
Abstract
Background
Hyperfibrinolysis is a critical complication in severe trauma. Hyperfibrinolysis is traditionally diagnosed via elevated D-dimer or fibrin/fibrinogen degradation product levels, and recently, using thromboelastometry. Although hyperfibrinolysis is observed in patients with severe isolated traumatic brain injury (TBI) on arrival at the emergency department (ED), it is unclear which factors induce hyperfibrinolysis. The present study aimed to investigate the factors associated with hyperfibrinolysis in patients with isolated severe TBI.
Methods
We conducted a multicentre retrospective review of data for adult trauma patients with an injury severity score ≥ 16, and selected patients with isolated TBI (TBI group) and extra-cranial trauma (non-TBI group). The TBI group included patients with an abbreviated injury score (AIS) for the head ≥ 4 and an extra-cranial AIS < 2. The non-TBI group included patients with an extra-cranial AIS ≥ 3 and head AIS < 2. Hyperfibrinolysis was defined as a D-dimer level ≥ 38 mg/L on arrival at the ED. We evaluated the relationships between hyperfibrinolysis and injury severity/tissue injury/tissue perfusion in TBI patients by comparing them with non-TBI patients.
Results
We enrolled 111 patients in the TBI group and 126 in the non-TBI group. In both groups, patients with hyperfibrinolysis had more severe injuries and received transfusion more frequently than patients without hyperfibrinolysis. Tissue injury, evaluated on the basis of lactate dehydrogenase and creatine kinase levels, was associated with hyperfibrinolysis in both groups. Among patients with TBI, the mortality rate was higher in those with hyperfibrinolysis than in those without hyperfibrinolysis. Tissue hypoperfusion, evaluated on the basis of lactate level, was associated with hyperfibrinolysis in only the non-TBI group. Although the increase in lactate level was correlated with the deterioration of coagulofibrinolytic variables (prolonged prothrombin time and activated partial thromboplastin time, decreased fibrinogen levels, and increased D-dimer levels) in the non-TBI group, no such correlation was observed in the TBI group.
Conclusions
Hyperfibrinolysis is associated with tissue injury and trauma severity in TBI and non-TBI patients. However, tissue hypoperfusion is associated with hyperfibrinolysis in non-TBI patients, but not in TBI patients. Tissue hypoperfusion may not be a prerequisite for the occurrence of hyperfibrinolysis in patients with isolated TBI.
Similar content being viewed by others
Background
The acute phase of severe trauma is a frequent setting for trauma-induced coagulopathy, which may develop into severe haemorrhage due to coagulation abnormalities and contribute to a poor outcome [1,2,3,4,5,6,7,8,9,10,11]. Hyperfibrinolysis is one of the distinctive features of trauma-induced coagulopathy [1,2,3,4,5,6,7,8,9,10,11]. Hyperfibrinolysis has been traditionally recognized by the presence of elevated D-dimer or fibrin/fibrinogen degradation product levels [2,3,4,5,6]. In addition, recent studies have reported that hyperfibrinolysis is detected as clot lysis on thromboelastometry [7,8,9,10,11]. In thromboelastometry, clot lysis is observed when the α2-antiplasmin cannot inhibit the action of free plasmin activated by tissue-plasminogen activator (t-PA) in blood samples [9]. On the other hand, elevation of the D-dimer level results from degradation of fibrin by plasmin, regardless of the marked increase in free t-PA level and consumption of α2-antiplasmin [12]. Therefore, elevated D-dimer levels may be a sensitive indicator of hyperfibrinolysis and a predictor of poor outcome in patients with severe trauma [3, 5, 9].
In patients with isolated severe traumatic brain injury (TBI), hyperfibrinolysis is often observed on arrival at the emergency department (ED), and is associated with intracranial haematoma enlargement [5, 6, 13,14,15,16]. Furthermore, hyperfibrinolysis contributes to the development of coagulopathy and a poor outcome in isolated severe TBI [5, 14,15,16]. Maegele proposed that hyperfibrinolysis was induced by tissue hypoperfusion in TBI [16]. Furthermore, Cohen at al. [17] reported that TBI alone does not cause hyperfibrinolysis and coagulopathy, but must be coupled with tissue hypoperfusion. Conversely, Lustenberger et al. [18] indicated that tissue hypoperfusion was not essential for the development of coagulation abnormalities in patients with severe TBI. Therefore, the relationship between tissue hypoperfusion and hyperfibrinolysis remains unclear in patients with isolated TBI.
As is the case with hypoperfusion, tissue injury is speculated to independently cause hyperfibrinolysis in severe trauma [1,2,3, 13, 14, 19, 20]. Immediately after trauma, microparticles exposing tissue factor [13, 19, 21,22,23] and damage-associated molecular patterns (DAMPs), such as histones [24, 25] and mitochondrial DNA [26], are released from damaged tissue into the systemic circulation and activate the coagulation system. The activation of the coagulation system triggers the activation of the fibrinolytic system, thus leading to the generation of plasmin [27]. Massive tissue injury induces excessive release of tissue factor and DAMPs and unregulated activation of the coagulation system [1, 3, 13, 14, 24,25,26], which induces increased plasmin generation and the consumption of alpha 2-plasmin inhibitor [13, 14, 20, 28]. Low levels of alpha 2-plasmin inhibitor may lead to accelerated fibrinolysis and result in hyperfibrinolysis [13, 14, 20, 28]. However, the pathophysiological role of tissue injury and hypoperfusion in the development of hyperfibrinolysis remains unclear in patients with and without TBI.
The present study aimed to investigate the factors associated with hyperfibrinolysis in patients with isolated severe TBI. We evaluated the relationships between hyperfibrinolysis and injury severity/tissue injury/tissue perfusion in patients with TBI by comparing them with non-TBI patients.
Methods
Patient selection and data collection
This retrospective study investigated coagulation disorders in patients with severe trauma at 15 tertiary emergency and critical care centres in Japan (Japanese Observational Study for Coagulation and Thrombolysis in Early Trauma, J-OCTET). The J-OCTET recruited consecutive patients with severe trauma with an injury severity score (ISS) ≥ 16, who were admitted to EDs from January 2012 through December 2012 for the purpose of investigating coagulation abnormalities, transfusion, and mortality [3, 29, 30]. Patients were excluded if they were younger than 18 years; pregnant; or exhibited cardiac arrest, burns, isolated cervical spine injury not caused by a high-energy accident, or liver cirrhosis. The laboratory test results, which were promptly obtained on arrival at the ED, clinical background, treatment, and outcome of the patients were retrospectively analysed.
In the present study, we selected patients with isolated TBI and patients with severe extra-cranial trauma without TBI from the J-OCTET database. Patients were excluded if: (1) they had used anti-coagulant or anti-platelet drugs before the accident; (2) they had received infusions before arrival in the ED; (3) they had penetrating trauma; or (4) the D-dimer level had not been measured on arrival at the ED.
Definitions
We defined patients with isolated TBI (the TBI group) as patients with an abbreviated injury score (AIS) for the head ≥ 4 and an extra-cranial AIS < 2. Patients with isolated TBI and with a head AIS of 3 and extra-cranial AIS < 2 were not included, because the J-OCTET recruited only patients with severe trauma, with an ISS ≥ 16. Patients with severe extra-cranial trauma without TBI (the non-TBI group) were defined as patients with an extra-cranial AIS ≥ 3 and head AIS < 2. Hyperfibrinolysis was defined as a D-dimer level ≥ 38 mg/L on arrival at the ED, on the basis of our previous study, which indicated that a high D-dimer level was a strong predictor of poor outcome [3]. Lactate was used as a surrogate marker of tissue hypoperfusion, similar to previous studies [2, 7, 11]. Lactate dehydrogenase (LDH) and creatine kinase (CK) were used as surrogate markers of tissue injury, because they are expressed by brain tissue and muscle as well as various other tissues [31, 32]. The patient’s outcome was evaluated according to mortality rates at 24 hours, 48 hours, and 28 days, and at the time of hospital discharge (in-hospital mortality).
Statistical analysis
All variables are expressed as the median and interquartile range (i.e., the first to third quartiles) or number (percentage). Intergroup comparisons were made using the Mann–Whitney U test or χ2 test. Multiple logistic regression analysis was used to assess relationships between hyperfibrinolysis and patient characteristics. Patient age and sex were used as covariates in the multiple logistic regression analysis. Correlations between two measurements were investigated via Pearson’s correlation analysis after logarithmic transformation. SPSS 22.0 J (SPSS Inc., Chicago, IL, USA) was used for all statistical analyses. The level of significance was set at P < 0.05; we did not conduct or adjust for multiple tests because of the retrospective, exploratory nature of this study.
Results
Patient characteristics
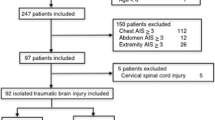
The TBI group included 111 patients and the non-TBI group included 126 patients (Fig. 1). The patient characteristics in the TBI and non-TBI groups are shown in Table 1. The patients in the TBI group were older than those in the non-TBI group. The revised trauma scores in the TBI group were worse than those in the non-TBI group. Although many variables differed between the two groups, the D-dimer levels on admission to the ED were not different.

Flow chart of the selection of patients in the traumatic brain injury (TBI) and non-TBI groups. The Japanese Observational Study for Coagulation and Thrombolysis in Early Trauma (J-OCTET) recruited 796 patients with severe trauma with an injury severity score ≥ 16, who were admitted to the emergency department (ED). Patients with isolated severe TBI were defined as patients with an abbreviated injury score (AIS) for the head ≥ 4 and an extra-cranial AIS < 2. Patients with severe extra-cranial trauma without TBI (non-TBI patients) were defined by an extra-cranial AIS ≥ 3 and head AIS < 2. Patients were excluded if they had used anti-coagulant or anti-platelet drugs before the accident or received an infusion before arrival at the ED. Patients with penetrating trauma and those whose D-dimer levels were not measured on arrival at the ED were also excluded
Table 2 presents the characteristics of patients with and without hyperfibrinolysis in each group. On arrival at the ED, 34 (31%) and 46 (37%) patients in the TBI and non-TBI groups, respectively, were diagnosed as having hyperfibrinolysis. The frequency of hyperfibrinolysis was not significantly different between the groups (P = 0.340). In both groups, the injury severity scores (ISS and revised trauma score) of the patients with hyperfibrinolysis were higher than those of patients without hyperfibrinolysis. Although the systolic blood pressure measurements in patients with TBI and hyperfibrinolysis were higher than those in patients with TBI and no hyperfibrinolysis, the systolic blood pressure measurements in non-TBI patients with hyperfibrinolysis were lower than those in non-TBI patients without hyperfibrinolysis. The levels of LDH and CK related to tissue injury in patients with hyperfibrinolysis were greater than those in patients without hyperfibrinolysis in both groups. The lactate levels did not differ between patients with and without hyperfibrinolysis in both groups.
Haemostatic treatments and amounts of transfusion
The haemostatic treatments and amounts of transfusion in patients with and without hyperfibrinolysis in the two groups are presented in Table 3. Patients with hyperfibrinolysis were transfused larger amounts of red blood cell concentrate and fresh frozen plasma than those without hyperfibrinolysis, in both groups. Moreover, massive transfusions (≥10 units) were frequently performed in hyperfibrinolytic patients in both groups.
Mortality rates
Mortality rates in patients with and without hyperfibrinolysis in the two groups are presented in Table 4. Mortality rates at 24 hours, 48 hours, and 28 days and in-hospital mortality rates were higher in hyperfibrinolytic patients than in those without hyperfibrinolysis in the TBI group only.
Factors associated with hyperfibrinolysis
Figure 2 shows the odds ratios for hyperfibrinolysis adjusted for age and sex. Associations between hyperfibrinolysis and injury severity (ISS and revised trauma score) were observed in both the TBI and non-TBI groups. Tissue injury evaluated on the basis of the LDH and CK levels was also associated with hyperfibrinolysis in both groups.

Adjusted odds ratios for hyperfibrinolysis. The odds ratios were adjusted for age and sex. Odds ratios (black squares) and 95% confidence intervals (bars). TBI traumatic brain injury, ISS injury severity score, RTS revised trauma score, BP blood pressure, LDH lactate dehydrogenase, CK creatine kinase
On analysing the relationship between hyperfibrinolysis and tissue perfusion, the systolic blood pressure measurements were associated with hyperfibrinolysis in both groups. However, hyperfibrinolysis was associated with elevated blood pressure in the TBI group, and decreased blood pressure in the non-TBI group. The lactate level, reflecting tissue hypoperfusion, was associated with hyperfibrinolysis in the non-TBI group only.
Associations between coagulofibrinolytic variables and lactate level
The results of Pearson’s correlation analysis between the coagulofibrinolytic variables and lactate levels in the two groups are presented in Table 5. In the non-TBI group, an increase in the lactate level was correlated with deterioration in each of the coagulofibrinolytic variables (prolongation of prothrombin time (PT) and activated partial thromboplastin time (APTT), decrease in fibrinogen levels, and elevation of D-dimer levels) on arrival at the ED. However, no correlation was observed between the lactate level and any of the coagulofibrinolytic variables in the TBI group.
Discussion
The present study demonstrated that tissue hypoperfusion evaluated on the basis of the lactate level was associated with hyperfibrinolysis and deterioration of coagulofibrinolytic variables (prolongation of prothrombin time (PT) and activated partial thromboplastin time (APTT), decrease in fibrinogen levels, and elevation of D-dimer levels) on arrival at the ED in the non-TBI group, but not in the TBI group. However, hyperfibrinolysis on arrival at the ED was related to injury severity and degree of tissue injury in both the TBI and non-TBI groups.
Previous studies indicate that tissue hypoperfusion may contribute to the development of hyperfibrinolysis, evaluated via thromboelastometry in areas outside the isolated TBI [7,8,9,10,11]. In the present study, hyperfibrinolysis was associated with hypoperfusion and decreased blood pressure in the non-TBI group (Fig. 2). Furthermore, increased lactate levels were correlated with deterioration of the coagulofibrinolytic variables in the non-TBI group. In the fibrinolytic system, one of the key enzymes is t-PA, which catalyses the conversion of plasminogen to plasmin [33]. Tissue hypoperfusion stimulates the endothelial cells and induces the release of t-PA from the endothelial cells into the systemic circulation [34, 35]. Furthermore, this acute and massive t-PA release induces hyperfibrinolysis [2, 7, 8, 10, 36,37,38,39]. Previous studies [17, 18] that investigated the relationships between hyperfibrinolysis and hypoperfusion in isolated TBI involved more than twice the number of patients with a base deficit (BD) > 6 mEq/L than that included in the present study. Although the present study was limited to patients with isolated blunt TBI, previous studies also included patients with penetrating brain injury [17, 18]. Moreover, injuries to organs other than the brain were more severe (AIS < 3) than those reported in our study (AIS < 2) [17, 18]. Therefore, the effects of injuries to organs other than the brain and/or penetrating brain injuries should have demonstrated a more pronounced effect on the elevation of BD in the previous studies, when compared with our findings.
We demonstrated that tissue injury, evaluated on the basis of the LDH and CK levels, was associated with hyperfibrinolysis in both the TBI and the non-TBI groups (Fig. 2) in the present study. LDH and CK have been reported to be associated with severity of multiple trauma [40], liver injury [41], TBI [42], and crash syndrome [43]; thus, LDH and CK appropriately represent the degree of trauma-induced cell destruction in various tissues. The destroyed cells release microparticles expressing tissue factor [13, 19, 21,22,23] and DAMPs [24,25,26], similar to LDH and CK, into the systemic circulation and activate the coagulation system during the early phase of trauma. The activation of the coagulation system results in fibrin formation and triggers the activation of the fibrinolytic system, thus leading to the generation of plasmin [27]. Consequently, the generated plasmin induces physiological fibrinolysis [27]. However, excessive release of microparticles and DAMPs results in unregulated activation of the coagulation system and formation of massive amounts of fibrin, eventually leading to consumption coagulopathy [13, 19, 21,22,23,24,25,26]. The massive fibrin formation simultaneously induces increased plasmin generation and the consumption of α2-plasmin inhibitor [13, 20, 44]. Low levels of the α2-plasmin inhibitor accelerate fibrinolysis and result in hyperfibrinolysis [13, 20, 44].
In the present study, the aetiological factors in hyperfibrinolysis differed between the TBI and non-TBI groups. Both severe tissue injury and hypoperfusion induce hyperfibrinolysis [2, 7, 8, 10, 13, 20, 36,37,38,39, 44]. In patients with injuries to the torso (non-TBI group), both severe tissue injury and hypoperfusion are frequent complications. Therefore, hyperfibrinolysis was found to be associated with both severe tissue injury and hypoperfusion in the non-TBI group. On the other hand, in patients with isolated TBI (TBI group), although tissue injury is a common complication, hypoperfusion rarely occurs. Therefore, in the TBI group, hyperfibrinolysis was determined to be associated with tissue injury only, and not tissue hypoperfusion.
Although hyperfibrinolysis in trauma patients is currently evaluated using thromboelastometry [7,8,9,10,11], we used D-dimer as a surrogate marker of hyperfibrinolysis in the present study. Thromboelastometry-indicated hyperfibrinolysis is observed when α2-antiplasmin cannot inhibit the action of free plasmin, which is newly activated by the t-PA in the blood sample subjected to thromboelastometry [9]. Raza et al. indicated that hyperfibrinolysis was not detected in more than 90% of trauma patients with marked generation of plasmin using thromboelastometry [9]. Therefore, thromboelastometry can detect hyperfibrinolysis only under the conditions of high free t-PA levels and significantly reduced α2-antiplasmin levels [9]. However, elevated D-dimer level is a more sensitive detection tool for hyperfibrinolysis than thromboelastometry, because elevation of the D-dimer level results from degradation of fibrin by plasmin, regardless of the marked increase in free t-PA level and consumption of α2-antiplasmin [12]. However, the appropriate cut-off level for D-dimer for diagnosing hyperfibrinolysis is unclear. We adopted ≥ 38 mg/L as the cut-off level for hyperfibrinolysis on the basis of the findings of our previous study, which indicated poor outcomes [3].
Previous studies have investigated the relationship between hyperfibrinolysis and tissue hypoperfusion in severe trauma [2, 7, 9,10,11, 17, 18]. In these studies, tissue hypoperfusion was assessed on the basis of base deficit [9,10,11, 17, 18] or lactate level [2, 7, 11]. However, the base deficit value is affected by the balance of sodium, chloride, albumin, and lactate levels [45]. Therefore, in the present study, we used only the lactate level to screen for tissue hypoperfusion because lactate reflects tissue perfusion more directly than does the base deficit [45, 46].
This retrospective multicentre study has several limitations. First, the number of patients with hyperfibrinolysis in the TBI group was not large. Statistical limitations may be involved. Second, the anatomical characteristics of patient injury (e.g., epidural haematoma, subdural haematoma, and cerebral contusion) in the TBI group were unclear. Therefore, the anatomical characteristics of brain injury have not been appropriately reflected in the present analysis. Third, although lactate level was used as a surrogate marker of tissue hypoperfusion in the present study, an increase in the lactate level was not caused only by tissue hypoperfusion, but also by epinephrine-induced β2-adrenoceptor stimulation [46]. In patients with severe trauma, serum epinephrine concentration frequently and significantly increases on arrival at the ED [47]. Therefore, lactate levels in patients with trauma may be affected by the epinephrine released from the adrenal gland due to trauma [46].
Conclusion
Hyperfibrinolysis is associated with tissue injury in both patients with TBI and in non-TBI patients. However, tissue hypoperfusion is associated with hyperfibrinolysis in non-TBI patients, but not in patients with TBI. Tissue hypoperfusion may therefore not be essential for the development of hyperfibrinolysis in patients with isolated TBI.
Abbreviations
- AIS:
-
Abbreviated injury score
- BD:
-
Base deficit
- CK:
-
Creatine kinase
- DAMPs:
-
Damage-associated molecular patterns
- ED:
-
Emergency department
- ISS:
-
Injury severity score
- J-OCTET:
-
Japanese Observational Study for Coagulation and Thrombolysis in Early Trauma
- LDH:
-
Lactate dehydrogenase
- TBI:
-
Traumatic brain injury
- t-PA:
-
Tissue-plasminogen activator
References
Gando S, Wada H, Thachil J, Scientific Standardization Committee on DIC of the International Society on Thrombosis and Haemostasis. Differentiating disseminated intravascular coagulation (DIC) with the fibrinolytic phenotype from coagulopathy of trauma and acute coagulopathy of trauma-shock (COT/ACOTS). J Thromb Haemost. 2013;11:826–35.
Sawamura A, Hayakawa M, Gando S, Kubota N, Sugano M, Wada T, et al. Disseminated intravascular coagulation with a fibrinolytic phenotype at an early phase of trauma predicts mortality. Thromb Res. 2009;124:608–13.
Hayakawa M, Maekawa K, Kushimoto S, Kato H, Sasaki J, Ogura H, et al. High D-dimer levels predict a poor outcome in patients with severe trauma, even with high fibrinogen levels on arrival: a multicenter retrospective study. Shock. 2016;45:308–14.
Vecht CJ, Sibinga CT, Minderhoud JM. Disseminated intravascular coagulation and head injury. J Neurol Neurosurg Psychiatry. 1975;38:567–71.
Kaufman HH, Moake JL, Olson JD, Miner ME, DuCret RP, Pruessner JL, et al. Delayed and recurrent intracranial hematomas related to disseminated intravascular clotting and fibrinolysis in head injury. Neurosurgery. 1980;7:445–9.
Kaufman HH, Hui KS, Mattson JC, Borit A, Childs TL, Hoots WK, et al. Clinicopathological correlations of disseminated intravascular coagulation in patients with head injury. Neurosurgery. 1984;15:34–42.
Kashuk JL, Moore EE, Sawyer M, Wohlauer M, Pezold M, Barnett C, et al. Primary fibrinolysis is integral in the pathogenesis of the acute coagulopathy of trauma. Ann Surg. 2010;252:434–42. discussion 43-4.
Schochl H, Frietsch T, Pavelka M, Jambor C. Hyperfibrinolysis after major trauma: differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J Trauma. 2009;67:125–31.
Raza I, Davenport R, Rourke C, Platton S, Manson J, Spoors C, et al. The incidence and magnitude of fibrinolytic activation in trauma patients. J Thromb Haemost. 2013;11:307–14.
Kutcher ME, Cripps MW, McCreery RC, Crane IM, Greenberg MD, Cachola LM, et al. Criteria for empiric treatment of hyperfibrinolysis after trauma. J Trauma Acute Care Surg. 2012;73:87–93.
Ostrowski SR, Sorensen AM, Larsen CF, Johansson PI. Thrombelastography and biomarker profiles in acute coagulopathy of trauma: a prospective study. Scand J Trauma Resusc Emerg Med. 2011;19:64.
Wada H, Sakuragawa N. Are fibrin-related markers useful for the diagnosis of thrombosis? Semin Thromb Hemost. 2008;34:33–8.
Gando S, Nanzaki S, Kemmotsu O. Coagulofibrinolytic changes after isolated head injury are not different from those in trauma patients without head injury. J Trauma. 1999;46:1070–6. discussion 6-7.
Kushimoto S, Shibata Y, Yamamoto Y. Implications of fibrinogenolysis in patients with closed head injury. J Neurotrauma. 2003;20:357–63.
Suehiro E, Koizumi H, Fujiyama Y, Yoneda H, Suzuki M. Predictors of deterioration indicating a requirement for surgery in mild to moderate traumatic brain injury. Clin Neurol Neurosurg. 2014;127:97–100.
Maegele M. Coagulopathy after traumatic brain injury: incidence, pathogenesis, and treatment options. Transfusion. 2013;53 Suppl 1:28S–37S.
Cohen MJ, Brohi K, Ganter MT, Manley GT, Mackersie RC, Pittet JF. Early coagulopathy after traumatic brain injury: the role of hypoperfusion and the protein C pathway. J Trauma. 2007;63:1254–61. discussion 61-2.
Lustenberger T, Talving P, Kobayashi L, Barmparas G, Inaba K, Lam L, et al. Early coagulopathy after isolated severe traumatic brain injury: relationship with hypoperfusion challenged. J Trauma. 2010;69:1410–4.
Gando S, Kameue T, Nanzaki S, Hayakawa T, Nakanishi Y. Participation of tissue factor and thrombin in posttraumatic systemic inflammatory syndrome. Crit Care Med. 1997;25:1820–6.
Gando S, Tedo I, Kubota M. Posttrauma coagulation and fibrinolysis. Crit Care Med. 1992;20:594–600.
Yasui H, Donahue DL, Walsh M, Castellino FJ, Ploplis VA. Early coagulation events induce acute lung injury in a rat model of blunt traumatic brain injury. Am J Physiol Lung Cell Mol Physiol. 2016;311:L74–86.
Tian Y, Salsbery B, Wang M, Yuan H, Yang J, Zhao Z, et al. Brain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injury. Blood. 2015;125:2151–9.
Nekludov M, Mobarrez F, Gryth D, Bellander BM, Wallen H. Formation of microparticles in the injured brain of patients with severe isolated traumatic brain injury. J Neurotrauma. 2014;31:1927–33.
Kutcher ME, Xu J, Vilardi RF, Ho C, Esmon CT, Cohen MJ. Extracellular histone release in response to traumatic injury: implications for a compensatory role of activated protein C. J Trauma Acute Care Surg. 2012;73:1389–94.
Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med. 2013;187:160–9.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7.
Medved L, Nieuwenhuizen W. Molecular mechanisms of initiation of fibrinolysis by fibrin. Thromb Haemost. 2003;89:409–19.
Risberg B, Medegard A, Heideman M, Gyzander E, Bundsen P, Oden M, et al. Early activation of humoral proteolytic systems in patients with multiple trauma. Crit Care Med. 1986;14:917–25.
Hagiwara A, Kushimoto S, Kato H, Sasaki J, Ogura H, Matsuoka T, et al. Can early aggressive administration of fresh frozen plasma improve outcomes in patients with severe blunt trauma? − A report by the Japanese Association for the Surgery of Trauma. Shock. 2016;45:495–501.
Kudo D, Kushimoto S, Shiraishi A, Ogura H, Hagiwara A, Saitoh D, et al. The impact of preinjury antithrombotic medication on hemostatic interventions in trauma patients. Am J Emerg Med. 2017;35:62–5.
Maekawa M. Lactate dehydrogenase isoenzymes. J Chromatogr. 1988;429:373–98.
Kanemitsu F, Okigaki T. Creatine kinase isoenzymes. J Chromatogr. 1988;429:399–417.
Huber D, Cramer EM, Kaufmann JE, Meda P, Masse JM, Kruithof EK, et al. Tissue-type plasminogen activator (t-PA) is stored in Weibel-Palade bodies in human endothelial cells both in vitro and in vivo. Blood. 2002;99:3637–45.
Lowenstein CJ, Morrell CN, Yamakuchi M. Regulation of Weibel-Palade body exocytosis. Trends Cardiovasc Med. 2005;15:302–8.
Kooistra T, Schrauwen Y, Arts J, Emeis JJ. Regulation of endothelial cell t-PA synthesis and release. Int J Hematol. 1994;59:233–55.
Gando S, Hayakawa M. Pathophysiology of trauma-induced coagulopathy and management of critical bleeding requiring massive transfusion. Semin Thromb Hemost. 2016;42:155–65.
Levrat A, Gros A, Rugeri L, Inaba K, Floccard B, Negrier C, et al. Evaluation of rotation thrombelastography for the diagnosis of hyperfibrinolysis in trauma patients. Br J Anaesth. 2008;100:792–7.
Theusinger OM, Baulig W, Seifert B, Muller SM, Mariotti S, Spahn DR. Changes in coagulation in standard laboratory tests and ROTEM in trauma patients between on-scene and arrival in the emergency department. Anesth Analg. 2015;120:627–35.
Theusinger OM, Wanner GA, Emmert MY, Billeter A, Eismon J, Seifert B, et al. Hyperfibrinolysis diagnosed by rotational thromboelastometry (ROTEM) is associated with higher mortality in patients with severe trauma. Anesth Analg. 2011;113:1003–12.
Karakus A, Kekec Z, Akcan R, Seydaoglu G. The relationship of trauma severity and mortality with cardiac enzymes and cytokines at multiple trauma patients. Ulus Travma Acil Cerrahi Derg. 2012;18:289–95.
Bilgic I, Gelecek S, Akgun AE, Ozmen MM. Predictive value of liver transaminases levels in abdominal trauma. Am J Emerg Med. 2014;32:705–8.
Rao CJ, Shukla PK, Mohanty S, Reddy YJ. Predictive value of serum lactate dehydrogenase in head injury. J Neurol Neurosurg Psychiatry. 1978;41:948–53.
Feng J, Zen P, Liu Y, Luo J, Jiang W, Peng G. Serum enzyme profile characteristics of victims following the Wenchuan earthquake in China. Clin Chem Lab Med. 2009;47:590–5.
Hayakawa M, Gando S, Ieko M, Honma Y, Homma T, Yanagida Y, et al. Massive amounts of tissue factor induce fibrinogenolysis without tissue hypoperfusion in rats. Shock. 2013;39:514–9.
Story DA, Morimatsu H, Bellomo R. Strong ions, weak acids and base excess: a simplified Fencl-Stewart approach to clinical acid-base disorders. Br J Anaesth. 2004;92:54–60.
Kraut JA, Madias NE. Lactic acidosis. N Engl J Med. 2014;371:2309–19.
Naganathar S, De’Ath HD, Wall J, Brohi K. Admission biomarkers of trauma-induced secondary cardiac injury predict adverse cardiac events and are associated with plasma catecholamine levels. J Trauma Acute Care Surg. 2015;79:71–7.
Acknowledgements
We would like to thank Editage (https://online.editage.jp/) for English language editing.
Funding
This study was supported in part by the Japan Society for the Promotion of Science KAKENHI (grant numbers 25462810 and 24592731).
Availability of data and materials
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
MH, S Kushimoto, HK, JS, HO, TM, TU, NM, HI, AH, MT, and NK designed the study. MH interpreted the data and drafted the manuscript. DS and AS supported the statistical analysis. MH, DK, TK, TS, HO, YH, TU, SF, YN, GM, AY, K Murata, S Kim, OT, and NK collected and assessed the data. All authors revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
This retrospective study was conducted at 15 tertiary emergency and critical care centres in Japan and was approved by the Institutional Review Board of each hospital (Additional file 1: Table S1). No patient consent was needed because of the retrospective study design.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1: Table S1.
Institutional Review Board of each hospital. (ODS 13 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hayakawa, M., Maekawa, K., Kushimoto, S. et al. Hyperfibrinolysis in severe isolated traumatic brain injury may occur without tissue hypoperfusion: a retrospective observational multicentre study. Crit Care 21, 222 (2017). https://doi.org/10.1186/s13054-017-1811-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-017-1811-1