
Abstract
Background
Ticks are blood-sucking ectoparasites that play a pivotal role in the transmission of various pathogens to humans and animals. In Korea, Haemaphysalis longicornis is the predominant tick species and is recognized as the vector of pathogens causing various diseases such as babesiosis, borreliosis, rickettsiosis, and severe fever with thrombocytopenia syndrome.
Methods
In this study, the targeted high-throughput sequencing of the 16S rRNA V4 region was performed using the state-of-the-art sequencing instrument, iSeq 100, to screen bacterial pathogens in H. longicornis, and the findings were compared with those using conventional PCR with specific primers. Microbiome analyses were performed with EzBioCloud, a commercially available ChunLab bioinformatics cloud platform. ANOVA-Like Differential Expression tool (ALDEx2) was used for differential abundance analysis.
Results
Rickettsia spp. were detected in 16 out of 37 samples using iSeq 100, and this was confirmed using a PCR assay. In the phylogenetic analysis using gltA and ompA sequences of the detected Rickettsia, the highest sequence similarity was found with ‘Candidatus Rickettsia jingxinensis’ isolate Xian-Hl-79, ‘Ca. R. jingxinensis’ isolate F18, and ‘Ca. R. longicornii‘ isolate ROK-HL727. In the microbiome study, Coxiella AB001519, a known tick symbiont, was detected in all 37 tick samples. Actinomycetospora chiangmaiensis was more abundant in Rickettsia-positive samples than in Rickettsia-negative samples.
Conclusions
In this study, iSeq 100 was used to investigate the microbiome of H. longicornis, and the potentially pathogenic Rickettsia strain was detected in 16 out of 37 ticks. We believe that this approach will aid in large-scale pathogen screening of arthropods to be used in vector-borne disease control programs.
Graphical Abstract

Similar content being viewed by others

Background
Ticks are blood-sucking ectoparasites that play a pivotal role in the transmission of a variety of pathogens to humans and animals [1, 2]. Ticks harbor numerous bacterial, protozoal, and viral pathogens and transfer these into the host’s body when sucking blood, causing serious infectious diseases [3, 4]. Tick-borne diseases such as anaplasmosis, ehrlichiosis, borreliosis, babesiosis, and rickettsiosis represent emerging threats to public and animal health worldwide [5, 6]. Each tick species has preferred environmental conditions, which determine the geographic distribution of the tick and, consequently, the risk areas for tick-borne diseases [1].
Ixodid ticks (including Haemaphysalis longicornis) have been recognized as vectors of pathogens causing diseases such as babesiosis, borreliosis, rickettsiosis, and severe fever with thrombocytopenia syndrome (SFTS) in Korea [7,8,9,10,11,12,13]. Rickettsia spp. have been isolated from infected animals, ticks, and the blood of human patients in Korea [13,14,15,16,17,18].
The main advantage of next-generation sequencing (NGS) is that a wide array of known and unknown pathogens can be identified simultaneously, without the need for designing individual specific bacterial primers [19]. iSeq 100, a recently released instrument, is capable of processing a targeted NGS library with accuracy comparable to that of MiSeq, the most widely used instrument in microbiome studies [20]. iSeq 100 also features shortened sequencing workflow preparation time and a total sequencing run time of under 24 h. The reduced capital and maintenance costs are also advantages of iSeq 100 over MiSeq [20].
In this study, we used iSeq 100 to detect bacterial pathogens in H. longicornis and compared the pathogen detection rates to those of pathogen-specific PCR test. In addition, we identified the strain of the detected potential pathogens using PCR with specific primers.
Methods
Sample collection and processing
Ticks were collected from the vegetation by flagging in Wonju (Gangwon-do Province; 37.389545, 127.801770) in July 2015. Species identification of collected ticks was performed by examination under a dissecting microscope according to the method of Yamaguti et al. [21]. Only nymph stage ticks were used in the study. The surface of each tick was sterilized using alcohol before DNA extraction, and whole ticks were used for DNA extraction. DNA was extracted from each tick using a NucleoSpin DNA Insect kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s instructions, and stored at −20 °C until use.
Illumina sequencing and bioinformatics
Total DNA was extracted using the NucleoSpin DNA Insect Kit (Macherey-Nagel, Düren, Germany) following the instructions of the manufacturer. The 16S rDNA V4 region was amplified by PCR using primers of 515F (5′-TCGTCGGCAGCGTCAGATG TGTATAAGAGACAGGTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GTCTCGTGGG CTCGGAGATGTGTATAAGAGACAGGGACTACHVGGGTWTCTAAT-3′) [22]. A limited-cycle (eight cycles) amplification step was performed to add multiplexing indices and Illumina sequencing adapters. Mixed amplicons were pooled, and the sequencing was performed with the Illumina iSeq 100 sequencing system according to the manufacturer’s instructions, utilizing an Illumina iSeq™ 100 i1 Reagent v2 kit (San Diego, CA, USA).
Processing raw reads started with a quality check and the filtering of low-quality (< Q25) reads by Trimmomatic ver. 0.321 [23]. After a quality control pass, sequence data were merged using the fastq_mergepairs command of VSEARCH version 2.13.42 with default parameters [24]. Primers were then trimmed with the alignment algorithm of Myers and Miller [25] at a similarity cut-off of 0.8. Non-specific amplicons that do not encode 16S rRNA were detected by nhmmer [26] in the HMMER software package ver. 3.2.1 with hmm profiles. Unique reads were extracted, and redundant reads were clustered with the unique reads by derep_fulllength command of VSEARCH2. The EzBioCloud 16S rRNA database [27] was used for taxonomic assignment using the usearch_global command of VSEARCH2, followed by more precise pairwise alignment [25]. Chimeric reads were filtered on reads with < 97% similarity by reference-based chimeric detection using the UCHIME algorithm [28] and the non-chimeric 16S rRNA database from EzBioCloud. After chimeric filtering, reads that were not identified to the species level (with < 97% similarity) in the EzBioCloud database were compiled, and the cluster_fast command was used to perform de novo clustering to generate additional operational taxonomic units (OTUs). Finally, OTUs with single reads (singletons) were omitted from further analysis. All subsequent analyses were performed with EzBioCloud, a commercially available ChunLab bioinformatics cloud platform for microbiome research (https://www.ezbiocloud.net/). The reads were normalized to 2500 to perform the analyses. We computed the Shannon index [29] and performed principal coordinate analysis (PCoA) [30]. The Wilcoxon rank-sum test was used to test the difference in the number of OTUs and the Shannon index between two groups. Significant differences in the relative abundances at the phylogenetic level between the two groups were assessed using the ANOVA-Like Differential Expression version 2 (ALDEx2) package and were visualized with the beeswarm package in R software (version 4.0.5) [31].
PCR, sequencing, and phylogenetic analysis
To detect the partial sequences of 17 kDa antigen-encoding gene of Rickettsia spp. in ticks, nested PCR was performed with two primer pairs: Rr17k.Ip (5′-TTTACAAAATTCTAAAAACCAT-3′) and Rr17k.539n (5′-TCAATTCACAACTTGCCATT-3′), which amplify a 539 bp fragment, and Rr17k.90p (5′-GCTCTTGCAACTTCTATGTT-3′) and Rr17k.539n (5′-TCAATTCACAACTTGCCATT-3′), which amplify a 450 bp fragment [32]. Sequencing of Rickettsia-positive nested PCR amplicons was conducted by Bionics Co. (Seoul, Korea). To identify the species using Rickettsia-positive nested PCR amplicons, the gltA gene and ompA gene were PCR-amplified. The sequences of the primers were as follows: gltA, 5′-GGCTAATGAAGCGGTAATAAATATGCTT-3′ (forward) and 5′-TTTGCGACGGTATAC C CATAGC-3′ (reverse); ompA, 5′-CACYACCTCAACCGCAGC-3′ (forward), and 5′-AAAGT TATATTTCCTAAACCYGTATAAKTATCRGC-3′ (reverse) [33].
A BLAST search was used to compare the obtained sequence of the gltA gene and ompA gene to those available in GenBank (USA). The obtained sequences were compared for similarity to sequences deposited in GenBank using BLAST. Gene sequences, excluding the primer regions, were aligned using the multisequence alignment program in Clustal Omega (Cambridgeshire, UK) [34]. The phylogenetic trees were constructed by Molecular Evolution Genetics Analysis (MEGA X) software using the maximum-likelihood method and employing the Tamura-nei model of nucleotide substitution with 1000 bootstrap replications [35].
Results
The microbiome composition of ticks analyzed with iSeq 100
The high-throughput sequencing of the 16S rRNA gene of 37 H. longicornis tick samples using iSeq 100 produced an average total read count of 7454 and 72 average number of OTUs from 37 tick samples. At the species level, all samples were dominated by Coxiella AB001519 (5.48–89.51% of the total community, average: 39.87%) (Fig. 1). Williamsia maris (0.04–41.83% of the total community, average: 14.56%), which was the second most abundant species, was also detected in all samples. Rickettsia rickettsii was detected in 16 out of 37 tick samples (Fig. 1).

The microbiome composition of each tick at the species level (n = 37). Species accounting for more than 1% of total reads are shown. Tick samples 1–16 (red) were found to harbor Rickettsia rickettsii (relative abundance > 0.1%)
Detection of rickettsiae using PCR
For the same set of samples, nested PCR of the 17 kDa surface protein gene determined 16 samples as positive for rickettsiae; the same samples were positive with NGS using iSeq 100. To identify the strain of Rickettsia spp. in our samples, species-specific PCR was performed using primers for partial sequences of gltA and ompA, and the resulting phylogenetic trees were obtained (Fig. 2). The PCR amplicon sequences of the gltA gene of all 16 Rickettsia-positive samples in the nested PCR demonstrated 100% homology to previously reported DNA sequences, which are ‘Candidatus Rickettsia jingxinensis’ isolate Xian-Hl-79 (MH932024), ‘Ca. R. jingxinensis’ isolate F18 (MN550898), and ‘Ca. Rickettsia longicornii’ isolate ROK-HL727 (MG906678) (Fig. 2a, Additional file 2: Figure S1). For ompA, DNA sequences from Rickettsia revealed 95.9–99.5% homology to ‘Ca. R. longicornii’ isolate ROK-HL727 (MG906676), ‘Ca. R. jingxinensis’ isolate Xian-Hl-79 (MH932069), and ‘Ca. R. jingxinensis’ isolate F18 (MN550905) (Fig. 2B, Additional file 3: Figure S2, Additional file 1: Table S1). Among the 16 Rickettsia-positive samples, 11 samples had 99.5% identity with those of ‘Ca. R. longicornii’ isolate ROK-HL727 (MG906676) and ‘Ca. R. jingxinensis’ isolate Xian-Hl-79 (MH932069) and 98.6% identity with the sequence of ‘Ca. R. jingxinensis’ isolate F18 (MN550905). Four samples showed relatively lower sequence identity (95.9–98.6%), which also showed poor sequencing quality (double peaks). One sample could not be sequenced (sample 13).

Phylogenetic trees based on partial sequences of gltA (a) and ompA (b) genes. Sequences of the Rickettsia spp. detected in the present study were aligned with those retrieved from the GenBank database. The phylogenetic trees were constructed in MEGA X software using the maximum-likelihood method, employing the Tamura-nei model of nucleotide substitution with 1000 bootstrap replications
The association between the presence of Rickettsia and the microbiome in ticks
We investigated the association between the presence of Rickettsia and the microbiome composition in ticks. The average of the total read counts for the Rickettsia-positive group (n = 16) and the Rickettsia-negative group (n = 21) were 8033 and 7012, respectively. The number of OTUs were not significantly different between the Rickettsia-positive and Rickettsia-negative groups (Fig. 3a). However, the Shannon index (bacterial diversity) was significantly different between the two groups (Fig. 3b). The results of PCoA (Fig. 3c) showed that the samples were not well organized as per Rickettsia positivity, but in those using permutational multivariate analysis of variance (PERMANOVA), Rickettsia positivity was found to be a significant factor in determining the tick microbiome composition (P = 0.001). Following this, we analyzed the bacterial composition between the two groups and found that Coxiella AB001519 accounted for 29.27% and 48.25% of the total reads in the Rickettsia-positive and Rickettsia-negative groups, respectively (Fig. 4). The differentially abundant bacterial species between the groups were identified using the ALDEx2 algorithm. The relative abundance of Coxiella AB001519 was higher in the Rickettsia-negative ticks than Rickettsia-positive ticks (the expected P value of Wilcoxon rank test = 0.022); conversely, the relative abundance of Actinomycetospora chiangmaiensis was higher in the Rickettsia-positive ticks than Rickettsia-negative ticks (the expected P value of Wilcoxon rank test = 0.008). This pattern was also seen in the swarm plots of the relative abundances (Fig. 5a, b).

Alpha and beta diversities of the microbiome of Rickettsia-positive groups and Rickettsia-negative groups. a The number of operational taxonomic units (OTUs) and b Shannon index between the Rickettsia-positive (n = 16) and Rickettsia-negative (n = 21) groups. c Microbiome composition of Rickettsia-positive and Rickettsia-negative groups are shown by principal coordinate analysis (PCoA)

The average microbiome composition of Rickettsia-positive ticks (n = 16) and Rickettsia-negative ticks (n = 21) at the species level. Species accounting for than 1% of total reads are shown

Swarm plots of the relative abundance of a Coxiella AB001519, and b Actinomycetospora chiangmaiensis in Rickettsia-positive ticks (n = 16), and Rickettsia-negative ticks (n = 21)
Discussion
Ticks can transmit bacterial, parasitic, and viral pathogens (often zoonotically) and often harbor more than one agent simultaneously [36]. Thus, obtaining broader information about pathogens in ticks is important from the perspective of proper diagnosis and treatment of these diseases [37]. In this study, we demonstrated that the recently released NGS instrument, iSeq 100, is useful for screening of bacteria in ticks. NGS approaches have the ability to identify a wide range of known or unknown pathogens or discover new organisms from a single test [19] without the need to design specific primers for each pathogen. This method makes it possible to identify pathogens immediately, not only in ticks, but also in arthropods that serve as vectors and reservoirs for pathogens, such as mosquitoes, tsetse flies, and sand flies.
Considering this advantage, in this study, we screened the tick-borne bacteria using iSeq 100 and found that 16 of the 37 ticks harbored pathogens of R. rickettsii. For an accurate taxonomic classification of the species of the detected Rickettsia spp., we used conventional PCR to amplify gltA and ompA sequences of the Rickettsia spp. and compared them with sequences deposited in GenBank using BLAST. In the phylogenetic analysis performed using MEGA X software, the sequence similarity to sequences of ‘Ca. R. jingxinensis’ isolate Xian-Hl-79, ‘Ca. R. jingxinensis’ isolate F18, and ‘Ca. R. longicornii’ isolate ROK-HL727 was 100% for gltA, indicating a close relationship between rickettsial isolates from H. longicornis from Korea with those from other East Asian countries. The close clustering of the Chinese and Korean strains of Rickettsia spp. may indicate a close epidemiological link between these strains.
Ixodid ticks (e.g., H. longicornis, H. flava, Ixodes persulcatus, and I. nipponensis) in Asia have the potential to be primary vectors/repositories of rickettsiae of medical and veterinary importance [15]. In 2006, the first case in Korea of R. japonica was isolated from a spotted fever patient [13]. Recent studies show a high prevalence of the emerging pathogen, Rickettsia raoultii, in canine ticks [38].
In our study, the sequences of gltA and ompA are identical or highly homologous to those of ‘Ca. R. jingxinensis’ isolate Xian-Hl-79, ‘Ca. R. jingxinensis’ isolate F18, and “Ca. R. longicornii’ isolate ROK-HL727. ‘Ca. R. jingxinensis,’ a novel Rickettsia species in Rhipicephalus microplus and H. longicornis ticks, was first discovered in China (Shenyang and Wuhan) [39,40,41] and subsequently reported in Korea (Chungnam, Jeonbuk, and Gwangju) [42, 43]. Many associated ‘Ca. R. jingxinensis’ sequences have been deposited in GenBank. Of these, a gltA sequence (KU853023) was recovered from a patient, suggesting its potential pathogenicity to humans [44]. The pathogenicity of ‘Ca. R. longicornii’ is yet to be determined. However, 99.6% identity was detected with the ‘Ca. R. longicornii’ ompA sequence of an isolate from rodent spleen tissue obtained in Korea, and 99.9% identity was detected with the ‘Ca. R. longicornii’ gltA sequence of an isolate from a human blood sample obtained in China [18, 44]. These results suggest that ‘Ca. R. longicornii’ has the potential to infect mammalian hosts, including humans [45].
Although not detected in our samples, Borrelia burgdorferi, B. afzelii, and B. garinii, which are implicated in Lyme disease, have been isolated from Ixodes persulcatus, I. nipponensis, and I. granulatus, which are distributed in parts of Korea [46, 47]. In addition, several Lyme disease (borreliosis) cases have been reported in Korea [48,49,50,51].
Coxiella AB001519, known as a Coxiella-like symbiont of H. longicornis, was detected in all 37 samples [52]. A bacterium belonging to Coxiella was reported as the primary endosymbiont of Amblyomma americanum ticks and was found to improve the reproductive health of the ticks [53,54,55]. Coxiella AB001519 was first identified in Japan in a phylogenetic association with the Coxiella-like endosymbiont of H. longicornis, and the Coxiella-like endosymbiont presenting more than 99% homology with Coxiella AB001519 in Thailand, China, and Korea was found in H. longicornis [56,57,58].
In the present study, the relative abundance of A. chiangmaiensis found to be significantly higher in Rickettsia spp.-positive samples than in Rickettsia spp.-negative samples. A. chiangmaiensis, a Gram-positive bacterium, was isolated from the soil of a tropical rainforest in northern Thailand in 2008 [59]. Actinomycetospora spp. are commonly found in the environment and are known to infect ticks [60].
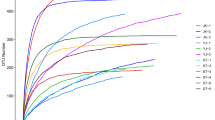
Although we confirmed the ability of iSeq 100 to detect potential pathogens using PCR, the rarefaction curves of some samples did not show a plateau in their read counts (Additional file 4: Figure S3). To cover the whole microbial diversity, sufficient reads should be sequenced. To this end, it would be helpful to load a small number of samples in one run or to use a machine with a larger sequencing capacity, such as MiSeq.
The targeted NGS using the bacterial 16S rRNA V4 region in this study cannot identify the exact strain of pathogens. However, this technique can theoretically detect all potentially pathogenic taxa in samples including Rickettsia spp., Coxiella spp., Borrelia spp., Bartonella spp., Francisella spp., and Anaplasma spp., and simultaneously analyze 96 samples. The cost of iSeq 100 reagents required for one run is approximately US $2000, and the sequencing takes 18 h to complete. Therefore, we believe that this method is useful in screening pathogens in arthropod vectors such as ticks, and can be followed by subsequent experiments to identify the exact strain of the suspected pathogens.
Conclusions
In this study, iSeq 100 was used to investigate the microbiome of H. longicornis and the potentially pathogenic Rickettsia strain was detected in 16 out of 37 ticks. We believe that this approach can be used for large-scale pathogen screening of arthropods, which can be used in vector-borne disease control programs.
Availability of data and materials
Raw sequence data are available in NCBI GenBank under BioProject PRJNA728802.
Abbreviations
- SFTS:
-
Severe fever with thrombocytopenia syndrome
- NGS:
-
Next-generation sequencing
- PCoA:
-
Principal coordinate analysis
- ALDEx2:
-
ANOVA-like differential expression version 2
- OTUs:
-
Operational taxonomic units
- PERMANOVA:
-
Permutational multivariate analysis of variance
- LDA:
-
Linear discriminant analysis
References
Parola P, Raoult D. Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Infect Dis. 2001;32:897–928.
Wikel SK. Tick modulation of host immunity: an important factor in pathogen transmission. Int J Parasitol. 1999;29:851–9.
Nuttall PA, Paesen GC, Lawrie CH, Wang H. Vector-host interactions in disease transmission. J Mol Microbiol Biotechnol. 2000;2:381–6.
Kazimírová M, Štibrániová I. Tick salivary compounds: their role in modulation of host defences and pathogen transmission. Front Cell Infect Microbiol. 2013;3:43.
Kernif T, Leulmi H, Raoult D, Parola P. Emerging tick-borne bacterial pathogens. Microbiol Spectr. 2016. https://doi.org/10.1128/microbiolspec.EI10-0012-2016.
Dantas-Torres F, Chomel BB, Otranto D. Ticks and tick-borne diseases: a one health perspective. Trends Parasitol. 2012;28:437–46.
Park SW, Song BG, Shin EH, Yun SM, Han MG, Park MY, Park C, Ryou J. Prevalence of severe fever with thrombocytopenia syndrome virus in Haemaphysalis longicornis ticks in South Korea. Ticks Tick-borne Dis. 2014;5:975–7.
Chae JB, Kang JG, Kim HC, Chong ST, Lee IY, Shin NS, Chae JS. Identification of tick species collected from wild boars and habitats of wild boars and domestic pigs in the Republic of Korea. Korean J Parasitol. 2017;55:185–91.
Chae JB, Cho YS, Cho YK, Kang JG, Shin NS, Chae JS. Epidemiological investigation of tick species from near domestic animal farms and cattle goat, and wild boar in Korea. Korean J Parasitol. 2019;57:319–24.
Kim-Jeon MD, Jegal S, Jun H, Jung H, Park SH, Ahn SK, Lee J, Gong YW, Joo K, Kwon MJ, Roh JY, Lee WG, Bahk YY, Kim TS. Four year surveillance of the vector hard ticks for SFTS, Ganghwa-do, Republic of Korea. Korean J Parasitol. 2019;57:691–8.
Kwon HY, Im JH, Park YK, Durey A, Lee JS, Baek JH. Two imported cases of babesiosis with complication or co-infection with Lyme disease in Republic of Korea. Korean J Parasitol. 2018;56:609–13.
Moon S, Gwack J, Hwang KJ, Kwon D, Kim S, Noh Y, Roh J, Shin EH, Jeong K, Seok W, Youn SK. Autochthonous Lyme borreliosis in humans and ticks in Korea. Osong Public Health Res Perspect. 2013;4:52–6.
Chung MH, Lee SH, Kim MJ, Lee JH, Kim ES, Lee JS, Kim MK, Park MY, Kang JS. Japanese spotted fever, South Korea. Emerg Infect Dis. 2006;12:1122–4.
Kim YS, Choi YJ, Lee KM, Ahn KJ, Kim HC, Klein T, Jiang J, Richards A, Park KH, Jang WJ. First isolation of Rickettsia monacensis from a patient in South Korea. Microbiol Immunol. 2017;61:258–63.
Noh Y, Lee YS, Kim HC, Chong ST, Klein TA, Jiang J, Richards AL, Lee HK, Kim SY. Molecular detection of Rickettsia species in ticks collected from the southwestern provinces of the Republic of Korea. Parasit Vectors. 2017;10:20.
Jiang J, Choi YJ, Kim J, Kim HC, Klein TA, Chong ST, Richards AL, Park HJ, Shin SH, Song D, Park KH, Jang WJ. Distribution of Rickettsia spp. in ticks from northwestern and southwestern provinces, Republic of Korea. Korean J Parasitol. 2019;57:161–6.
Ma JS. Rickettsioses in South Korea, data analysis. Emerg Infect Dis. 2006;12:531–2.
Kim CM, Yi YH, Yu DH, Lee MJ, Cho MR, Desai AR, Shringi S, Klein TA, Kim HC, Song JW, Baek LJ, Chong ST, O’guinn ML, Lee JS, Lee IY, Park JH, Foley J, Chae JS. Tick-borne rickettsial pathogens in ticks and small mammals in Korea. Appl Environ Microbiol. 2006;72:5766–76.
Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol. 2019;24:319–38.
Colman RE, Mace A, Seifert M, Hetzel J, Mshaiel H, Suresh A, Lemmer D, Engelthaler DM, Catanzaro DG, Young AG, Denkinger CM, Rodwell TC. Whole-genome and targeted sequencing of drug-resistant Mycobacterium tuberculosis on the iSeq100 and MiSeq: a performance, ease-of-use, and cost evaluation. PLoS Med. 2019;30:e1002794.
Yamaguti N, Tipton VJ, Keegan HL, Toshioka H. Ticks of Japan, Korea, and the Ryukyu islands. Brigham Young Univ Sci Bull Bio Ser. 1971;15:1–227.
Bugenyi AW, Cho HS, Heo J. Association between oropharyngeal microbiome and weight gain in piglets during pre and post weaning life. J Anim Sci Technol. 2020;62:247–62.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
Myers EW, Miller W. Optimal alignments in linear space. Bioinformatics. 1988;4:11–7.
Wheeler TJ, Eddy SR. nhmmer: DNA homology search with profile HMMs. Bioinformatics. 2013;29:2487–9.
Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol. 2017;67:1613–7.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200.
Magurran AE. Measuring biological diversity. Hoboken: Wiley; 2013.
Gower JC. Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika. 1966;1966(53):325–38.
Fernandes AD, Reid JN, Macklaim JM, McMurrough TA, Edgell DR, Gloor GB. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome. 2014;2:15.
Ishikura M, Ando S, Shinagawa Y, Matsuura K, Hasegawa S, Nakayama T, Fujita H, Watanabe M. Phylogenetic analysis of spotted fever group rickettsiae based on gltA, 17-kDa, and rOmpA genes amplified by nested PCR from ticks in Japan. Microbiol Immunol. 2003;47:823–32.
Phan JN, Lu CR, Bender WG, Smoak RM, Zhong J. Molecular detection and identification of Rickettsia species in Ixodes pacificus in California. Vector-Borne Zoonotic Dis. 2011;11:957–61.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soeding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol. 2011;7:539.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;2018(35):1547–9.
Parola P, Paddock CD, Socolovschi C, Labruna MB, Mediannikov O, Kernif T, Abdad MY, Stenos J, Bitam I, Fournier PE, Raoult D. Update on tick-borne rickettsioses around the world: a geographic approach. Clin Microbiol Rev. 2013;26:657–702.
Madison-Antenucci S, Kramer LD, Gebhardt LL, Kauffman E. Emerging tick-borne diseases. Clin Microbiol Rev. 2020;2020(33):e00083-18.
Seo MG, Kwon OD, Kwak D. High prevalence of Rickettsia raoultii and associated pathogens in canine ticks, South Korea. Emerg Infect Dis. 2020;26:2530–2.
Dong X, Chen XP, Liu N, Dumler SJ, Zhang YZ. Co-circulation of multiple species of Rickettsiales bacteria in one single species of hard ticks in Shenyang, China. Ticks Tick Borne Dis. 2014;5:727–33.
Lu M, Tian JH, Yu B, Guo WP, Holmes EC, Zhang YZ. Extensive diversity of Rickettsiales bacteria in ticks from Wuhan, China. Ticks Tick Borne Dis. 2017;8:574–80.
Liu H, Li Q, Zhang X, Li Z, Wang Z, Song M, Wei F, Wang S, Liu Q. Characterization of rickettsiae in ticks in northeastern China. Parasit Vectors. 2016;13:498.
Park HJ, Kim J, Choi YJ, Kim HC, Klein TA, Chong ST, Jiang J, Richards AL, Jang WJ. Tick-borne rickettsiae in midwestern region of Republic of Korea. Acta Trop. 2021;215:105794.
Park JW, Lee SH, Lee GS, Seo JJ, Chung JK. Epidemiological characteristics of field tick-borne pathogens in Gwang-ju metropolitan area, South Korea, from 2014 to 2018. Osong Public Health Res Perspect. 2020;11:177–84.
Guo WP, Wang YH, Lu Q, Xu G, Luo Y, Ni X, Zhou EM. Molecular detection of spotted fever group rickettsiae in hard ticks, northern China. Transbound Emerg Dis. 2019;2019(66):1587–96.
Jiang J, An H, Lee JS, O’Guinn ML, Kim HC, Chong ST, Zhang Y, Song D, Burrus RG, Bao Y, Klein TA, Richards AL. Molecular characterization of Haemaphysalis longicornis-borne rickettsiae, Republic of Korea and China. Ticks Tick Borne Dis. 2018;9:1606–13.
Park KH, Lee SH, Won WJ, Park MY. Isolation of Borrelia burgdorferi, the causative agent of Lyme disease, from Ixodes ticks in Korea. J Korean Soc Microbiol. 1992;1992(27):307–12.
Kee S, Hwang KJ, Oh HB, Park KS. Identification of Borrelia burgdorferi isolated in Korea using outer surface protein A (OspA) serotyping system. Microbiol Immunol. 1994;38:989–93.
Kim TH, Choi EH, Lee MG, Ahn SK. Serologically diagnosed Lyme disease manifesting erythema migrans in Korea. J Korean Med Sci. 1999;14:85–8.
Lee CN, Mo HJ, Kim JE, Park HJ, Lee JY, Cho BK, Lee IY. A case of Lyme disease presenting with erythema (chronicum) migrans. Korean J Dermatol. 2003;41:1202–5.
Kim JW, Kim JS. A case of Lyme disease with unusual cutaneous manifestations. Korean J Dermatol. 2005;43:501–6.
Lee D, Kim SH, Hong SK, Seo JK, Sung HS, Hwang SW. A case of Lyme disease with various general symptoms. Korean J Dermatol. 2008;46:1112–6.
Noda H, Munderloh UG, Kurtti TJ. Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl Environ Microbiol. 1997;63:3926–32.
Zhong J, Jasinskas A, Barbour AG. Antibiotic treatment of the tick vector Amblyomma americanum reduced reproductive fitness. PLoS ONE. 2007;2:e4.
Smith TA, Driscoll T, Gillespie JJ, Raghavan R. A Coxiella-like endosymbiont is a potential vitamin source for the lone star tick. Genome Biol Evol. 2015;23:831–8.
Machado-Ferreira E, Dietrich G, Hojgaard A, Levin M, Piesman J, Zeidner NS, Soares CAG. Coxiella symbionts in the cayenne tick Amblyomma cajennense. Microb Ecol. 2011;62:134–42.
Machado-Ferreira E, Vizzoni VF, Balsemão-Pires E, Moerbeck L, Gazeta GS, Piesman J, Voloch CM, Soares CA. Coxiella symbionts are widespread into hard ticks. Parasitol Res. 2016;115:4691–9.
Liu LM, Liu JN, Liu Z, Yu ZJ, Xu SQ, Yang XH, Li T, Li SS, Guo LD, Liu JZ. Microbial communities and symbionts in the hard tick Haemaphysalis longicornis (Acari: Ixodidae) from north China. Parasit Vectors. 2013;28:310.
Seo MG, Lee SH, Ouh IO, Lee GH, Goo YK, Kim S, Kwon OD, Kwak D. Molecular detection and genotyping of Coxiella-like endosymbionts in ticks that infest horses in South Korea. PLoS ONE. 2016;28:e0165784.
Jiang Y, Wiese J, Tang SK, Xu LH, Imhoff JF, Jiang CL. Actinomycetospora chiangmaiensis gen. nov., sp. nov., a new member of the family Pseudonocardiaceae. Int J Syst Evol Microbiol. 2008;58:408–13.
Lejal E, Chiquet J, Aubert J, Robin S, Estrada-Peña A, Rue O, Midoux C, Mariadassou M, Bailly X, Cougoul A, Gasqui P, Cosson JF, Chalvet-Monfray K, Vayssier-Taussat M, Pollet T. Temporal patterns in Ixodes ricinus microbial communities: an insight into tick-borne microbe interactions. bioRxiv. 2020. https://doi.org/10.1101/2020.09.26.314179.
Acknowledgements
Not applicable.
Funding
This study was supported by a National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MEST; Numbers NRF-2019R1A2B5B01069843, 2020R1I1A2074562 and 2020R1I1A1A0105358211).
Author information
Authors and Affiliations
Contributions
JYK and TSY designed this study. MHY and AASM performed the laboratory experiments. JYK, MHY and TSY analyzed and interpreted the data and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Homology of ompA gene sequence between the PCR amplicon and the gene of three known Rickettsia spp.
Additional file 2: Figure S1.
The PCR amplicon sequence alignment of gltA gene nucleotide sequences from Rickettsia spp.
Additional file 3: Figure S2.
The PCR amplicon sequence alignment of the ompA gene nucleotide sequences from Rickettsia spp.
Additional file 4: Figure S3.
Rarefaction curves for the number of operational taxonomic units (OTUs) of the 37 tick samples.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kim, J.Y., Yi, Mh., Mahdi, A.A.S. et al. iSeq 100 for metagenomic pathogen screening in ticks. Parasites Vectors 14, 346 (2021). https://doi.org/10.1186/s13071-021-04852-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-04852-w