
Abstract
Background
The RAS/MAPK pathway has been intensively studied in cancer. Constitutive activation of ERK1 and ERK2 is frequently found in cancer cells from a variety of tissues. In clinical practice and clinical trials, small molecules targeting receptor tyrosine kinases or components in the MAPK cascade are used for treatment. MEK1 and MEK2 are ideal targets because these enzymes are physiologically important and have narrow substrate specificities and distinctive structural characteristics. Despite success in pre-clinical testing, only two MEK inhibitors, trametinib and cobimetinib, have been approved, both for treatment of BRAF-mutant melanoma. Surprisingly, the efficacy of MEK inhibitors in other tumors has been disappointing. These facts suggest the need for a different approach. We here consider transcription factor ETS1 and ETS2 as alternate therapeutic targets because they are major MAPK downstream effectors.
Main text
The lack of clinical efficacy of MEK inhibitors is attributed mostly to a subsequent loss of negative feedback regulation in the MAPK pathway. To overcome this obstacle, second-generation MEK inhibitors, so-called “feedback busters,” have been developed. However, their efficacy is still unsatisfactory in the majority of cancers. To substitute ETS-targeted therapy, therapeutic strategies to modulate the transcription factor in cancer must be considered. Chemical targeting of ETS1 for proteolysis is a promising strategy; Src and USP9X inhibitors might achieve this by accelerating ETS1 protein turnover. Targeting the ETS1 interface might have great therapeutic value because ETS1 dimerizes itself or with other transcription factors to regulate target genes. In addition, transcriptional cofactors, including CBP/p300 and BRD4, represent intriguing targets for both ETS1 and ETS2.
Conclusions
ETS-targeted therapy appears to be promising. However, it may have a potential problem. It might inhibit autoregulatory negative feedback loops in the MAPK pathway, with consequent resistance to cell death by ERK1 and ERK2 activation. Further research is warranted to explore clinically applicable ways to inhibit ETS1 and ETS2.
Similar content being viewed by others

Introduction
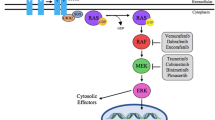
Mitogen-activated protein kinases (MAPKs) are serine (Ser) or threonine (Thr) protein kinases that respond to stimulation from extracellular growth factors through specific cell surface receptors [1,2,3,4]. They are a part of major signaling cascades. Among MAPKs, extracellular signal-regulated kinases 1 and 2 (ERK1 and ERK2) have been most characterized (Fig. 1). Activated receptor tyrosine kinases (RTKs) recruit adaptor proteins and guanine nucleotide exchange factors (GEFs: SOS1 and SOS2) to trigger RAS (HRAS, KRAS, or NRAS), which drives the formation of high-activity homo- or heterodimers of RAF (also known as MAPKKK: ARAF, BRAF, or CRAF), causing phosphorylation and activation of MEK1 and MEK2 (MAPKK) with consequent ERK1 and ERK2 (classical MAPK) stimulation. Numerous proteins are identified as their substrates, among which transcription factors ETS1, ETS2, and AP-1 are particularly important [1, 4]. They regulate expression of matrix metalloproteases, BCL-2 family members, and D-type cyclins to mediate cellular invasion and migration, cell survival and anti-apoptosis, and entry into the S phase from the G1 phase in the cell cycle [5,6,7,8,9,10,11,12].

Negative feedback regulates the RAS/MAPK pathway. Although MAPK signaling was previously considered linear, autoregulatory negative feedback loops precisely control signaling output. MEK inhibitors reduce activity of ERK1 and ERK2 and then relieve the feedback inhibition of RAF, resulting in enhancement of RAF kinase activity. Likewise, ETS1-targeted therapy may activate ERK1 and ERK2 by inhibiting DUSP6, which is an ETS1 transcriptional target
The RAS/MAPK pathway has been intensively studied [1,2,3,4], with constitutive activation of ERK1 and ERK2 found frequently in human cancer cells from a variety of tissues (e.g., lung, pancreas, colon, ovary, kidney, skin, and thyroid) [13]. Amplification, overexpression, or mutations in RTKs and genetic alterations in upstream components of the MAPK pathway, including KRAS, NRAS, HRAS, CRAF, BRAF, MEK1, and MEK2, alter cell signaling in tumors. In clinical practice and clinical trials, small molecules targeting RTKs or components in the MAPK cascade are used to treat cancer [1, 3, 4]. MEK1 and MEK2 are ideal targets; not only do they play a key role in tumor development and progression [3, 4], they have narrow substrate specificities and distinctive structural characteristics.
MEK activation through the MAPK signaling cascade is necessary for mammalian cell transformation, and constitutively active MEK mutants promote transformation of fibroblast cells [14, 15]. Furthermore, MEK inhibitors inhibit growth of xenografted human and murine colon carcinomas [16], a mechanism we have studied [11]. Treating colon cancer cells with small-molecule MEK inhibitors blocked both CDK4 and CDK2 kinase activity and arrested G1 growth. MEK1 and MEK2 also have a crucial role in inhibition of apoptosis [17, 18]: their downstream effectors ERK1 and ERK2 phosphorylate a BH3-only protein Bim at Ser69, targeting it for degradation via the proteasome-ubiquitin pathway.
ERK1 and ERK2 are the only known physiological substrates of MEK1 and MEK2 [1,2,3,4]—a narrow specificity that allows the development of MEK inhibitors with fewer off-target side effects. MEK1 and MEK2 are dual-specificity protein kinases and phosphorylate both tyrosine (Tyr) and Thr residues of ERK1 and ERK2—Tyr204 and Thr202 in ERK1 and Tyr187 and Thr185 in ERK2—rendering them active [19]. On the other hand, MEK1 and MEK2 themselves are phosphorylated and activated by RAF in two key Ser residues in the regulatory loop—Ser218 and Ser222 in MEK1 and Ser222 and Ser226 in MEK2 [20].
MEK inhibitors are divided into two groups, ATP-competitive and -noncompetitive [21], although most of the known MEK inhibitors are the latter [21]. By not competing directly for the ATP-binding site, they avoid competition with high intracellular ATP levels [22]. Structural analysis of MEK1 and MEK2 with PD184352, a putative non-competitive inhibitor, has revealed that the molecules possess a unique allosteric inhibitor-binding pocket adjacent to, but separate from, the ATP-binding site [23]. Once the MEK inhibitor binds the pocket, several conformational changes follow, causing MEK1 and MEK2 to be locked into a catalytically inactive state. These facts may provide an explanation for why this class of MEK inhibitors has shown keen specificity and high potency.
However, despite promising drug targets and success in pre-clinical testing, only two MEK inhibitors, trametinib (Mekinist) and cobimetinib (Cotellic), have been approved, both for the treatment of BRAF-mutant melanoma [3, 4, 24, 25]. The efficacy of MEK1/2 inhibitors in other tumors has been more disappointing [3, 4]. These facts suggest the need for an alternative approach. To this end, we present our perspective on ETS-targeted therapy.
Most MEK inhibitors have limited clinical efficacy
Numerous potent, selective allosteric MEK inhibitors have been developed and have undergone clinical evaluation of their ability to inhibit tumor growth [3, 4]. Preclinical studies showed efficient inhibition of phosphorylation of ERK1 and ERK2, which correlates with potent growth inhibition in cancer cell lines with mutant BRAF or RAS with elevated phosphorylation of MEK1 and MEK2 [3, 4, 26, 27]. However, most MEK inhibitors have demonstrated limited clinical efficacy as single-agent therapies [3, 4]. Only trametinib (see above) showed improved progression-free and overall survival both as a single agent and in combination with the BRAF inhibitor, dabrafenib [28,29,30]. More recently, another MEK inhibitor, cobimetinib—when used in combination with the BRAF-inhibitor, vemurafenib—was reported to improve progression-free survival among patients with BRAF V600-mutated metastatic melanoma [25]. These facts underscore the challenge of bringing MEK inhibitors from bench to bedside.
Loss of negative feedback regulation may reduce the clinical efficacy of MEK inhibitors
Loss of negative feedback regulation in the MAPK pathway after MEK inhibition could be a major cause for the lack of clinical efficacy [3, 4]. MAPK signaling was once considered a linear and relatively simple receptor-to-nucleus pathway [31,32,33]. However, we now know that autoregulatory negative feedback loops in the MAPK pathway precisely control signaling output (Fig. 1) [33]. In cancer, both inhibition and hyperactivation of ERK 1 and ERK2 cause growth inhibition or senescence [34]. Thus, their activity is maintained within a narrow threshold range. For example, ERK1 and ERK2 feedback phosphorylates CRAF and BRAF, which impairs their ability to bind to RAS in the plasma membrane and/or disrupts heterodimeric association of BRAF with CRAF, causing attenuation of RAF protein kinase activity [33]. In contrast, MEK inhibitors reduce the activity of ERK1 and ERK2 and then relieve the feedback inhibition of RAF, resulting in enhancement of RAF kinase activity [35]. Consequently, RAF’s downstream effectors MEK1 and MEK2, and then ERK1 and ERK2, are paradoxically activated. This was recently identified as adaptive drug resistance, a subtype of primary (intrinsic or innate) drug resistance [36]. Targeted therapy inhibits the oncogenic pathway but also relieves the negative feedback [37]. Consequently, the targeted cell signaling is paradoxically activated. This rebound effect appears immediately after exposure of cancer cells to the inhibitor, which enables them to survive showing little primary response [38].
Second-generation MEK inhibitors are feedback busters
To overcome adaptive drug resistance, second-generation MEK inhibitors, so-called “feedback busters,” were developed: trametinib, GDC-0623, and RO5126766 (CH5126766) [3, 4]. The compounds inhibit not only the ability of MEK1 and MEK2 to elevate ERK1 and ERK2, but also impair the ability of RAF to phosphorylate MEK1 and MEK2 by disrupting the conformation of the activation loop of MEK1 and MEK2. Among them, only trametinib (see above) has been approved. Other MEK inhibitors, including GDC-0623 and RO5126766, require further study to support their safety and efficacy [3, 39, 40]. In BRAF-mutated melanoma, CRAF and RAS activities are diminished by activated BRAF [3, 4]. Thus, the combination of BRAF and MEK inhibitors (dabrafenib and trametinib) is remarkably effective [28,29,30]. Likewise, although cobimetinib is a first-generation MEK inhibitor, its addition to the BRAF inhibitor vemurafenib significantly increases the durable response rate over single-agent BRAF-inhibitor therapy in patients with BRAF V600-mutated metastatic melanoma [25].
Previous studies have uncovered MEK-independent but kinase-dependent functions of CRAF. For example, CRAF was found directly to phosphorylate and inactivate retinoblastoma protein (RB), leading to cell-cycle progression [41]. Likewise, phosphorylated-CRAF at Ser338 was reported to localize to the mitotic spindle to promote mitosis in cancer cells [42]. Thus, a combination of MEK and CRAF inhibitors may also prevent the non-MAPK pathways downstream of CRAF. Although the biological phenomena are less relevant to RAF protein kinase activation after MEK inhibition, CRAF was reported to have kinase- and MEK-independent functions in cancer cells [43].
Cyclin D1 plays a critical role as a downstream effector molecule in the MAPK pathway
Cyclin D1 is a major transcriptional target of ERK1 and ERK2. Given the importance of the MAPK signaling pathway for G1 to S cell-cycle transition [11, 12, 44], it could serve as a biomarker to determine the clinical efficacy of MAPK-targeted therapy.
The critical role of cyclin D1 for MAPK-mediated oncogenesis was established first in the murine model for skin cancer [45] and later for breast cancer [46]. In HER2-driven or oncogenic HRAS-driven breast cancer in mice, tumor development was specifically protected by depleting cyclin D1 or CDK4 or expressing the CDK4- or CDK6-specific inhibitor INK4A or a dominant-negative mutant form of K112E cyclin D1 [46,47,48,49,50].
This finding led to a new therapeutic opportunity targeting the cyclin D1-CDK4/6 complex by a CDK4/6 inhibitor, palbociclib (PD0332991) [51,52,53]. This inhibits proliferation of estrogen receptor (ER)-positive luminal breast cancer cell lines in the presence or absence of HER2 amplification [51]. In fact, ER-positive breast cancer cells highly elevate the activity of ERK1 and ERK2 through HER2- and PKCδ-mediated RAS activation [52]. Consistent with the preclinical study, patients with ER-positive breast cancer showed the best clinical responses to palbociclib in combination with endocrine therapy [54]. Although other clinical trials have shown single-agent activity in mantle-cell lymphoma, liposarcoma, and teratoma with a manageable toxicity profile [53], expanding the use of CDK4/6 inhibitors beyond ER-positive breast cancer is challenging—a circumstance that may be attributable to redundancy in function among different CDKs [11].
Mouse embryonic fibroblasts can proliferate with CDK1 as the sole cell cycle-associated CDK [55, 56]. Likewise, most cell types from CDK4 and CDK6 double-knockout mice proliferate normally [57]. Moreover, D-cyclins can associate with CDK2 to drive G1-S cell-cycle transition [57]. These facts may suggest redundancy among CDKs, and that selection of appropriate target groups for CDK4/6 inhibition relies on successful identification of the tumor type [53]. Thus, tumors that do not depend on CDK4/6 for G1-S transition and/or can rescue CDK4/6 inhibition by the activity of other CDKs may require alternative approaches, including the use of compounds that affect cyclin D1 transcription or protein turnover and combination therapies that target multiple endpoints of cyclin D1 action simultaneously [50].
ETS1 and ETS2 transcription factors are major downstream effectors of RAS/MAPK signaling and cooperate with AP-1 transcription factor to regulate target genes
Studies with the polyomavirus enhancer region revealed that ETS family transcription factors are major downstream effectors of RAS/MAPK signaling [58]. The RAS/MAPK response sequence in the polyomavirus enhancer region consists of adjacent binding sites for ETS and AP-1 family transcription factors [59,60,61]. Also, ETS1 or ETS2 cooperates with c-Fos and c-Jun (components of AP-1) to activate transcription from the polyomavirus enhancer domain [62]. Likewise, downstream target genes of RAS/MAPK in humans often have AP-1 DNA binding sequence adjacent to ETS binding sites in the promoter regions. For example, ETS1, which autoregulates its transcript, has ETS and AP-1 binding sites in the ETS1 promoter regulatory region [63, 64]. Similarly, the minimum 5′ sequence of CCND1 (encoding cyclin D1) that retains responsiveness to RAS has both ETS and AP-1 binding sites [9, 10, 44].
There are 28 characterized ETS family members in humans [65]. Among them, 21 are phosphorylated by ERK2 in vitro [66], although not all equally. For example, ERG, FLI1, ETV1 and ETV4 are barely expressed in normal epithelial cells and most of RAS-transformed cells [58, 66, 67]. In contrast, ETS1 and ETS2 are particularly important, and their deletion has been shown to inhibit transformation caused by G12V HRAS in mouse embryonic fibroblasts [68]. ETS1 and ETS2 are respectively phosphorylated by ERK1/2 at Thr38 and Thr72; this enhances association with p300 or CREB binding protein (CBP) transcriptional co-activator, resulting in an increase in the transactivational activity of their target genes [69,70,71]. These observations suggest that ETS1 and ETS2 are major effectors of RAS/MAPK signaling, and thus can be alternative targets for the RAS/MAPK pathway. In support of this, because the inhibition of ERK1/2 by MEK inhibitors triggers an adaptive drug response, we may, by targeting ETS1 and ETS2—the downstream effectors of ERK1/2—avert this limitation.
ETS1 and ETS2 have both distinct and redundant roles
ETS1 and ETS2 expression is ubiquitous, although cell-type-dependent [67]. ETS1 is expressed at higher levels in the spleen and thymus; ETS2 is elevated in the brain, fetal liver, muscle, and uterus. In the lung, both genes are expressed at the same elevated level. In mice ETS2 gene manipulation causes embryonic death before 8.5 days’ gestation owing to defects in extraembryonic tissue rather than to a major embryonic anomaly [72]. In contrast, ETS1-deficient mice are viable, but demonstrate abnormalities in the differentiation of all lymphoid lineages [73, 74]. These observations suggest that ETS1 and ETS2 have distinct roles.
Likewise, a recent study demonstrated that ETS1, but not ETS2, is necessary for cell migration after RAS/MAPK activation in DU145 prostate cancer cells [58]. Similar observations were reported in ETS1-mediated epithelial cell morphogenesis after activation of MET-RAS-MAPK signaling by hepatocyte growth factor [75].
However, ETS1 and ETS2 depletion synergistically inhibits the RAS-induced cellular transformation in mouse embryonic fibroblast cells [68]. Thus, ETS1 and ETS2 would have a redundant role for the transforming effects of oncogenic RAS.
Inhibiting or modulating ETS1 transcription factor
Phosphorylated Tyr283 protects ETS1, but not ETS2, from proteolysis, which may allow the development of ETS1-specific small-molecule inhibitors [76, 77].
ETS1 protein is destabilized after phosphorylation at Ser276 and Ser282 by calcium/calmodulin-dependent protein kinase II (CAMK2) (Fig. 2) [78, 79]. The phosphorylation enhances an association with ring finger and WD repeat domain 2 protein (RFWD2 or COP1), which is an E3 ubiquitin ligase [80]. Subsequently, phosphorylated-ETS1 is degraded through the ubiquitin–proteasome pathway. Conversely, c-Src or YES1 phosphorylates ETS1 at Tyr283, which reverses CAMK2-dependent destabilization of ETS1 protein [79]. Thus, Src inhibitors such as dasatinib and saracatinib may accelerate rapid turnover of ETS1 protein and might prevent transactivation of ETS1 target genes (Fig. 2). WP1130 is a partly selective deubiquitinase inhibitor for USP9X, USP5, USP14, and UCH37 [81]. A recent report demonstrated that USP9X prevents ETS1 ubiquitination and thereby stabilizes the protein [77]. Thus, WP1130 may accelerate ETS1 protein degradation (Fig. 2) and inhibit transactivation of ETS1 target genes.

The complex containing RFWD2/COP1 mediates ubiquitination of ETS1. The E3 ubiquitin ligase RFWD2/COP1 recognizes ETS1 in a CAMK2-mediated Ser276 and Ser282 phosphorylation-dependent manner. Src or YES1 phosphorylates ETS1 at Tyr283, which reverses CAMK2-dependent destabilization of ETS1 protein. The deubiquitinase USP9X prevents EST1 ubiquitination and stabilizes the protein. Thus, dasatinib and WP1130 (Src and USP9X inhibitors, respectively) might target ETS1 for proteolysis
Targeting the ETS1 interface might be another approach [82]. One of the early attempts was an inhibition of ETS1-DNA binding interaction by the use of oligonucleotides that mimic the ETS1-binding sites [83]. However, the core ETS binding sequence is shared by various ETS family members, and thus this strategy may not be specific for ETS1 inhibition. In contrast, targeting ETS1 protein–protein interaction might be. ETS1 dimerizes itself or other transcription factors [84,85,86,87,88]. Homodimeric interactions through the ETS domain may play a role in cooperative binding to repeated DNA elements [86], whereas heterodimeric interactions might regulate tissue-specific gene expression [87, 88]. Although ETS1 protein–protein Interactions have great therapeutic promise, further studies are warranted to develop small-molecule inhibitors targeting them.
Targeting transcriptional co-factors of ETS1 and ETS2
Transcriptional cofactors might represent fascinating therapeutic targets for both ETS1 and ETS2 [76]. These assemble on transcription-binding DNA sequences with transcription factors to influence RNA polymerase II activity (Fig. 3). ERK1/2-phosphorylated ETS1 at Thr38 and Ser41, and ETS2 at Thr72 and Ser75, directly interact with two closely related transcriptional co-activating proteins, CBP and p300 (Fig. 3) [89]. The association promotes the assembly of RNA polymerase II and the basal machinery at the initiation of transcription. Indeed, the recruitment of CBP or p300 enhances transactivation of ETS1 and ETS2 target genes more than 20 times [89]. Thus, the RAS/MAPK signaling pathway activates ETS1 and ETS2 by promoting a unique binding interface with p300 or CBP. Conversely, inhibition of ERK1/2 by a MEK inhibitor disrupts the interface with p300/CBP, decreasing transcriptional activity of ETS1 and ETS2 (Fig. 3).

ETS1/2 interacts with CBP/p300 to transactivate target genes. ERK1/2-phosphorylated ETS1 at Thr38 and Ser41, or ETS2 at Thr72 and Ser75, which enhances an association with two closely related transcriptional co-activating proteins, CBP and p300. The binding promotes the assembly of RNA polymerase II and the basal machinery at the initiation of transcription. AKT is likely to activate the ETS1/2-containing transcription factor-enhancer complex by phosphorylating CBP or p300, histone acetyltransferases (HAT) that possess bromodomain (BRD). BRD4 is often co-localized with ETS1 and probably with ETS2 in transcription factor-enhancer complex. Thus, we may inhibit ETS1/2 activity by trametinib (MEK inhibitor), apitolisib (PI3K/mTOR inhibitor), C646 (CBP HAT inhibitor), PF-CBP1 or SGC-CBP30 (CBP BRD inhibitor), or JQ1 or OTX015 (BRD4 inhibitor)
Likewise, AKT might activate the ETS1- or ETS2-containing transcription factor-enhancer complex by phosphorylating its co-activator p300 or CBP (Fig. 3) [44, 90,91,92,93,94]. In fact, AKT phosphorylates p300 at Ser1834, which is essential for its transcription from the promoter of intercellular adhesion molecule-1 [95], whose transcription is also activated by ETS1 and ETS2 [96, 97]. This possibility was supported by our protein motif analysis [44]. CBP has highly stringent potential AKT phosphorylation sites at Ser381, Ser1733 and Thr1833, all of which are in CBP’s CH1 and CH2/CH3 domains, which interact with ETS1 [98]. Thus, inhibition of AKT might attenuate CBP/p300 activity, resulting in reduction of transactivation of ETS1 and ETS2 target genes (Fig. 3).
CBP and p300 are not only transcriptional co-activators but also histone acetyl-transferases (HAT) that acetylate both histone and non-histone proteins (Fig. 3) [99, 100]. Moreover, they possess bromodomain (BRD), which recognizes acetylated-lysine residues on proteins (Fig. 3) [100]. Thus, if we could decrease the HAT activity of CBP/p300 or perturb BRD-mediated protein–protein interactions, this might prevent chromatin remodeling and ETS1/2 binding to DNA and transcriptional regulatory complexes (Fig. 3). Recently, highly selective small-molecule inhibitors for CBP/p300, including SGC-CBP30 and PF-CBP1, were developed with a structure-based design to inhibit CBP BRD [101, 102].
Likewise, a recent study demonstrated that BRD-containing protein 4 (BRD4) is highly enriched at enhancers associated with genes involved in multiple profibrotic pathways, where BRD4 is co-localized with profibrotic transcription factors, including ETS1, SRF, SMAD3, and NF-κb/p65 (Fig. 3) [103]. Thus, it may be possible to target ETS1/2 by inhibiting BRD4 with small-molecule inhibitors such as JQ1 and OTX015 [104, 105].
Hormonal therapy may augment ETS-targeted therapy
Given that selection of appropriate target groups for ETS1/2 inhibition relies on successful identification of tumor type, ETS-targeted therapy may have a synergistic benefit with standard therapies.
The transcriptional function of the androgen receptor (AR) is essential for the genesis and development of prostate cancer [106]. A recent report demonstrated that ETS1-binding sequences were specifically enriched in AR-targeted genes [107]. Likewise, estrogen receptor α (ERα) associates directly with ETS1 to stimulate estradiol-dependent growth in breast cancer and neuroblastoma cells [108, 109]. These observations suggest that simultaneous treatments of these cancers with ETS1 and hormonal therapy may enhance clinical outcomes.
A potential problem of ETS-targeted therapy
As described above, the ETS1-containing transcription factor-enhancer complex could be disrupted with small-molecule inhibitors by specifically targeting ETS1 for proteolysis. However, the redundancy between ETS1 and ETS2 is a possible drawback of an ETS1-specific therapy, as ETS2 might subsequently compensate for ETS1. Likewise, it is possible that ETS1/2-targeted therapy may inhibit autoregulatory negative feedback loops, causing paradoxical activation of ERK1 and ERK2.
We recently studied the mechanism of ERK1/2 activation after EGFR inhibition in non-small cell lung cancer (NSCLC) harboring EGFR mutations [44, 90,91,92,93,94]. Our study demonstrated that EGFR inhibition in lung cancer cells generates a drug-tolerant subpopulation by blocking AKT activity and thus inactivating ETS1 function. The remaining cells enter a dormant, non-dividing state because of the inhibited transactivation of ETS1 target genes, cyclins D1, D3, and E2. Moreover, ETS1 inactivation inhibits transcription of dual specificity phosphatase 6 (DUSP6), a negative regulator specific for ERK1/2. The resulting activation of ERK1/2 combines with c-Src to renew activation of the RAS/MAPK pathway, causing increased cell survival by accelerating BIM protein turnover. By analogy, ETS-targeted therapy may activate ERK1 and ERK2 by inhibiting DUSP6.
However, conflicting data exist regarding the impact of ETS1 inhibition on MAPK signaling. A recent report has shown that ETS1 knockdown in DU145 prostate cancer cells activates dual specificity phosphatase 4 (DUSP4), DUSP6, and sprouty RTK-signaling antagonist 4 (SPRY4) [58]. Because DUSP4/6 and SPRY4 are negative regulators for ERK1/2 and RTK, respectively [31, 61, 110], these observations may suggest that ETS1 is required for robust RAS/ERK pathway activation, and reducing its activity would attenuate the activity of ERK1 and ERK2. This is an area where further study is warranted.
Conclusions
Efficacy of MEK inhibitors has been unsatisfactory in most tumors, despite successful pre-clinical testing. This fact has prompted us to develop an alternative approach. In this article, we have proposed transcription factor ETS1 and ETS2 as alternate therapeutic targets because they are major effectors of RAS/MAPK signaling. Chemical targeting of ETS1 for proteolysis would be among the few curative strategies in cancer therapeutics. Src and USP9X inhibitors might accomplish this by accelerating ETS1 protein turnover. Likewise, targeting ETS1 interface might have great therapeutic promise because ETS1 dimerizes itself or other transcription factors to regulate transcriptional target genes. Also, transcriptional cofactors of ETS1 and ETS2, including CBP/p300 and BRD4, may represent the other fascinating therapeutic targets around the transcription factor-enhancer complex. However, there exists a potential issue in ETS-targeted therapy. The remedy may inhibit autoregulatory negative feedback loops in the MAPK pathway, which might cause resistance to cell death by paradoxically activating ERK1 and ERK2. Further research is warranted to explore ways to inhibit ETS1 and ETS2 for clinical application.
Abbreviations
- MAPK:
-
mitogen-activated protein kinase
- Ser:
-
serine
- Thr:
-
threonine
- Tyr:
-
tyrosine
- ERK1 and ERK2:
-
extracellular signal-regulated kinase 1 and 2
- RTK:
-
receptor tyrosine kinase
- RB:
-
retinoblastoma protein
- ER:
-
estrogen receptor
- PKC:
-
protein kinase C
- CDK:
-
cyclin-dependent kinase
- CBP:
-
CREB binding protein
- CAMK2:
-
calcium/calmodulin dependent protein kinase II
- RFWD2:
-
ring finger and WD repeat domain 2
- HAT:
-
histone acetyltransferase
- BRD:
-
bromodomain
- BRD4:
-
bromodomain-containing protein 4
- NSCLC:
-
non-small cell lung cancer
- EGFR:
-
epidermal growth factor receptor
- DUSP6:
-
dual specificity phosphatase 6
- DUSP4:
-
dual specificity phosphatase 4
- SPRY4:
-
sprouty RTK signaling antagonist 4
References
Rodriguez-Viciana P, Tetsu O, Oda K et al (2005) Cancer targets in the Ras pathway. In: Cold Spring Harb Symp Quant Biol, vol 70. pp 461–467
Senawong T, Phuchareon J, Ohara O et al (2008) Germline mutations of MEK in cardio-facio-cutaneous syndrome are sensitive to MEK and RAF inhibition: implications for therapeutic options. Hum Mol Genet 17:419–430
Zhao Y, Adjei AA (2014) The clinical development of MEK inhibitors. Nat Rev Clin Oncol 11:385–400
Caunt CJ, Sale MJ, Smith PD et al (2015) MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer 15:577–592
Gavrilov D, Kenzior O, Evans M et al (2001) Expression of urokinase plasminogen activator and receptor in conjunction with the ets family and AP-1 complex transcription factors in high grade prostate cancers. Eur J Cancer 37:1033–1040
Gutman A, Wasylyk B (1990) The collagenase gene promoter contains a TPA and oncogene-responsive unit encompassing the PEA3 and AP-1 binding sites. EMBO J 9:2241–2246
D’Orazio D, Besser D, Marksitzer R et al (1997) Cooperation of two PEA3/AP1 sites in uPA gene induction by TPA and FGF-2. Gene 201:179–187
Wei G, Srinivasan R, Cantemir-Stone CZ et al (2009) Ets1 and Ets2 are required for endothelial cell survival during embryonic angiogenesis. Blood 114:1123–1130
Albanese C, Johnson J, Watanabe G et al (1995) Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem 270:23589–23597
Tetsu O, McCormick F (1999) Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398:422–426
Tetsu O, McCormick F (2003) Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 3:233–245
Meloche S, Pouyssegur J (2007) The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 26:3227–3239
Hoshino R, Chatani Y, Yamori T et al (1999) Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 18:813–822
Mansour SJ, Matten WT, Hermann AS et al (1994) Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265:966–970
Cowley S, Paterson H, Kemp P et al (1994) Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 77:841–852
Klein PJ, Schmidt CM, Wiesenauer CA et al (2006) The effects of a novel MEK inhibitor PD184161 on MEK-ERK signaling and growth in human liver cancer. Neoplasia 8:1–8
Luciano F, Jacquel A, Colosetti P et al (2003) Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene 22:6785–6793
Ley R, Balmanno K, Hadfield K et al (2003) Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem 278:18811–18816
Roskoski R Jr (2012) MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem Biophys Res Commun 417:5–10
Cargnello M, Roux PP (2012) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75:50–83
Akinleye A, Furqan M, Mukhi N et al (2013) MEK and the inhibitors: from bench to bedside. J Hematol Oncol 6:27
Garuti L, Roberti M, Bottegoni G (2010) Non-ATP competitive protein kinase inhibitors. Curr Med Chem 17:2804–2821
Ohren JF, Chen H, Pavlovsky A et al (2004) Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol 11:1192–1197
Gilmartin AG, Bleam MR, Groy A et al (2011) GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res 17:989–1000
Ascierto PA, McArthur GA, Dreno B et al (2016) Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol 17:1248–1260
Solit DB, Garraway LA, Pratilas CA et al (2006) BRAF mutation predicts sensitivity to MEK inhibition. Nature 439:358–362
Yeh JJ, Routh ED, Rubinas T et al (2009) KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Mol Cancer Ther 8:834–843
Long GV, Stroyakovskiy D, Gogas H et al (2014) Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371:1877–1888
Menzies AM, Long GV (2014) Dabrafenib and trametinib, alone and in combination for BRAF-mutant metastatic melanoma. Clin Cancer Res 20:2035–2043
Robert C, Karaszewska B, Schachter J et al (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372:30–39
Caunt CJ, Keyse SM (2013) Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J 280:489–504
Kidger AM, Keyse SM (2016) The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin Cell Dev Biol 50:125–132
Lake D, Correa SA, Muller J (2016) Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci 73:4397–4413
Kidger AM, Rushworth LK, Stellzig J et al (2017) Dual-specificity phosphatase 5 controls the localized inhibition, propagation, and transforming potential of ERK signaling. Proc Natl Acad Sci USA 114:E317–E326
Holderfield M, Deuker MM et al (2014) Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 14:455–467
Sun C, Wang L, Huang S et al (2014) Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508:118–122
Chandarlapaty S (2012) Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer Discov 2:311–319
Vivanco I (2014) Targeting molecular addictions in cancer. Br J Cancer 111:2033–2038
El-Khoueiry A, Kurkjian C, Semrad T et al (2013) A first in-human phase I study to evaluate the MEK1/2 inhibitor GDC-0623 in patients with advanced solid tumors. Mol Cancer Ther 11(Supplement):B75
Martinez-Garcia M, Banerji U, Albanell J et al (2012) First-in-human, phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of RO5126766, a first-in-class dual MEK/RAF inhibitor in patients with solid tumors. Clin Cancer Res 18:4806–4819
Wang S, Ghosh RN, Chellappan SP (1998) Raf-1 physically interacts with Rb and regulates its function: a link between mitogenic signaling and cell cycle regulation. Mol Cell Biol 18:7487–7498
Mielgo A, Seguin L, Huang M et al (2011) A MEK-independent role for CRAF in mitosis and tumor progression. Nat Med 17:1641–1645
Advani SJ, Camargo MF, Seguin L et al (2015) Kinase-independent role for CRAF-driving tumour radioresistance via CHK2. Nat Commun 6:8154
Phuchareon J, McCormick F, Eisele DW et al (2015) EGFR inhibition evokes innate drug resistance in lung cancer cells by preventing Akt activity and thus inactivating Ets-1 function. Proc Natl Acad Sci USA 112:E3855–E3863
Robles AI, Rodriguez-Puebla ML, Glick AB et al (1998) Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev 12:2469–2474
Yu Q, Geng Y, Sicinski P (2001) Specific protection against breast cancers by cyclin D1 ablation. Nature 411:1017–1021
Yang C, Ionescu-Tiba V, Burns K et al (2004) The role of the cyclin D1-dependent kinases in ErbB2-mediated breast cancer. Am J Pathol 164:1031–1038
Yu Q, Sicinska E, Geng Y et al (2006) Requirement for CDK4 kinase function in breast cancer. Cancer Cell 9:23–32
Landis MW, Pawlyk BS, Li T et al (2006) Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell 9:13–22
Musgrove EA, Caldon CE, Barraclough J et al (2011) Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 11:558–572
Finn RS, Dering J, Conklin D et al (2009) PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 11:R77
Keshamouni VG, Mattingly RR, Reddy KB (2002) Mechanism of 17-beta-estradiol-induced Erk1/2 activation in breast cancer cells. A role for HER2 AND PKC-delta. J Biol Chem 277:22558–22565
O’Leary B, Finn RS, Turner NC (2016) Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 13:417–430
Finn RS, Crown JP, Lang I et al (2015) The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 16:25–35
Santamaria D, Barriere C, Cerqueira A et al (2007) Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448:811–815
Moore JD (2013) In the wrong place at the wrong time: does cyclin mislocalization drive oncogenic transformation? Nat Rev Cancer 13:201–208
Malumbres M, Sotillo R, Santamaria D et al (2004) Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118:493–504
Plotnik JP, Budka JA, Ferris MW et al (2014) ETS1 is a genome-wide effector of RAS/ERK signaling in epithelial cells. Nucleic Acids Res 42:11928–11940
Imler JL, Schatz C, Wasylyk C et al (1988) A Harvey-ras responsive transcription element is also responsive to a tumour-promoter and to serum. Nature 332:275–278
Martin ME, Piette J, Yaniv M et al (1988) Activation of the polyomavirus enhancer by a murine activator protein 1 (AP1) homolog and two contiguous proteins. Proc Natl Acad Sci USA 85:5839–5843
Xin JH, Cowie A, Lachance P et al (1992) Molecular cloning and characterization of PEA3, a new member of the Ets oncogene family that is differentially expressed in mouse embryonic cells. Genes Dev 6:481–496
Wasylyk B, Wasylyk C, Flores P et al (1990) The c-ets proto-oncogenes encode transcription factors that cooperate with c-Fos and c-Jun for transcriptional activation. Nature 346:191–193
Oka T, Rairkar A, Chen JH (1991) Structural and functional analysis of the regulatory sequences of the ets-1 gene. Oncogene 6:2077–2083
Majerus MA, Bibollet-Ruche F, Telliez JB et al (1992) Serum, AP-1 and Ets-1 stimulate the human ets-1 promoter. Nucleic Acids Res 20:2699–2703
Hollenhorst PC, McIntosh LP, Graves BJ (2011) Genomic and biochemical insights into the specificity of ETS transcription factors. Annu Rev Biochem 80:437–471
Selvaraj N, Kedage V, Hollenhorst PC (2015) Comparison of MAPK specificity across the ETS transcription factor family identifies a high-affinity ERK interaction required for ERG function in prostate cells. Cell Commun Signal 13:12
Hollenhorst PC, Jones DA, Graves BJ (2004) Expression profiles frame the promoter specificity dilemma of the ETS family of transcription factors. Nucleic Acids Res 32:5693–5702
Kabbout M, Dakhlallah D, Sharma S et al (2014) MicroRNA 17-92 cluster mediates ETS1 and ETS2-dependent RAS-oncogenic transformation. PLoS ONE 9:e100693
Yang BS, Hauser CA, Henkel G et al (1996) Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol Cell Biol 16:538–547
Slupsky CM, Gentile LN, Donaldson LW et al (1998) Structure of the Ets-1 pointed domain and mitogen-activated protein kinase phosphorylation site. Proc Natl Acad Sci USA 95:12129–12134
Nelson ML, Kang HS, Lee GM et al (2010) Ras signaling requires dynamic properties of Ets1 for phosphorylation-enhanced binding to coactivator CBP. Proc Natl Acad Sci USA 107:10026–10031
Yamamoto H, Flannery ML, Kupriyanov S et al (1998) Defective trophoblast function in mice with a targeted mutation of Ets2. Genes Dev 12:1315–1326
Muthusamy N, Barton K, Leiden JM (1995) Defective activation and survival of T cells lacking the Ets-1 transcription factor. Nature 377:639–642
Bories JC, Willerford DM, Grevin D et al (1995) T-cell apoptosis and terminal B-cell differentiation induced by inactivation of the Ets-1 proto-oncogene. Nature 377:635–638
Paumelle R, Tulasne D, Kherrouche Z et al (2002) Hepatocyte growth factor/scatter factor activates the ETS1 transcription factor by a RAS-RAF-MEK-ERK signaling pathway. Oncogene 21:2309–2319
Bhagwat AS, Vakoc CR (2015) Targeting transcription factors in cancer. Trends Cancer 1:53–65
Potu H, Peterson LF, Kandarpa M et al (2017) Usp9x regulates Ets-1 ubiquitination and stability to control NRAS expression and tumorigenicity in melanoma. Nat Commun 8:14449
Yu JC, Chen JR, Lin CH et al (2009) Tensile strain-induced Ets-2 phosphorylation by CaMKII and the homeostasis of cranial sutures. Plast Reconstr Surg 123(2 Suppl):83S–93S
Lu G, Zhang Q, Huang Y, Song J et al (2014) Phosphorylation of ETS1 by Src family kinases prevents its recognition by the COP1 tumor suppressor. Cancer Cell 26:222–234
Marine JC (2012) Spotlight on the role of COP1 in tumorigenesis. Nat Rev Cancer 12:455–464
Bartholomeusz GA, Talpaz M, Kapuria V et al (2007) Activation of a novel Bcr/Abl destruction pathway by WP1130 induces apoptosis of chronic myelogenous leukemia cells. Blood 109:3470–3478
Cooper CD, Newman JA, Gileadi O (2014) Recent advances in the structural molecular biology of Ets transcription factors: interactions, interfaces and inhibition. Biochem Soc Trans 42:130–138
Taniguchi H, Fujiwara Y, Doki Y et al (2007) Gene therapy using ets-1 transcription factor decoy for peritoneal dissemination of gastric cancer. Int J Cancer 121:1609–1617
Lamber EP, Vanhille L, Textor LC et al (2008) Regulation of the transcription factor Ets-1 by DNA-mediated homo-dimerization. EMBO J 27:2006–2017
Babayeva ND, Wilder PJ, Shiina M et al (2010) Structural basis of Ets1 cooperative binding to palindromic sequences on stromelysin-1 promoter DNA. Cell Cycle 9:3054–3062
Babayeva ND, Baranovskaya OI, Tahirov TH (2012) Structural basis of Ets1 cooperative binding to widely separated sites on promoter DNA. PLoS ONE 7:e33698
Garvie CW, Hagman J, Wolberger C (2001) Structural studies of Ets-1/Pax5 complex formation on DNA. Mol Cell 8:1267–1276
Kim WY, Sieweke M, Ogawa E et al (1999) Mutual activation of Ets-1 and AML1 DNA binding by direct interaction of their autoinhibitory domains. EMBO J 18:1609–1620
Foulds CE, Nelson ML, Blaszczak AG et al (2004) Ras/mitogen-activated protein kinase signaling activates Ets-1 and Ets-2 by CBP/p300 recruitment. Mol Cell Biol 24:10954–10964
Tetsu O, Phuchareon J, Eisele DW et al (2015) ETS1 inactivation causes innate drug resistance to EGFR inhibitors. Mol Cell Oncol 3:e1078924
Tetsu O, Eisele DW, McCormick F (2015) Resistance to EGFR-targeted therapy by Ets-1 inactivation. Cell Cycle 14:3211–3212
Tetsu O, Phuchareon J, Eisele DW et al (2015) AKT inactivation causes persistent drug tolerance to EGFR inhibitors. Pharmacol Res 102:132–137
Phuchareon J, McCormick F, Eisele DW et al (2015) EGFR inhibition generates drug-tolerant persister cells by blocking AKT activity. Cancer Cell Microenviron 2:e1045
Tetsu O, Hangauer MJ, Phuchareon J et al (2016) Drug resistance to EGFR inhibitors in lung cancer. Chemotherapy 61:223–235
Huang WC, Chen CC (2005) Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol 25:6592–6602
de Launoit Y, Audette M, Pelczar H et al (1998) The transcription of the intercellular adhesion molecule-1 is regulated by Ets transcription factors. Oncogene 16:2065–2073
Yockell-Lelievre J, Spriet C, Cantin P et al (2009) Functional cooperation between Stat-1 and ets-1 to optimize icam-1 gene transcription. Biochem Cell Biol 87:905–918
Yang C, Shapiro LH, Rivera M et al (1998) A role for CREB binding protein and p300 transcriptional coactivators in Ets-1 transactivation functions. Mol Cell Biol 18:2218–2229
Ogryzko VV, Schiltz RL, Russanova V et al (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87:953–959
Filippakopoulos P, Knapp S (2014) Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov 13:337–356
Hay DA, Fedorov O, Martin S et al (2014) Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J Am Chem Soc 136:9308–9319
Chekler EL, Pellegrino JA, Lanz TA et al (2015) Transcriptional profiling of a selective CREB binding protein bromodomain inhibitor highlights therapeutic opportunities. Chem Biol 22:1588–1596
Ding N, Hah N, Yu RT et al (2015) BRD4 is a novel therapeutic target for liver fibrosis. Proc Natl Acad Sci USA 112:15713–15718
Filippakopoulos P, Qi J, Picaud S, Shen Y et al (2010) Selective inhibition of BET bromodomains. Nature 468:1067–1073
Boi M, Gaudio E, Bonetti P et al (2015) The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin Cancer Res 21:1628–1638
Lonergan PE, Tindall DJ (2011) Androgen receptor signaling in prostate cancer development and progression. J Carcinog 10:20
Massie CE, Adryan B, Barbosa-Morais NL et al (2007) New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep 8:871–878
Kalet BT, Anglin SR, Handschy A et al (2013) Transcription factor Ets1 cooperates with estrogen receptor α to stimulate estradiol-dependent growth in breast cancer cells and tumors. PLoS ONE 8:e68815
Cao P, Feng F, Dong G et al (2015) Estrogen receptor α enhances the transcriptional activity of ETS-1 and promotes the proliferation, migration and invasion of neuroblastoma cell in a ligand dependent manner. BMC Cancer 15:491
Cabrita MA, Christofori G (2008) Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis 11:53–62
Authors’ contributions
Both authors were involved in the conception, design and drafting of the manuscript. Both authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by grants from the Joan and Irwin Jacobs Fund of the Jewish Community Foundation to O.T. and the National Cancer Institute (1R35CA197709) to F.M.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tetsu, O., McCormick, F. ETS-targeted therapy: can it substitute for MEK inhibitors?. Clin Trans Med 6, 16 (2017). https://doi.org/10.1186/s40169-017-0147-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40169-017-0147-4