
Abstract
Chromosome organisation is increasingly recognised as an essential component of genome regulation, cell fate and cell health. Within the realm of transposable elements (TEs) however, the spatial information of how genomes are folded is still only rarely integrated in experimental studies or accounted for in modelling. Whilst polymer physics is recognised as an important tool to understand the mechanisms of genome folding, in this commentary we discuss its potential applicability to aspects of TE biology. Based on recent works on the relationship between genome organisation and TE integration, we argue that existing polymer models may be extended to create a predictive framework for the study of TE integration patterns. We suggest that these models may offer orthogonal and generic insights into the integration profiles (or “topography”) of TEs across organisms. In addition, we provide simple polymer physics arguments and preliminary molecular dynamics simulations of TEs inserting into heterogeneously flexible polymers. By considering this simple model, we show how polymer folding and local flexibility may generically affect TE integration patterns. The preliminary discussion reported in this commentary is aimed to lay the foundations for a large-scale analysis of TE integration dynamics and topography as a function of the three-dimensional host genome.
Similar content being viewed by others


Background
Transposable elements (TEs) are DNA sequences that can move from one location of the genome to another. By being able to spread their own DNA across the genome independent of the cell’s replication cycle [1], TEs represent the majority of genomic content in most eukaryotes. For example, they comprise 85% of the maize genome [2] and up to 50% of primate genomes [3]. As such, TE activity is a major driver of phenotypic and genotypic evolution [4, 5] and affects key biological processes from meiosis and transcription to immunological responses [6]. At the same time, TEs have been associated with various diseases and cancer in humans [7].
Most TEs transpose via cut-and-paste or copy-and-paste mechanisms that can both result in a net increase of the TE copy number [8]. Amplification phases, or bursts, of TEs can occur multiple times in the evolutionary history of the host and may produce hundreds if not thousands of new copies within short time windows [5, 9, 10].
Most TEs exhibit some level of integration site selection, from very specific target sites [11] to non-random but more dispersed genomic biases [12–14]. Short DNA motifs, epigenetic marks and nuclear proteins have been associated with such integration site preferences. For example, yeast Ty1 retrotransposons integrate upstream of Pol III-transcribed genes through a direct interaction between the integrase complex and the AC40 subunit of Pol III [15, 16]. In contrast, in plants and fungi, the integrase of certain Gypsy retrotransposons contains a chromodomain that can bind to repressive histone marks and aid insertion into heterochromatin [17].
While the role of protein tethering and DNA motifs in TE integration is well established by now, it remains elusive how the three-dimensional (3D) structure of chromosomes and the nuclear environment is affecting TE spreading in host genomes. Chromosome folding and nuclear organisation have been shown to play key roles in all major DNA related processes [18–25], from transcription and replication to DNA repair, and it is thus natural to expect that transposition will also be affected by the 3D organisation of the genome.
Main text
Roles of TEs in 3D genome organisation
A number of recent reports have highlighted that TE activity is involved in shaping 3D chromosome structure. For example, TEs of diverse families have been implicated in the establishment and maintenance of insulator boundaries between so-called “topologically associated domains” (TADs) [26–31]. Furthermore, TE amplification is suggested to account for a significant amount of binding motifs for the CTCF [32–34] protein, a key regulator of 3D chromosome organisation [35]. The involvement of TEs in the establishment of evolutionarily conserved long-range chromosomal interactions has been shown in different organisms [36, 37] and some of these TE-mediated interactions appear to be of functional importance in gene regulation. In the plant Arabidopsis thaliana, TEs are enriched at genomic hubs of long-range chromosomal interactions with anticipated functional roles in silencing of foreign DNA elements [38].
Arguably, how TEs contribute to genome folding will certainly receive more attention in the future, but it is an equally fundamental question for both genome and TE biology to understand how genome folding affects TE integration preferences. For example, depending on the 3D organisation of chromosomes, a new TE copy that enters the nucleus from the cytoplasm will come across distinct parts of the genome in terms of their accessibility and organisation compared to a preexisting TE copy that relocates to a new genomic locus without exiting the nucleus. Intriguingly, retroelements (including retrotransposons and retroviruses) and DNA transposons have different replication and transposition pathways [39], which implies that genome architecture may have a different impact in each TE type.
There is a clear gap in the experimental and theoretical work on understanding the impact of genome architecture on integration of TEs. Models of TE amplification dynamics have traditionally been based on population-based approaches [40], which typically set up systems of (stochastic) ordinary differential equations accounting for generic competing elements during TE expansion [41–43]. Few works, instead, have considered the 1D distribution of nucleosomes along the genome in order to predict preferential sites of HIV integration [44]. Both these classes of models necessarily neglect the multi-scale 3D organisation of the genome, i.e. from nucleosomes to TADs and from compartments to chromosome territories [45, 46]. Because of this, they are not suited to predict the “topography” of TEs, i.e. the pattern of genomic sites in which TEs will preferentially integrate.
In this commentary, we introduce and discuss a computational model based on principles of polymer physics, which aims to dissect the interplay between genome organisation and biases in TE integration. We first briefly review the existing framework of polymer models – which have been proved to be very successful tools to rationalise 3D genome folding [47–53] – we then discuss a recent development of such models to understand the physical principles of HIV integration [54], and finally present preliminary data obtained by extending these models to the case of TEs (Fig. 3). We conclude this commentary by discussing potential future directions in this unexplored line of research.
Biophysical principles of genome folding
While genomes are, biologically speaking, the carrier of genetic information they also are, physically speaking, long polymers [55]. Polymers are well-known objects that have been studied for several decades in particular in relation to industrial applications, such as rubbers [56]. Pioneers in polymer physics realised a long time ago that they obey “universal” laws that are independent of their chemical composition [57]. For instance, the way a long polystyrene molecule folds in space must be identical, statistically speaking, to that of a long DNA molecule in the same solvent conditions. Because of this, polymer physicists typically employ “coarse-grained” approaches, which blur the chemical details and only retain the necessary ingredients that allow the formulation of simple and generic (universal) frameworks [58]. Universality then implies that these coarse-grained models have predictive power for a broad range of systems with different chemistry.
Coarse-graining several base-pairs and groups of atoms into mesoscopic beads (see Fig. 1a), while retaining the salient physical behaviour of DNA, allows the formulation of computational models that can reliably predict the spatial organisation of whole chromosomes from minimal input – such as epigenetic patterns and generic binding proteins [51, 53, 59–64] – and disentangle the contribution of different classes of proteins to genome folding [61, 65, 66].

a Coarse graining of microscopic details of double stranded DNA into a bead-spring polymer. b A polymer model for the nucleosome: highlighted are the features of DNA stiffness (set by penalising large angles θ between consecutive pairs of monomers) and connectivity (set by penalising large extensions x between consecutive beads). We also account for excluded volume interactions and pair-attraction represented by the wrapping of the orange segment around the histone octamer (here a blue spherical bead). c Schematics showing that integration events on DNA deform the substrate. d Snapshots from molecular dynamics simulations showing an integration event within a nucleosome. Color scheme: orange = wrapped host DNA, green= viral DNA, grey = non-wrapped host DNA. Adapted from Ref. [54]
These computational models, coupled to Chromosome Conformation Capture (and its higher order variants, such as HiC) experiments [18, 19], are providing new information on the spatio-temporal organisation of the genome in different conditions, such as healthy [62, 67], senescent [68] and diseased [52] cells, or even during cell-fate decisions [25] and reprogramming [69]. For instance, polymer models can rationalise features such as TADs [64, 70], compartments [51, 59] and loops [53, 64] seen in experimental HiC maps [71]. Importantly, these works are proving that traditionally physical phenomena such as liquid-liquid and polymer-polymer phase separation [49, 72–76], gelation [77, 78], emulsification [79] and viscoelasticity [80, 81] may be found in ubiquitous and key biological processes such as transcription, replication, mitosis, RNA splicing and V(D)J recombination to name a few. Polymer models are thus providing the community with a physical lens through which they may interpret complex data, and a quantitative framework to generate de novo predictions. In light of this, we here propose that the use of polymer models may shed new light into the relationship between TE transposition and 3D organisation. Earlier this year one of us made a first step in this direction by formalising a polymer-based model for understanding the site selection features displayed by HIV integration in the human genome [54]. Below, we briefly review this work, which will then be used as a stepping stone to formalise a polymer model for TE expansion.
A polymer model for HIV integration
One of the least understood features of HIV integration is that its integration patterns display markedly non-random distributions both along the genome [82] and within the 3D nuclear environment [83]. HIV displays a bias for nucleosomes [84, 85], gene-rich regions [82] and super-enhancer hotspots [86] that has defied comprehension for the past three decades. Clearly, from the perspective of a retrovirus such as HIV, integrating in frequently transcribed regions is evolutionary advantageous. But how is this precise targeting achieved?
For the past decades, the working hypothesis to address this important question was that there must exist specialised factors or protein chaperones that guide HIV integration site selection. Prompted by this hypothesis, much work has been devoted to discover and identify such proteins [14]. Some factors, such as the lens-epithelium-derived growth factor [87] (LEDGF/p75) have been proposed as potential candidates for this role but even knocking-down their expression could not completely remove the bias for gene-rich regions [88]. Additionally, the preference of HIV to integrate in nucleosomes – oppositely to naked DNA – was shown in vitro using minimal reaction mixtures [85, 89–91].
We recently put forward a different working hypothesis to address the bias of HIV integration site selection: could there be universal (non-system specific) physical principles that – at least partially – can contribute to biasing the site-selection of HIV integration? [54] Prompted by this hypothesis, we decided to propose a polymer-based model in which retroviral integration occurs via a stochastic and quasi-equilibrium topological reconnection between 3D proximal polymer segments [54]. In other words, whenever two polymer segments (one of the host and one of the invading DNA) are nearby in 3D we assign a certain probability for these two segments to reconnect, based on the difference in energy between the old and new configurations. This strategy is known as a Metropolis criterion and it satisfies detailed balance, thus ensuring that the system is sensitive to the underlying free energy [92]. [Note that in vitro HIV integrase works without the need of ATP [85], and we therefore assume that the integration process is in (or near) equilibrium.] Finally, we impose that the viral DNA is stuck once integrated in the selected location and cannot be excised within the simulation time.
Within this simple model, we discovered that geometry alone may be responsible for a bias in the integration of HIV in nucleosomes (Fig. 1b-d adapted from Ref. [54]). This is because the pre-bent conformation of DNA wrapped around the histone octamer lowers the energy barrier against DNA deformation required to integrate the viral DNA into the host (Fig. 1b, see also Refs. [89–91]). While the preference of HIV for pre-bent DNA conformations was suggested before [85, 93, 94], it had not been explained and formalised within a physical and mathematical framework that could generate quantitative predictions. Further, by considering a longer region of a human chromosome folded as predicted by the above mentioned polymer models [49, 50, 59], we discovered that at larger scales HIV integration sites obtained from experiments [82] are predominantly determined by chromatin accessibility. Thus, by accounting for DNA elasticity and chromatin accessibility – two universal and cell unspecific features of genome organisation – our model could predict HIV integration patterns remarkably similar to those observed in experiments in vitro [84, 85] and in vivo [82].
Extension to DNA transposition
In light of the success of this polymer model, we now propose to extend it to understand the distribution of integration sites across the TE phylogeny. Importantly, different TEs have different integration strategies, which suggests that they are likely to interact differently with the 3D genome organization within the nucleus. TEs can be primarily distinguished based on the mechanism through which they proliferate, i.e. via “copy and paste” (class I or retrotransposons) or “cut and paste” (class II, DNA transposons). The former require an RNA intermediate to proliferate and thus exit the nuclear environment, whereas the latter simply relocate their DNA via endonuclease excision [8, 39].
Retroviruses, like HIV, are very similar to retrotransposons with the addition that they can exit to the extracellular space and invade other cells. Thus, a new copy of a retrotransposon (class I or copy-and-paste) or a retrovirus must travel from the periphery to the nuclear interior while the genome is “scanned” from the outside-in for integration sites (Fig. 2a). This implies that the global, nuclear-scale genome architecture is expected to be important for this re-integration process. For instance, Lamin Aassociated Domains (LAD) [21] positioning with respect to nuclear pores, inverted versus conventional organisation [95], compartments [71] and enhancer hot-spots [86] will likely play major roles for retrotransposons. On the contrary, a DNA transposon (class II or cut-and-paste) probes the genome in the immediate surrounding of its excision site and will diffuse from the inside-out (Fig. 2b-c). As a result of this, the mesoscale (∼1 Mbp) organisation of the genome may have a profound effect on the 1D genomic distribution of DNA transposons. For example, heterochromatin-rich chromatin is thought to be collapsed [49, 96] with a typical overall size that depends on the genomic length as R∼L1/3; on the other hand, euchromatin-rich compartments [97] are more open [98] and their size may be more similar to that of a random walk, i.e. scaling as R∼L1/2. The contact probability of two genomic loci at distance s can be estimated to scale as Pc∼s−3ν [47] where ν is 1/3 for collapsed polymers (such as heterochromatin), 1/2 for ideal ones (such as euchromatin [96]) and 3/5 for self-avoiding walks [56]. Thus, a crude calculation would predict that a DNA transposon should re-integrate at distance s with a probability Pc(s)∼s−3ν that depends (through the exponent ν) on the folding of the genome at these (TAD-size) length-scales. A similar effect is at play in the enhancement of long range contacts in oncogene-induced senescent cells [99].

a Copy-and-paste transposition explores the nuclear space by diffusing from the periphery towards the interior, i.e. outside-in. The large-scale nuclear architecture, i.e. inverted or conventional [95], Lamin Associated Domains (LADs) [21], compartments [71] and enhancers hot-spots [83, 86], are expected to play the biggest roles in the integration site selection. b–c Cut-and-paste transposition explores the nuclear interior inside-out. In this case, TAD-scale (∼1 Mbp) genome folding is expected to dominate and in particular open conformations will yield short range de novo re-integration whereas collapsed ones will lead to longer range re-integration. Duplication of the transposon is also possible by homologous DNA repair of the broken strands

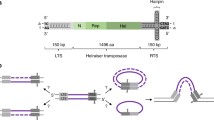
a Sketch of the original simulations performed in this work where we consider a segment of DNA 1.6 kbp long (or N=200 beads, each bead representing 8 bp) with rigidity lp=150 bp. The DNA is interspersed with “soft” sites which display a different rigidity lf. The length of these weak sites is 8 bp, or 1 bead. b We compute the frequency of integration events per each segment of the substrate by counting the number of events occurring at a specific locus over the total integration events. We average over 1000 independent simulations. One can notice that the patterns, which are roughly uniform for lf=lp become more and more periodic and reflecting the positions of the soft sites (denoted by the black arrows) when we reduce lf. The dotted line shows the expected frequency for random events 1/N, with N=200 the length of the substrate. For clarity we report only the segment 0.5–1 kbp. c Integration enhancement in soft sites over the expected random frequency. Each box represents a different value of the rigidity of the soft sites lf. Recall that lf=lp=150 bp reflects a uniformly stiff substrate and indeed we recover the expected value (unity) for the enhancement
It should be noted that the arguments above assume that the chromosomes can be seen as polymers in the melt [55, 100]. In such a picture, chromosome folding at the level of TADs (100kbp-10Mbp) and territories (>10Mbp) takes place on heteromorphic chromatin fibres, which can assume a range of local packaging at the scale of 10–30 nm (1–100kbp) [62, 101, 102].
In addition to this contribution coming from large- and meso-scale folding, one may argue that there ought to be other complementary effects such as specific features of the integrase [90, 103] or tethering [17] enzymes. These orthogonal elements are more local and are expected to equally affect both DNA transposons and retrotransposons. To investigate the role of local chromatin features on a generic integration event, we here perform some original, yet preliminary, simulations on a heterogeneously flexible polymer that crudely mimics heterogeneous chromatin in vivo (Fig. 3a). Specifically, we consider a stretch of 1.6kbp long DNA with persistence length lp=150bp=50nm and regularly interspersed with soft sites that display a lower bending rigidity lf. This lower local DNA rigidity may be due to, for instance, to denaturation bubbles [104], R-loops [105] or replication stress [106]. In these conditions – which may be reproduced in vitro by considering DNA with a sequence of bases that modulates its local flexibility [84] – we ask what is the integration pattern displayed by an invading DNA element by counting the number of integration events in each segment of the polymer over many (1000) independent simulations. We observe that, by varying the value of the rigidity parameter from lf=lp=150 bp to lf=60 bp, the integration patterns become less uniform, more periodic and reflecting the distribution of soft sites (Fig. 3b).
From these patterns we can compute the enhancement of integration in susceptible sites due to their different flexibility. This is simply the sum of integration frequencies in all soft sites divided by the one expected for a random distribution of events, i.e. n/N where n is the number of soft sites and N the length of the polymer. This calculation is reported in Fig. 3c and shows that the enhancement increases with the flexibility of the susceptible sites. For small deviations from lp=150 bp, this increase can be fitted as a single exponential as expected for a Metropolis Monte Carlo algorithm. For lp=60 bp the increase is faster than exponential and may be indicative of non linear effects coming from the polymer folding in presence of soft sites.
The output of these simulations may be readily measured in experiments in vitro on designed DNA and chromatin templates as done for HIV [89, 91], and may thus inform the mechanistic principles leading to DNA integration. Perhaps more importantly, however, these simulations suggest that the heterogeneity of the DNA (or chromatin) substrate in both mechanics and folding may affect TE expansion with potentially important and far-reaching consequences on the evolutionary paths and proliferative success of certain TEs in vivo.
It should be finally mentioned that other classes of transposases have been found to display enhanced efficiency on bent or geometrically deformed substrates. Most notably the DNA-bending class of proteins HMGB is found to enhance the efficiency of V(D)J recombination by RAG1-RAG2 [107, 108] and Sleeping Beauty transposition [109]. This suggests that the model introduced in Ref. [54] and described here could have a broader relevance to other classes of transposition and recombination.
Conclusions
It is now becoming increasingly clear that cell function, health and fate are correlated to 3D genome folding [25, 45]. TEs are intrinsically linked to 3D organisation as they are “living elements” within a complex multi-scale environment. In the last few years, there have been a handful of studies that started to interrogate how TEs shape genome organisation, from demarcating TAD boundaries [29–31] to harboring binding sites for architectural proteins [34]. It is thus now realized that TEs have profound implications in the fate and health of a cell – not only via the traditional pathway of genomic instability and epigenetic silencing – but also through the global regulation of genome folding.
Now, while this crucial relationship will certainly receive more attention in the future, in this commentary we argue that the other direction of the relationship, i.e. how 3D structure affects de novo TE insertions, is also of utmost importance. For example, biases in insertion patterns due to tissue-specific genome organisation in the germline (versus, for example, somatic cells) may create preferential pathways for genome evolution. In mammals, while the overall genome organisation is preserved in the germ line, the strength of specific features such as compartments and TADs varies [110–112]; far less is known in plants and significant differences between chromosome organization in germ line and somatic cells have been reported [113]. We suggest that the dissection of the interplay between 3D organisation and TE integration could be done by employing a “perturb-and-measure” strategy, i.e. by inducing TE expansion in a cell line whilst obtaining information on the 3D genome organisation and epigenetic states pre and post expansion. This approach may determine – also through the use of polymer physics models – which 3D and/or epigenetic features are associated with de novo TE insertions and thus detect insertion biases. Consequently, it will allow the generation of “topographical maps” of TE insertions in a given tissue-specific 3D genome organisation. Ultimately, understanding the preferential insertion of TEs may lead to a better understanding of genome and TE evolution or even inform better strategies to drive genomic variations in crops.
Availability of data and materials
Please contact corresponding author for data and code requests.
References
Deniz Ö, Frost JM, Branco MR. Regulation of transposable elements by DNA modifications. Nat Rev Genet. 2019; 20(14710064). https://doi.org/10.1038/s41576-019-0106-6.
Schnable PS, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, Minx P, Reily AD, Courtney L, Kruchowski SS, Tomlinson C, Strong C, Delehaunty K, Fronick C, Courtney B, Rock SM, Belter E, Du F, Kim K, Abbott RM, Cotton M, Levy A, Marchetto P, Ochoa K, Jackson SM, Gillam B, Chen W, Yan L, Higginbotham J, Cardenas M, Waligorski J, Applebaum E, Phelps L, Falcone J, Kanchi K, Thane T, Scimone A, Thane N, Henke J, Wang T, Ruppert J, Shah N, Rotter K, Hodges J, Ingenthron E, Cordes M, Kohlberg S, Sgro J, Delgado B, Mead K, Chinwalla A, Leonard S, Crouse K, Collura K, Kudrna D, Currie J, He R, Angelova A, Rajasekar S, Mueller T, Lomeli R, Scara G, Ko A, Delaney K, Wissotski M, Lopez G, Campos D, Braidotti M, Ashley E, Golser W, Kim H, Lee S, Lin J, Dujmic Z, Kim W, Talag J, Zuccolo A, Fan C, Sebastian A, Kramer M, Spiegel L, Nascimento L, Zutavern T, Miller B, Ambroise C, Muller S, Spooner W, Narechania A, Ren L, Wei S, Kumari S. The B73 Maize Genome: Complexity, Diversity, and Dynamics. J Sci. 2009; 326:1112.
Lee HE, Ayarpadikannan S, Kim HS. Role of transposable elements in genomic rearrangement, evolution, gene regulation and epigenetics in primates. Genes Genet Syst. 2016; 90(18805779):245. https://doi.org/10.1266/ggs.15-00016.
Chuong EB, Elde NC, Feschotte C. Regulatory activities of transposable elements: From conflicts to benefits. Nat Rev Genet. 2017; 18(14710064):71. https://doi.org/10.1038/nrg.2016.139.
Bousios A, Gaut BS. ScienceDirect Mechanistic and evolutionary questions about epigenetic conflicts between transposable elements and their plant hosts. Curr Opin Plant Biol. 2016; 30(1369-5266):123. https://doi.org/10.1016/j.pbi.2016.02.009.
Schatz DG, Ji Y. Nat Rev Immunol. 2011; 11(14741733):251. https://doi.org/10.1038/nri2941.
Payer LM, Burns KH. Transposable elements in human genetic disease. Nat Rev Genet. 2019; 1471-0056. https://doi.org/10.1038/s41576-019-0165-8.
Wicker T, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, Sanmiguel P, Schulman AH. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007; 8:973. https://doi.org/10.1038/nrg2165.
Lu L, Chen J, Robb SM, Okumoto Y, Stajich JE, Wessler SR. Tracking the genome-wide outcomes of a transposable element burst over decades of amplification. Proc Natl Acad Sci U S A. 2017; 114(10916490):E10550. https://doi.org/10.1073/pnas.1716459114.
Sanchez DH, Gaubert H, Drost HG, Zabet NR, Paszkowski J. High-frequency recombination between members of an LTR retrotransposon family during transposition bursts. Nat Commun. 2017; 8(20411723):1. https://doi.org/10.1038/s41467-017-01374-x.
Eickbush TH, Eickbush DG. Long-Term Stability of R2 Retrotransposons. Microbiol Spectr. 2014; 3:1. https://doi.org/10.1128/microbiolspec.MDNA3-0011.
Schroder ARW, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell. 2002; 110:521.
Craigie R, Bushman FD. HIV DNA integration. Cold Spring Harb Perspect Med. 2012; 2(21571422):1. https://doi.org/10.1101/cshperspect.a006890. http://arxiv.org/abs/arXiv:1011.1669v3.
Kvaratskhelia M, Sharma A, Larue RC, Serrao E, Engelman A. Molecular mechanisms of retroviral integration site selection. Nucleic Acids Res. 2014; 42(16):10209–25. https://doi.org/10.1093/nar/gku769.
Cheung S, Ma L, Chan PHW, Hu HL, Mayor T, Chen HT, Measday V. Ty1 integrase interacts with RNA Polymerase III-specific Subcomplexes to Promote Insertion of Ty1 Elements Upstream of Polymerase (Pol) III-transcribed Genes. J Biol Chem. 2016; 291(12):6396–411. https://doi.org/10.1074/jbc.M115.686840.
Bridier-Nahmias A, Tchalikian-Cosson A, Baller JA, Menouni R, Fayol H, Flores A, Saïb A, Werner M, Voytas DF, Lesage P. An RNA polymerase III subunit determines sites of retrotransposon integration. Science. 2015; 348(6234):585–8. https://doi.org/10.1126/science.1259114.
Gao X, Hou Y, Ebina H, Levin HL, Voytas DF. Chromodomains direct integration of retrotransposons to heterochromatin. Genome Res. 2008; 18(3):359–69. https://doi.org/10.1101/gr.7146408.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender Ma, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny La, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome,. Science. 2009; 326(october):289–93. https://doi.org/10.1126/science.1181369.
Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, Mirny LA, O’Shea CC, Park PJ, Ren B, Ritland Politz JC, Shendure J, Zhong S. The 4D nucleome project. Nature. 2017; 549(7671):219–26. https://doi.org/10.1038/nature23884.
Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure,. Nature. 2013; 502(7469):59–64. https://doi.org/10.1038/nature12593. NIHMS150003.
Kind J, Pagie L, Ortabozkoyun H, Boyle S, De Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, Van Steensel B. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 2013; 153(1):178–92. https://doi.org/10.1016/j.cell.2013.02.028.
Flyamer IM, Gassler J, Imakaev M, Ulyanov SV, Abdennur N, Razin SV, Mirny L, Tachibana-Konwalski K. Single-cell Hi-C reveals unique chromatin reorganization at oocyte-tozygote transition. Nature. 2017; 544(7648):1–17. https://doi.org/10.1038/nature21711.
Nozawa R-s, Boteva L, Soares DC, Naughton C, Dun AR, Ramsahoye B, Bruton PC, Saleeb RS, Arnedo M, Hill B, Duncan R, Maciver SK, Gilbert N. SAF-A regulates interphase chromosome structure through oligomerisation with chromatin- associated RNAs. Cell. 2017; 169(7):1214–122718. https://doi.org/10.1016/j.cell.2017.05.029.
Dong P, Tu X, Chu PY, Lü P, Zhu N, Grierson D, Du B, Li P, Zhong S. 3D Chromatin Architecture of Large Plant Genomes Determined by Local A/B Compartments. Mol Plant. 2017; 10(12):1497–509. https://doi.org/10.1016/j.molp.2017.11.005.
Stadhouders R, Filion GJ, Graf T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature. 2019; 569(7756):345–54. https://doi.org/10.1038/s41586-019-1182-7.
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485(7398):376–80. https://doi.org/10.1038/nature11082.
Wang J, Vicente-García C, Seruggia D, Moltó E, Fernandez-Miñán A, Neto A, Lee E, Gómez-Skarmeta JL, Montoliu L, Lunyak VV, Jordan IK, Craig NL. MIR retrotransposon sequences provide insulators to the human genome. Proc Natl Acad Sci U S A. 2015; 112(32):4428–37. https://doi.org/10.1073/pnas.1507253112.
Raviram R, Rocha PP, Luo VM, Swanzey E, Miraldi ER, Chuong EB, Feschotte C, Bonneau R, Skok JA. Analysis of 3D genomic interactions identifies candidate host genes that transposable elements potentially regulate. Genome Biol. 2018; 28(216):1–19. https://doi.org/10.1186/s13059-018-1598-7.
Zhang Y, Li T, Preissl S, Amaral ML, Grinstein JD, Farah EN, Destici E, Qiu Y, Hu R, Lee AY, Chee S, Ma K, Ye Z, Zhu Q, Huang H, Fang R, Yu L, Izpisua Belmonte JC, Wu J, Evans SM, Chi NC, Ren B. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat Genet. 2019; 51(9):1380–8. https://doi.org/10.1038/s41588-019-0479-7.
Kruse K, Diaz N, Enriquez-Gasca R, Gaume X, Torres-Padilla M-E, Vaquerizas JM. Transposable elements drive reorganisation of 3D chromatin during early embryogenesis. bioRxiv. 2019:523712. https://doi.org/10.1101/523712.
Sun JH, Zhou L, Emerson DJ, Phyo SA, Titus KR, Gong W, Gilgenast TG, Beagan JA, Davidson BL, Tassone F, Phillips-Cremins JE. Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell. 2018; 175(1):224–23815. https://doi.org/10.1016/j.cell.2018.08.005.
Kunarso G, Chia N-YY, Jeyakani J, Hwang C, Lu X, Chan Y-SS, Ng H-HH, Bourque G. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet. 2010; 42(7):631–34. https://doi.org/10.1038/ng.600.
Schmidt D, Schwalie PC, Wilson MD, Ballester B, Gonalves Â, Kutter C, Brown GD, Marshall A, Flicek P, Odom DT. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell. 2012; 148(1-2):335–48. https://doi.org/10.1016/j.cell.2011.11.058.
Choudhary MN, Friedman RZ, Wang JT, Jang HS, Zhuo X, Wang T. Co-opted transposons help perpetuate conserved higher-order chromosomal structures. bioRxiv. 2018; 314:485342. https://doi.org/10.1101/485342.
Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell. 2017; 169(5):930–94422. https://doi.org/10.1016/j.cell.2017.05.004. 095802.
Cournac A, Koszul R, Mozziconacci J. The 3D folding of metazoan genomes correlates with the association of similar repetitive elements. Nucleic Acids Res. 2016; 44(1):245–55. https://doi.org/10.1093/nar/gkv1292.
Winter DJ, Ganley ARD, Young CA, Liachko I, Schardl CL, Dupont PY, Berry D, Ram A, Scott B, Cox MP. Repeat elements organise 3D genome structure and mediate transcription in the filamentous fungus Epichloë festucae. PLoS Genet. 2018; 14(10):1–29. https://doi.org/10.1371/journal.pgen.1007467.
Grob S, Grossniklaus U. Invasive DNA elements modify the nuclear architecture of their insertion site by KNOT-linked silencing in Arabidopsis thaliana. Genome Biol. 2019; 20(1):1–15. https://doi.org/10.1186/s13059-019-1722-3.
Sultana T, Zamborlini A, Cristofari G, Lesage P. Integration site selection by retroviruses and transposable elements in eukaryotes. Nat Rev Genet. 2017; 18(5):292–308. https://doi.org/10.1038/nrg.2017.7.
Charlesworth B, Charlesworth D. The population dynamics of transposable elements. Genet Res Camb. 1983; 42:1–27. https://doi.org/10.1017/S0016672300021455.
Xue C, Goldenfeld N. Stochastic predator-prey dynamics of transposons in the human genome. Phys Rev Lett. 2016; 117(20):1–5. https://doi.org/10.1103/PhysRevLett.117.208101.
Le Rouzic A, Boutin TS, Capy P. Long-term evolution of transposable elements. Proc Natl Acad Sci U S A. 2007; 104(49):19375–80. https://doi.org/10.1073/pnas.0705238104.
Roessler K, Bousios A, Meca E, Gaut BS. Modeling interactions between transposable elements and the plant epigenetic response: A surprising reliance on element retention. Genome Biol Evol. 2018; 10(3):803–15. https://doi.org/10.1093/gbe/evy043.
Naughtin M, Haftek-Terreau Z, Xavier J, Meyer S, Silvain M, Jaszczyszyn Y, Levy N, Miele V, Benleulmi MS, Ruff M, Parissi V, Vaillant C, Lavigne M. DNA physical properties and nucleosome positions are major determinants of HIV-1 integrase selectivity. PLoS ONE. 2015; 10(6):1–28. https://doi.org/10.1371/journal.pone.0129427.
Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, Cavalli G. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell. 2017; 171(3):557–57224. https://doi.org/10.1016/j.cell.2017.09.043.
Cremer T, Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells,. Nat Rev Genet. 2001; 2(4):292–301. https://doi.org/10.1038/35066075.
Mirny LA. The fractal globule as a model of chromatin architecture in the cell,. Chromosome Res. 2011; 19(1):37–51. https://doi.org/10.1007/s10577-010-9177-0.
Brackley CA, Cates ME, Marenduzzo D. Facilitated Diffusion on Mobile DNA: Configurational Traps and Sequence Heterogeneity. Phys Rev Lett. 2012; 109(16):168103. https://doi.org/10.1103/PhysRevLett.109.168103. http://arxiv.org/abs/arXiv:1212.1601v1.
Michieletto D, Orlandini E, Marenduzzo D. Polymer model with Epigenetic Recoloring Reveals a Pathway for the de novo Establishment and 3D Organization of Chromatin Domains. Phys Rev X. 2016; 6(4):041047. https://doi.org/10.1103/PhysRevX.6.041047. http://arxiv.org/abs/1606.04653.
Michieletto D, Chiang M, Colì D, Papantonis A, Orlandini E, Cook PR, Marenduzzo D. Shaping epigenetic memory via genomic bookmarking. Nucleic Acids Res. 2018; 46(1):83–93. http://dx.doi.org/10.1093/nar/gkx1200. http://arxiv.org/abs/1709.01322.
Jost D, Carrivain P, Cavalli G, Vaillant C. Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res. 2014; 42(15):1–9. https://doi.org/10.1093/nar/gku698.
Bianco S, Lupiáñez DG, Chiariello AM, Annunziatella C, Kraft K, Schöpflin R, Wittler L, Andrey G, Vingron M, Pombo A, Mundlos S, Nicodemi M. Polymer physics predicts the effects of structural variants on chromatin architecture. Nat Genet. 2018; 50(5):662–7. https://doi.org/10.1038/s41588-018-0098-8.
Sanborn AL, Rao SSP, Huang S-C, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, Geeting KP, Gnirke A, Melnikov A, McKenna D, Stamenova EK, Lander ES, Aiden EL. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci. 2015; 112(47):201518552. https://doi.org/10.1073/pnas.1518552112.
Michieletto D, Lusic M, Marenduzzo D, Orlandini E. Physical principles of retroviral integration in the human genome. Nat Commun. 2019; 10(1):575. https://doi.org/10.1038/s41467-019-08333-8.
Eltsov M, MacLellan KM, Maeshima K, Frangakis AS, Dubochet J. Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc Natl Acad Sci U S A. 2008; 105(50):19732–7. https://doi.org/10.1073/pnas.0810057105.
Gennes PGD. Scaling Concepts in Polymer Physics: Cornell University Press; 1979. http://books.google.com/books?hl=en{&}lr={&}id=ApzfJ2LYwGUC{&}oi=fnd{&}pg=PA13{&}dq=Scaling+concepts+in+polymer+physics{&}ots=JbY95ezUU7{&}sig=9HVI12ZPi0MuFf0a1v{_}ZsrGhoqg.
Flory PJ. Principles of Polymer Chemistry. Ithaca: Cornell University Press; 1953.
Hafner AE, Krausser J, Šarić A. Minimal coarse-grained models for molecular self-organisation in biology. Curr Opin Struct Biol. 2019; 58:43–52. https://doi.org/10.1016/j.sbi.2019.05.018. http://arxiv.org/abs/1906.09349.
Brackley CA, Johnson J, Kelly S, Cook PR, Marenduzzo D. Simulated binding of transcription factors to active and inactive regions folds human chromosomes into loops, rosettes and topological domains. Nucleic Acids Res. 2016; 44(8):3503–12. http://dx.doi.org/10.1093/nar/gkw135. http://arxiv.org/abs/1511.01848.
Di Pierro M, Zhang B, Aiden EL, Wolynes PG, Onuchic JN. Transferable model for chromosome architecture. Proc Natl Acad Sci. 2016; 113(43):12168–73. https://doi.org/10.1073/pnas.1613607113.
Nuebler J, Fudenberg G, Imakaev M, Abdennur N, Mirny LA. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc Natl Acad Sci. 2018; 115(29):6697–706. https://doi.org/10.1073/pnas.1717730115.
Buckle A, Brackley CA, Boyle S, Marenduzzo D, Gilbert N. Polymer Simulations of Heteromorphic Chromatin Predict the 3D Folding of Complex Genomic Loci,. Mol Cell. 2018; 72(4):786–79711. https://doi.org/10.1016/j.molcel.2018.09.016.
Brackley CA, Johnson J, Michieletto D, Morozov AN, Nicodemi M, Cook PR, Marenduzzo D. Nonequilibrium Chromosome Looping via Molecular Slip Links. Phys Rev Lett. 2017; 119(13):138101. https://doi.org/10.1103/PhysRevLett.119.138101.
Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016; 15(9):2038–49. https://doi.org/10.1016/j.celrep.2016.04.085. 15334406.
Orlandini E, Marenduzzo D, Michieletto D. Synergy of topoisomerase and structural-maintenance-of-chromosomes proteins creates a universal pathway to simplify genome topology. Proc Natl Acad Sci. 2019; 116(17):8149–54. https://doi.org/10.1073/pnas.1815394116.
Pereira MCF, Brackley CA, Michieletto D, Annunziatella C, Bianco S, Chiariello AndreaM, Nicodemi M, Marenduzzo D. Complementary chromosome folding by transcription factors and cohesin. bioRxiv. 2018:32–34. https://doi.org/10.1101/305359.
Brackley CA, Brown JM, Waithe D, Babbs C, Davies J, Hughes JR, Buckle VJ, Marenduzzo D. Predicting the three-dimensional folding of cis -regulatory regions in mammalian genomes using bioinformatic data and polymer models. Genome Biol. 2016; 17(59):31–6. https://doi.org/10.1186/s13059-016-0909-0. http://arxiv.org/abs/arXiv:1601.02822v1.
Chiang M, Michieletto D, Brackley CA, Rattanavirotkul N, Mohammed H, Marenduzzo D, Chandra T. Lamina and Heterochromatin Direct Chromosome Organisation in Senescence and Progeria. bioRxiv. 2018:468561. https://doi.org/10.1101/468561.
Stadhouders R, Vidal E, Serra F, Di Stefano B, Le Dily F, Quilez J, Gomez A, Collombet S, Berenguer C, Cuartero Y, Hecht J, Filion GJ, Beato M, Marti-Renom MA, Graf T. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet. 2018; 50(2):238–49. https://doi.org/10.1038/s41588-017-0030-7.
Benedetti F, Dorier J, Burnier Y, Stasiak A. Models that include supercoiling of topological domains reproduce several known features of interphase chromosomes. Nucleic Acids Res. 2014; 42(5):2848–55. https://doi.org/10.1093/nar/gkt1353.
Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159(7):1665–80. https://doi.org/10.1016/j.cell.2014.11.021. http://arxiv.org/abs/1206.5533.
Brangwynne CP, Koenderink GH, MacKintosh FC, Weitz DA. Intracellular transport by active diffusion. Trends Cell Biol. 2009; 19(9):423–27. https://doi.org/10.1016/j.tcb.2009.04.004.
Caragine CM, Haley SC, Zidovska A. Surface Fluctuations and Coalescence of Nucleolar Droplets in the Human Cell Nucleus. Phys Rev Lett. 2018; 121(14):148101. https://doi.org/10.1103/PhysRevLett.121.148101.
Cho W-K, Spille J-H, Hecht M, Lee C, Li C, Grube V, Cisse II. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018; 361(6400):412–5. https://doi.org/10.1126/science.aar4199.
Brangwynne CP, Tompa P, Pappu RV. Polymer physics of intracellular phase transitions. Nat Phys. 2015; 11(11):899–904. https://doi.org/10.1038/nphys3532.
Erdel F, Rippe K. Formation of Chromatin Subcompartments by Phase Separation. Biophys J. 2018; 114(10):2262–70. https://doi.org/10.1016/j.bpj.2018.03.011.
Michieletto D, Gilbert N. Role of nuclear RNA in regulating chromatin structure and transcription. Curr Opin Cell Biol. 2019; 58:120–5. https://doi.org/10.1016/j.ceb.2019.03.007.
Khanna N, Zhang Y, Lucas JS, Dudko OK, Murre C. Chromosome dynamics near the sol-gel phase transition dictate the timing of remote genomic interactions. Nat Commun. 2019; 10(1):1–13. https://doi.org/10.1038/s41467-019-10628-9.
Hilbert L, Sato Y, Kimura H, Jülicher F, Honigmann A, Zaburdaev V, Vastenhouw N. Transcription organizes euchromatin similar to an active microemulsion. bioRxiv. 2018; 2:234112. https://doi.org/10.1101/234112.
Lucas JS, Zhang Y, Dudko OK, Murre C. 3D trajectories adopted by coding and regulatory DNA elements: First-passage times for genomic interactions. Cell. 2014; 158(2):339–52. https://doi.org/10.1016/j.cell.2014.05.036.
Shin Y, Chang YC, Lee DSW, Berry J, Sanders DW, Ronceray P, Wingreen NS, Haataja M, Brangwynne CP. Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell. 2018; 175(6):1481–149113. https://doi.org/10.1016/j.cell.2018.10.057.
Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. HIV integration site selection : Analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007; 17:1186–94. https://doi.org/10.1101/gr.6286907.2006b.
Marini B, Kertesz-Farkas A, Ali H, Lucic B, Lisek K, Manganaro L, Pongor S, Luzzati R, Recchia A, Mavilio F, Giacca M, Lusic M. Nuclear architecture dictates HIV-1 integration site selection. Nature. 2015; 521(7551):227–31. https://doi.org/10.1038/nature14226.
Pruss D, Bushman F, Wolffe A. Human immunodeficiency virus integrase directs integration to sites of severe DNA distortion within the nucleosome core. Proc Natl Acad Sci USA. 1994; 91(June):5913–7.
Pruss D, Reeves R, Bushman F, Wolffe A. The influence of DNA and nucleosome structure on integration events directed by HIV integrase,. J Biol Chem. 1994; 269(40):25031–41.
Lucic B, Chen H-C, Kuzman M, Zorita E, Wegner J, Minneker V, Wang W, Fronza R, Laufs S, Schmidt M, Stadhouders R, Roukos V, Vlahovicek K, Filion GJ, Lusic M. Spatially clustered loci with multiple enhancers are frequent targets of HIV-1 integration. Nat Commun. 2019; 10(1). https://doi.org/10.1038/s41467-019-12046-3.
Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med. 2005; 11(12):1287–9. https://doi.org/10.1038/nm1329.
Schrijvers R, Vets S, De Rijck J, Malani N, Bushman FD, Debyser Z, Gijsbers R. HRP-2 determines HIV-1 integration site selection in LEDGF/p75 depleted cells. Retrovirology. 2012; 9:1–7. https://doi.org/10.1186/1742-4690-9-84.
Benleulmi M, Matysiak J, Henriquez D, Vaillant C, Lesbats P, Calmels C, Naughtin M, Leon O, Skalka A, Ruff M, Lavigne M, Andreola M-L, Parissi V. Intasome architecture and chromatin density modulate retroviral integration into nucleosome. Retrovirology. 2015; 12(1):13. https://doi.org/10.1186/s12977-015-0145-9.
Serrao E, Krishnan L, Shun MC, Li X, Cherepanov P, Engelman A, Maertens GN. Integrase residues that determine nucleotide preferences at sites of HIV-1 integration: Implications for the mechanism of target DNA binding. Nucleic Acids Res. 2014; 42(8):5164–76. https://doi.org/10.1093/nar/gku136.
Pasi M, Mornico D, Volant S, Juchet A, Batisse J, Bouchier C, Parissi V, Ruff M, Lavery R, Lavigne M. DNA minicircles clarify the specific role of DNA structure on retroviral integration. Nucleic Acids Res. 2016; 44(16):7830–47. https://doi.org/10.1093/nar/gkw651.
Frenkel D, Smit B. Understanding Molecular Simulation: from Algorithms to Applications: Academic Press; 2001. http://www.theeuropeanlibrary.org/tel4/record/3000092731230?subject={~}Modelos+matematicos.http://books.google.com/books?hl=en{&}lr={&}id=5qTzldS9ROIC{&}oi=fnd{&}pg=PP2{&}dq=Understanding+molecular+simulation+from+Algorithms+to+Applications{&}ots=nFMN-qY9Qn{&}sig=7-KnLCa1uO7dF3RnqGcFhG378Pc.
Pryciak PM, Varmus HE. Nucleosomes, DNA-binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell. 1992; 69(5):769–80. https://doi.org/10.1016/0092-8674(92)90289-O.
Wolffe AP. Chromatin: Structure and Function. Cambridge: Elsevier Science; 2012. https://books.google.co.uk/books?id=cY2WLTwo0EIC.
Solovei I, Kreysing M, Lanctôt C, Kösem S, Peichl L, Cremer T, Guck J, Joffe B. Nuclear Architecture of Rod Photoreceptor Cells Adapts to Vision in Mammalian Evolution. Cell. 2009; 137(2):356–68. https://doi.org/10.1016/j.cell.2009.01.052.
Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu C-t, Zhuang X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016; 529(7586):418–22. https://doi.org/10.1038/nature16496.
Rao SSP, Huang S-C, Hilaire BGS, Engreitz JM, Perez EM, Kieffer-Kwon K-R, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, Huang X, Shamim MS, Shin J, Turner D, Ye Z, Omer AD, Robinson JT, Schlick T, Bernstein BE, Casellas R, Lander ES, Aiden EL. Cohesin Loss Eliminates All Loop Domains. Cell. 2017; 171(2):305–32024. https://doi.org/10.1016/j.cell.2017.09.026.
Gilbert N, Boyle S, Fiegler H, Woodfine K, Carter NP, Bickmore WA. Chromatin architecture of the human genome: Gene-rich domains are enriched in open chromatin fibers. Cell. 2004; 118(5):555–66. https://doi.org/10.1016/j.cell.2004.08.011.
Chiang M, Michieletto D, Brackley CA, Rattanavirotkul N, Mohammed H, Marenduzzo D, Chandra T. Polymer Modeling Predicts Chromosome Reorganization in Senescence. Cell Rep. 2019; 28(12):3212–236. https://doi.org/10.1016/j.celrep.2019.08.045.
Halverson JD, Smrek J, Kremer K, Grosberg AY. From a melt of rings to chromosome territories: The role of topological constraints in genome folding. Rep Prog Phys. 2014; 77(2). http://dx.doi.org/10.1088/0034-4885/77/2/022601. http://arxiv.org/abs/1311.5262.
Collepardo-Guevara R, Schlick T. Chromatin fiber polymorphism triggered by variations of DNA linker lengths. Proc Natl Acad Sci U S A. 2014; 111(22):8061–6. https://doi.org/10.1073/pnas.1315872111.
Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, O’Shea CC. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017; 357(6349). https://doi.org/10.1126/science.aag0025. 15334406.
Taganov KD, Cuesta I, Cirillo LA, Katz RA, Zaret KS, Skalka AM. Integrase-Specific Enhancement and Suppression of Retroviral DNA Integration by Compacted Chromatin Structure In Vitro. J Virol. 2004; 78(11):5848–55. https://doi.org/10.1128/JVI.78.11.5848.
Fosado YAG, Michieletto D, Marenduzzo D. Dynamical Scaling and Phase Coexistence in Topologically-Constrained DNA Melting. Phys Rev Lett. 2017; 119(118002):1–14. https://doi.org/10.1103/PhysRevLett.119.118002. http://arxiv.org/abs/1703.08367.
Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nat Rev Genet. 2015; 16(10):583–97. https://doi.org/10.1038/nrg3961.
Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009; 11(6):753–60. https://doi.org/10.1038/ncb1882.
Ciubotaru M, Trexler AJ, Spiridon LN, Surleac MD, Rhoades E, Petrescu AJ, Schatz DG. RAG and HMGB1 create a large bend in the 23RSS in the V(D)J recombination synaptic complexes. Nucleic Acids Res. 2013; 41(4):2437–54. https://doi.org/10.1093/nar/gks1294.
Aidinis V, Bonaldi T, Beltrame M, Santagata S, Bianchi ME, Spanopoulou E. The RAG1 Homeodomain Recruits HMG1 and HMG2 To Facilitate Recombination Signal Sequence Binding and To Enhance the Intrinsic DNA-Bending Activity of RAG1-RAG2. Mol Cell Biol. 1999; 19(10):6532–42. https://doi.org/10.1128/mcb.19.10.6532.
Zayed H, Izsvák Z, Khare D, Heinemann U, Ivics Z. The DNA-bending protein HMGB1 is a cellular cofactor of Sleeping Beauty transposition. Nucleic Acids Res. 2003; 31(9):2313–22. https://doi.org/10.1093/nar/gkg341.
Battulin N, Fishman VS, Mazur AM, Pomaznoy M, Khabarova AA, Afonnikov DA, Prokhortchouk EB, Serov OL. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol. 2015; 16(1). https://doi.org/10.1186/s13059-015-0642-0.
Vara C, Paytuví-Gallart A, Cuartero Y, Le Dily F, Garcia F, Salvà-Castro J, Gómez-H L, Julià E, Moutinho C, Aiese Cigliano R, Sanseverino W, Fornas O, Pendás AM, Heyn H, Waters PD, Marti-Renom MA, Ruiz-Herrera A. Three-Dimensional Genomic Structure and Cohesin Occupancy Correlate with Transcriptional Activity during Spermatogenesis. Cell Rep. 2019; 28(2):352–3679. https://doi.org/10.1016/j.celrep.2019.06.037.
Alavattam KG, Maezawa S, Sakashita A, Khoury H, Barski A, Kaplan N, Namekawa SH. Attenuated chromatin compartmentalization in meiosis and its maturation in sperm development. Nat Struct Mol Biol. 2019; 26(3):175–84. https://doi.org/10.1038/s41594-019-0189-y.
Zhou S, Jiang W, Zhao Y, Zhou DX. Single-cell three-dimensional genome structures of rice gametes and unicellular zygotes. Nat Plants. 2019; 5(8):795–800. https://doi.org/10.1038/s41477-019-0471-3.
Acknowledgements
The authors thank Brandon Gaut for reading the manuscript and the anonymous referees for excellent feedback and comments.
Funding
HWN, AB and DM acknowledge support of EPSRC and BBSRC through the Physics of Life Network under the form of a sandpit grant. HWN, DB and DM would also like to acknowledge the networking support by the Cooperation in Science and Technology (COST) action “European Topology Interdisciplinary Action” (EUTOPIA) CA17139. HWN is supported by The Royal Society (award numbers UF160138). AB is supported by The Royal Society (award numbers UF160222 and RGF/R1/180006).
Author information
Authors and Affiliations
Contributions
All authors wrote the paper. DM performed simulations. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bousios, A., Nützmann, HW., Buck, D. et al. Integrating transposable elements in the 3D genome. Mobile DNA 11, 8 (2020). https://doi.org/10.1186/s13100-020-0202-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13100-020-0202-3