pathol. 2010;34:1830–41.
Skala SL, Hristov B, Hristov AC. Primary cutaneous follicle center lymphoma. Arch Pathol Lab Med. 2018;142:1313–21.
Travaglino A, et al. Borrelia burgdorferi in primary cutaneous lymphomas: a systematic review and meta-analysis. JDDG J German Soc Dermatol. 2020. https://doi.org/10.1111/ddg.14289.
Goodlad JR, et al. Primary cutaneous B-cell lymphoma and Borrelia burgdorferi infection in patients from the highlands of Scotland. Am J Surg Pathol. 2000;24:1279–85.
Suárez AL, et al. Primary cutaneous B-cell lymphomas: Part II. Therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11.
Holm F, et al. Ocular adnexal lymphoma in Denmark: a nationwide study of 387 cases from 1980 to 2017. Br J Ophthalmol. 2020;85:220.
Rehn S, et al. Radiotherapy dose and volume de-escalation in ocular adnexal lymphoma. Anticancer Res. 2020;40:4041–6.
Ferreri AJM, et al. Evidence for an association between Chlamydia psittaci and ocular adnexal lymphomas. J Natl Cancer Inst. 2004;96:586–94.
Ferreri AJM, et al. Regression of ocular adnexal lymphoma after Chlamydia psittaci-eradicating antibiotic therapy. J Clin Oncol. 2005;23:5067–73.
Casulo C, et al. Early relapse of follicular lymphoma after rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone defines patients at high risk for death: an analysis from the National LymphoCare Study. J Clin Oncol. 2015;33:2516–22.
Seymour JF, et al. Association of early disease progression and very poor survival in the GALLIUM study in follicular lymphoma: benefit of obinutuzumab in reducing the rate of early progression. Haematologica. 2019;104:1202–8.
Maurer MJ, et al. Early event status informs subsequent outcome in newly diagnosed follicular lymphoma. Am J Hematol. 2016;91:1096–101.
Freeman CL, et al. Early progression after bendamustine-rituximab is associated with high risk of transformation in advanced stage follicular lymphoma. Blood. 2019;134:761–4.
Luminari S, et al. Early progression as a predictor of survival in marginal zone lymphomas: an analysis from the FIL-NF10 study. Blood. 2019;134:798–801.
Ghielmini M, et al. Prolonged treatment with rituximab in patients with follicular lymphoma significantly increases event-free survival and response duration compared with the standard weekly × 4 schedule. Blood. 2004;103:4416–23.
Sehn LH, et al. Randomized phase II trial comparing obinutuzumab (GA101) with rituximab in patients with relapsed CD20+ indolent B-Cell non-hodgkin lymphoma: final analysis of the GAUSS study. J Clin Oncol. 2015;33:3467–74.
Rummel M, et al. Bendamustine plus rituximab versus fludarabine plus rituximab for patients with relapsed indolent and mantle-cell lymphomas: a multicentre, randomised, open-label, non-inferiority phase 3 trial. Lancet Oncol. 2016;17:57–66.
von Keudell G, Moskowitz AJ. The role of PI3K inhibition in lymphoid malignancies. Curr Hematol Malig Rep. 2019;14:405–13.
Flinn IW, et al. DYNAMO: a Phase II study of duvelisib (IPI-145) in patients with refractory indolent non-hodgkin lymphoma. J Clin Oncol. 2019a;37:912–22.
Dreyling M, et al. Phosphatidylinositol 3-kinase inhibition by Copanlisib in relapsed or refractory indolent lymphoma. J Clin Oncol. 2017;35:3898–905.
Flinn IW, et al. Idelalisib, a selective inhibitor of phosphatidylinositol 3-kinase-δ, as therapy for previously treated indolent non-Hodgkin lymphoma. Blood. 2014b;123:3406–13.
Lampson BL, et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood. 2016;128:195–203.
Coutre S, et al. Safety of idelalisib in B-cell malignancies: Integrated analysis of eight clinical trials. J Clin Oncol. 2015;33:e18030–e18030.
O’Brien SM, et al. A phase 2 study of idelalisib plus rituximab in treatment-naïve older patients with chronic lymphocytic leukemia. Blood. 2015;126:2686–94.
Burris HA, et al. Umbralisib, a novel PI3Kδ and casein kinase-1ε inhibitor, in relapsed or refractory chronic lymphocytic leukaemia and lymphoma: an open-label, phase 1, dose-escalation, first-in-human study. Lancet Oncol. 2018;19:486–96.
Pagel J, et al. (2020) The PI3KΔ inhibitor ME-401 is well-tolerated on intermittent… EHA library. Pagel J. 2020;2020:293663.
Forero-Torres A, et al. Parsaclisib, a potent and highly selective PI3Kd inhibitor, in patients with relapsed or refractory B-cell malignancies. Blood. 2019;133:1742–52.
Noy A, et al. Targeting Bruton tyrosine kinase with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood. 2017;129:2224–32.
Gachard N, et al. IGHV gene features and MYD88 L265P mutation separate the three marginal zone lymphoma entities and Waldenström macroglobulinemia/lymphoplasmacytic lymphomas. Leukemia. 2013;27:183–99.
Noy A, et al. Durable ibrutinib responses in relapsed/refractory marginal zone lymphoma: long-term follow-up and biomarker analysis. Blood Adv. 2020;4:5773–84.
Wu C, et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: impact of recurrent CARD11 mutations. Oncotarget. 2016;7:38180–90.
Bartlett NL, et al. Single-agent ibrutinib in relapsed or refractory follicular lymphoma: a phase 2 consortium trial. Blood. 2018;131:182–90.
Davids MS, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-hodgkin lymphoma. J Clin Oncol. 2017;35:826–33.
Castillo J, et al. Multicenter prospective phase II study of venetoclax in patients with previously treated Waldenstrom macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2019;19:e39–40.
Seymour JF, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107–20.
Green MR. Chromatin modifying gene mutations in follicular lymphoma. Blood. 2018;131:595–604.
Morschhauser F, et al. Phase 2 multicenter study of tazemetostat, an EZH2 inhibitor, in patients with relapsed or refractory follicular lymphoma. Blood. 2019a;134:123–123.
Dornan D, et al. Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood. 2009;114:2721–9.
Sehn LH, et al. Randomized phase 2 trial of polatuzumab vedotin (pola) with bendamustine and rituximab (BR) in relapsed/refractory (r/r) FL and DLBCL. J Clin Oncol. 2018;36:7507–7507.
Morschhauser F, et al. Polatuzumab vedotin or pinatuzumab vedotin plus rituximab in patients with relapsed or refractory non-Hodgkin lymphoma: final results from a phase 2 randomised study (ROMULUS). Lancet Haematol. 2019b;6:e254–65.
Schuster SJ, et al. Mosunetuzumab induces complete remissions in poor prognosis non-hodgkin lymphoma patients, including those who are resistant to or relapsing after chimeric antigen receptor T-cell (CAR-T) therapies, and is active in treatment through multiple lines. Blood. 2019;134:6.
Advani R, et al. The first-in-class anti-CD47 antibody HU5F9-G4 + rituximab induces durable responses in relapsed/refractory DLBCL and indolent lymphoma: interim phase 1B/2 results. Hematol Oncol. 2019;37:89–90.
Emmanouilides C, et al. Safety and efficacy of yttrium-90 ibritumomab tiuxetan in older patients with non-Hodgkin’s lymphoma. Cancer Biother Radiopharm. 2007;22:684–91.
McDonald GB, et al. Survival, nonrelapse mortality, and relapse-related mortality after allogeneic hematopoietic cell transplantation: comparing 2003–2007 versus 2013–2017 cohorts. Ann Intern Med. 2020;172:229–39.
Jurinovic V, et al. Autologous stem cell transplantation for patients with early progression of follicular lymphoma: a follow-up study of 2 randomized trials from the german low grade lymphoma study group. Biol Blood Marrow Transplant. 2018;24:1172–9.
Smith SM, et al. Autologous transplantation versus allogeneic transplantation in patients with follicular lymphoma experiencing early treatment failure. Cancer. 2018;124:2541–51.
Casulo C, et al. Autologous transplantation in follicular lymphoma with early therapy failure: a national lymphocare study and center for international blood and marrow transplant research analysis. Biol Blood Marrow Transplant. 2018;24:1163–71.
Ladetto M, et al. Prospective, multicenter randomized GITMO/IIL trial comparing intensive (R-HDS) versus conventional (CHOP-R) chemoimmunotherapy in high-risk follicular lymphoma at diagnosis: the superior disease control of R-HDS does not translate into an overall surviva. Blood. 2008;111:4004–18.
Gyan E, et al. High-dose therapy followed by autologous purged stem cell transplantation and doxorubicin-based chemotherapy in patients with advanced follicular lymphoma: a randomized multicenter study by the GOELAMS with final results after a median follow-up of 9 year. Blood. 2009;113:995–1001.
Dahi PB, et al. Favorable outcomes in elderly patients undergoing high-dose therapy and autologous stem cell transplantation for non-Hodgkin lymphoma. Biol Blood Marrow Transplant. 2014;20:2004–9.
Majhail NS, et al. Indications for autologous and allogeneic hematopoietic cell transplantation: guidelines from the American society for blood and marrow transplantation. Biol Blood Marrow Transplant. 2015;21:1–7.
Hamadani M, Horowitz MM. Allogeneic transplantation for follicular lymphoma: does one size fit all? J Oncol Pract. 2017;13:798–806.
Sureda A, et al. Allogeneic hematopoietic stem cell transplantation for relapsed follicular lymphoma: a combined analysis on behalf of the Lymphoma Working Party of the EBMT and the Lymphoma Committee of the CIBMTR. Cancer. 2018;124:1733–42.
Shah NN, et al. Outcomes of Medicare-age eligible NHL patients receiving RIC allogeneic transplantation: a CIBMTR analysis. Blood Adv. 2018;2:933–40.
Kanate AS, et al. Reduced-intensity transplantation for lymphomas using haploidentical related donors vs HLA-matched unrelated donors. Blood. 2016;127:938–47.
Ghosh N, et al. Reduced-intensity transplantation for lymphomas using haploidentical related donors versus HLA-matched sibling donors: a center for international blood and marrow transplant research analysis. J Clin Oncol. 2016;34:3141–9.
Sauter CS, et al. A Phase II study of a nonmyeloablative allogeneic stem cell transplant with peritransplant rituximab in patients with BCell lymphoid malignancies: favorably durable event-free survival in chemosensitive patients. Biol Blood Marrow Transplant. 2014;20:354–60.
Neelapu SS, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–44.
Schuster SJ, et al. Chimeric antigen receptor T Cells in refractory B-Cell lymphomas. N Engl J Med. 2017;377:2545–54.
Hirayama AV, et al. High rate of durable complete remission in follicular lymphoma after CD19 CAR-T cell immunotherapy. Blood. 2019;134:636–40.
Jacobson CA, et al. Interim analysis of ZUMA-5: a phase II study of axicabtagene ciloleucel (axi-cel) in patients (pts) with relapsed/refractory indolent non-Hodgkin lymphoma (R/R iNHL). J Clin Oncol. 2020;38:8008.
Lee DW, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25:625–38.
Brudno JN, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for lymphoma. Nat Rev Clin Oncol. 2017;15:31–46.
Herfarth K, et al. Rituximab with involved field irradiation for early-stage nodal follicular lymphoma. HemaSphere. 2018;1:e160.
Flinn IW, et al. First-line treatment of patients with indolent non-hodgkin lymphoma or mantle-cell lymphoma with bendamustine plus rituximab versus R-CHOP or R-CVP: results of the BRIGHT 5-year follow-up study. J Clin Oncol. 2019b;37:984–91.
Sehn LH, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17:1081–93.
Acknowledgements
None.
Funding
This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Author information
Authors and Affiliations
Contributions
All authors contributed to the literature review and writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lumish, M., Falchi, L., Imber, B.S. et al. How we treat mature B-cell neoplasms (indolent B-cell lymphomas). J Hematol Oncol 14, 5 (2021). https://doi.org/10.1186/s13045-020-01018-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-01018-6