
Abstract
The respiratory system’s complex cellular heterogeneity presents unique challenges to researchers in this field. Although bulk RNA sequencing and single-cell RNA sequencing (scRNA-seq) have provided insights into cell types and heterogeneity in the respiratory system, the relevant specific spatial localization and cellular interactions have not been clearly elucidated. Spatial transcriptomics (ST) has filled this gap and has been widely used in respiratory studies. This review focuses on the latest iterative technology of ST in recent years, summarizing how ST can be applied to the physiological and pathological processes of the respiratory system, with emphasis on the lungs. Finally, the current challenges and potential development directions are proposed, including high-throughput full-length transcriptome, integration of multi-omics, temporal and spatial omics, bioinformatics analysis, etc. These viewpoints are expected to advance the study of systematic mechanisms, including respiratory studies.
Similar content being viewed by others


Background
The respiratory system comprises the respiratory tract and lungs and is one of the organs that directly interface with the external environment [1]. The complexity of its structure and function hinders our understanding of the physiological and pathological processes involved [2]. The pathogenesis of most common respiratory diseases is complicated, a significant public health problem. Lung cancer is one of the most common diseases in clinical practice [3], with its morbidity and mortality ranking first among all tumor types. The complexity of the lung tumor microenvironment (TME) is the main factor leading to misdiagnosis. Chronic respiratory diseases, such as asthma, emphysema, and bronchitis, are still diagnosed based on respiratory symptoms, medical imaging, and lung function parameters, but they are highly heterogeneous and often overlap [4]. This vague description of underlying disease mechanisms leads to non-specific treatment schemes that may ultimately decrease the effectiveness of treatment for these diseases. In addition, tuberculosis and pneumonia place a substantial economic burden on the patients’ families and society. Therefore, revealing the pathogenesis of respiratory diseases, searching for specific biomarkers, and introducing new therapeutic targets are important strategies to improve the current diagnosis and treatment.
Transcriptomics is a significant advance in combining high-throughput sequencing and bioinformatics to explore biological mechanisms [5]. However, sequencing analysis of bulk tissue obscured individual cell phenotypic and functional differences and could not identify the molecular features of single-cell resolution [6]. With the development of single-cell RNA sequencing (scRNA-seq), an increasing number of cell types and subtypes are being detected and clarified that allow defining the multiple cell types and associated molecular characteristics of the lung, providing a valuable tool for studying the respiratory system [7, 8]. Nevertheless, due to the loss of spatial information caused by cell dissociation in single-cell sequencing, the interactions and functional changes of adjacent cells cannot be described in lung anatomy [9, 10]. Moreover, the relationship between cell state and different cell positions should be clearly elucidated [11].
Since it was proposed in 2016, spatial transcriptomics (ST) has provided a new perspective to decipher physiological and pathological bases [12]. While maintaining the original spatial context, quantitative transcriptome analysis allows the resulting gene expression signatures to correlate with cell spatial localization, physiology, and histology. In addition, ST studies can reveal subcellular RNA distribution patterns to help understanding the biological processes of spatial labelling and regulation. With the development of techniques to collect and process samples for ST, the simultaneous reduction in reagent and sequencing costs, and the increasing potency of computing platforms, the potential to tackle fundamental biological inquiries is steadily expanding. Some related applications of ST have been reviewed [13,14,15]. However, compared to brain neuroscience, embryonic development, and heart and other organ tissues, ST has rarely been reviewed in respiratory and lung disease research.
This review focuses on the latest iterative technology of ST in recent years and summarizes how these techniques can be applied to the physiological and pathological processes of the respiratory system, such as lung development, lung atlas, lung cancer, and lung injury. Finally, the current challenges and potential development directions are proposed, including high-throughput full-length transcriptome, multi-omics and spatiotemporal omics integration, bioinformatics analysis, etc. By presenting a unique combination of comprehensive disease coverage, in-depth exploration of disease mechanisms, emphasis on spatial heterogeneity, and future directions, this review can provide a distinct and valuable contribution to the field of ST application in respiratory diseases.
Developments and limitations of ST
Spatial transcriptome methods have developed and emerged rapidly and can be categorized into imaging-based and sequencing-based methods according to detection strategies. Previous reviews have elaborated on the principles and classification of ST [10, 16, 17]. This article focuses on the innovative forms and variants of these technologies.
Imaging-based ST strategies
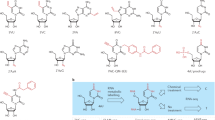
Image-based ST technologies include fluorescence in situ hybridization (ISH)- and in situ sequencing (ISS)-based methods. The advent of high-resolution microscopy and single-molecule fluorescence in situ hybridization (smFISH) has made it possible to quantify the subcellular resolution of transcripts in situ [18]. A unique fluorescently labelled probe binds to RNA, allowing the localization of individual molecules. The variant RNA scope of this technology is commercially available [19], and strategies such as multi-round hybridization, imaging, and probe dissection have widely been applied, such as sequential FISH (seqFISH) [20, 21] (Fig. 1a). Hybridization chain reactions (HCR) based on isothermal amplification have also been applied to solve the problem of high autofluorescence, namely smHCR [22] (Fig. 1b). To overcome probe hybridization errors and read errors, a barcode allocation scheme, multiplexed error-robust FISH (MERFISH), has been developed and widely used in single-cell transcription localization and ST at the tissue level (Fig. 1c). On this basis, sequential fluorescence in situ hybridization (seq-FISH +) [23], ouroboros smFISH (osmFISH) [24], and signal amplification by exchange reaction (SABER) [25] are proposed to solve the molecular crowding in the imaging process. Among them, seq-FISH + used 20 probes and 3 excitation lights to analyse 10,000 genes (Fig. 1d). Enhanced electric FISH (EEL FISH) combines electrophoresis-aided large tissue RNA sampling and multiplexed FISH for transcription imaging of thick tissue, reducing data collection time [26]. Recently, expansion FISH (exFISH) [27] and expansion-assisted iterative fluorescence in situ hybridization (EASI-FISH) [28] were used for the three-dimensional (3D) resolution of gene expression in tissues using hydrogel expansion. Overall, due to the expensive and time-consuming nature of hybridization techniques and additional challenges, such as background fluorescence in tissues, ISH-based methods have thus far been limited to research on cell and tissue culture.

Imaging-based ST strategies. a seqFISH decodes transcripts in space by sequential staining/imaging cycles. b Compared to seqFISH, smHCR can achieve ~ 20 fold signal amplification to detect single mRNA in situ. c MERFISH implements thousands of RNA imaging using a combinatorial FISH labeling with encoding schemes that correct errors. d seqFISH+ performs in situ RNA imaging using 20 probes in four-wheel coding. e ISS-methods by sequencing by ligation (ISS, IISS). f Electro-seq combines bioelectronics with ISS enabling electrophysiological and gene expression profiling. g Non-targeted sequencing, such as FISSEQ and ExSeq, enables unbiased covering sequencing of the whole transcriptome. ST spatial transcriptomic, FISH fluorescence in situ hybridization, seqFISH+ sequential FISH+, smHCR single-molecule hybridization chain reaction, MERFISH multiplexed error-robust FISH, ISH in situ hybridization, ISS in situ sequencing, IISS improved in situ sequencing, FISSEQ fluorescent in situ sequencing, Electro-seq in situ electro-sequencing
ISS-based methods are categorized into targeted and untargeted mRNA detection. One of the earliest targeted in situ sequencing techniques, ISS (Fig. 1e), generated signals through padlock probes and rolling circle amplification (RCA), enabling the expression of 256 RNA transcripts in single-round hybridization and was commercialized as Cartana [29]. Then BOLORAMIS (barcoded oligonucleotides ligated on RNA amplified for multiplexed and parallel in situ analyses) solved the problem of low detection efficiency of ISS by introducing the DNA ligase SplintR ligase [19]. Hybridization-based ISS (HybISS) allows a barcoding system to improve in situ detection and removes the limitations of the sequence-by-ligation chemistry of ISS. BaristaSeq utilizes synthetic chemical sequencing technology to display higher signal-to-noise ratios through multiple rounds of imaging and perform specific detection of RCA products [30]. In a preprint study in 2023, Tang et al. [31] proposed improved ISS (IISS), which developed an improved combinatorial probe anchor ligation chemistry using a 2-base encoding strategy for barcode interrogation, improving the signal strength and specificity of ISS (Fig. 1e). Moreover, a recent study integrated electrochemistry and ISS (Electro-seq) to correlate cell electrophysiology with gene expression at the single-cell level and identify changes in the gene expression profile during myocardial cell development [32] (Fig. 1f).
On the other hand, spatially-resolved transcript amplicon readout mapping (STARmap) bypasses cDNA synthesis and uses SNAIL probes and sequencing with error-reduction by dynamic annealing and ligation (SEDAL) to identify gene identifiers. By combining hydrogel histochemistry, it analyses tissue samples in 3D tissues rather than a single 2D pattern [33]. This year, the team further updated STARmap, or STARmap PLUS, and realized the joint detection of transcriptome and protein in a mouse model of Alzheimer’s disease [34].
Fluorescent in situ sequencing (FISSEQ) is a representative method in non-targeted sequencing that can achieve unbiased coverage of the whole transcriptome. However, its random primers lead to low detection efficiency and involve complex enzymatic reactions [35] (Fig. 1g). Relevant reagents and instruments have been produced and commercialized [36]. Based on FISSEQ, untargeted expansion sequencing (ExSeq) combines expansion microscopy with ISS, using an amplified hydrogel to anchor RNA and generate optical barcodes, which have been used for gene analysis in Drosophila embryos and mouse brains [37] (Fig. 1f).
Sequencing-based ST strategies
Combining the next-generation sequencing (NGS) platform and spatial information significantly improves the throughput of ST and unbiased retrieval of transcripts compared to image-based methods. A method based on laser capture microdissection (LCM) and scRNA-seq was developed to allow a spatially unbiased analysis of the transcriptome and classification and sequencing of regions of interest (ROI) under microscopic guidance (Fig. 2a). In addition, Tomo-seq [38], geographical position sequencing (Geo-seq) [39], and proximID [40] were developed to explain the heterogeneity and spatial differences of a small number of cell transcriptomes. At present, optical markers have replaced the traditional physical anatomy, such as NICHE-seq [41], transcriptome in vivo analysis (TIVA) [42], ZipSeq [43] (Fig. 2b), which use patterned illumination and photocaged oligonucleotides to mark ROIs; GeoMx digital spatial profiling utilizes cleavable oligonucleotide tags to quantify the abundance of RNA or proteins in ROIs [44]. A technique, called Image-seq, allows the harvesting of location-specific live cells for sequencing using a living microscope with high sensitivity and transcription coverage but at the cost of reduced throughput [45] (Fig. 2c).

Sequencing-based ST strategies. a LCM-seq integrates LCM and scRNA-seq to realize regional sequencing. b ZipSeq uses light activation tags for labeling, isolation, and scRNA-seq of ROIs. c Image-seq allows location-specific live cells to be harvested for sequencing by living microscope. d Stereo-seq utilizes DNA nanoballs with spatial barcodes for transcription spatial localization. e sci-Space uses spatial barcodes for imaging, labeling and transcriptome sequencing of nuclei in tissue slices. f Seq-Scope and Pixel-seq based on illumina clustering and sequence reading. g DBiT-seq utilizes microfluidic channels for orthogonal coding, and similar techniques include xDBiT, CBSST-seq and Matrix-seq. h STRS utilizes the 10 × Visium platform for total transcriptome analysis. ST spatial transcriptomic, LCM laser capture microdissection, scRNA-seq single cell RNA sequencing, ROI regions of interest, Stereo-seq spatio-temporal enhanced resolution omics-sequencing, Pixel-seq polony-indexed library-sequencing, DBiT-seq deterministic barcoding in tissue for spatial omics sequencing, CBSST-seq cross-amplified barcodes on slides for spatial transcriptomics sequencing, STRS spatial total RNA-sequencing, FACS fluorescence activated cell sorting, UMI unique molecular identifier, MID molecular identifiers, CID coordinate identity, mRNA messenger RNA, lncRNA long noncoding RNA, miRNA microRNA, snoRNA small nucleolar RNA, tRNA transfer RNA, ATP adenosine triphosphate, yPAP yeast poly(A) polymerase, xDBiT multiplexed deterministic barcoding in tissue
Given the low throughput and capture rate issues of imaging and microdissection technologies, researchers are gradually considering the in situ spatial indexing methods. ST was developed, mRNA location information and expression levels were mapped using spatial barcode and unique molecular identifier (UMI), which was acquired by 10 × Genomics (100 μm), and its capture efficiency (10 × Visium, 55 μm) was further improved [12]. Slide-seq [46] (10 μm) and high-definition ST [47] (HDST, 2 μm) utilizing random barcode beads have been proposed for higher resolution. Slide-seqV2 (10 μm) optimizes library construction and array indexing and demonstrates high capture efficiency for ST sequencing at near-cellular resolution [48]. However, low sensitivity and bead pre-decoding limit their application. Spatial enhanced resolution omics sequencing (Stereo-seq, 0.22 μm) uses random barcode DNA nanospheres deposited in array mode for nanoscale resolution. It has been applied to construct a spatiotemporal transcription atlas of organogenesis [49, 50] (Fig. 2d). sci-Space [51] (200 μm) and XYZeq (a workflow that encodes spatial metadata into scRNA-seq libraries) [52] (500 μm) analyze cells and nuclear spatial coordinates at large scales. However, they cannot provide actual spatial single-cell profiles because they lose cytoplasmic transcription information (Fig. 2e). Seq-Scope (0.5–0.8 μm) directly uses illumina NGS chips to generate spatial barcode arrays, achieving subcellular resolution of spatial barcodes for visualizing the nuclei [53] (Fig. 2f). Similar illumina chemistry, poly-indexed library-sequencing (Pixel-seq, 1 μm), reduces costs 35-fold through repeatable enzyme replication of barcode-patterned gels and improves resolution 200-fold compared to existing methods [54] (Fig. 2f).
Microfluidic channel-based approaches have also been integrated for spatial localization [55]. Deterministic barcoding in tissue for spatial omics sequencing (DBiT-seq) (10 μm) can spatially encode tissues by cross-coding, allowing transcriptome and protein analysis [56] (Fig. 2g). Inspired by this method, multiplexed deterministic barcoding in tissue (xDBiT) [57], cross-amplified barcodes on slides for spatial transcriptomics sequencing (CBSST-seq) [58] and Matrix-seq (a microfluidics-based barcoding strategy) [59] have also been developed and applied, and extended to spatial multi-omics (SM-omics), such as microfluidic indexing based spatial ATAC and RNA sequencing (MISAR-seq) [60] and spatial co-indexing of transcriptomes and epitopes (spatial-CITE-seq) [61]. In addition, spatial total RNA sequencing (STRS) utilizes the 10 × Visium platform to enable the detection of full-spectrum RNA rather than just polyadenylated RNA transcripts [62] (Fig. 2h). A more general challenge for ST based on spatial indexing methods is how to balance mRNA capture efficiency and lateral diffusion. Moreover, large-scale hybridization reverse transcription may lead to the distortion of gene expression.
Applications of ST to respiratory research
ST has been widely used in the study of lung development and respiratory disease mechanisms due to the great interest in the molecular structure of the respiratory system. This section summarizes the emerging applications of the respiratory system, such as lung development, lung atlas, lung cancer, and lung injury (Fig. 3) [63,64,65,66,67,68,69].

Applications of ST to respiratory research. ST is used to construct lungs spatial atlas and identify cellular composition. In lung cancer, the tumor microenvironment [63] and heterogeneity [64], evolution and metastasis [65], and further localization of intratumoral microbiota origin [66] have been revealed by ST. Other applications of ST include gene expression patterns in lung development [67], and the pathogenesis of pulmonary fibrosis [68] and COVID-19 pneumonia [69]. ST spatial transcriptomic, COVID-19 corona virus disease 2019, NK cell natural killer cell
Lung development
Ljungberg et al. [67] using spatial in situ hybridization attempted to elucidate gene expression patterns in prenatal and postnatal murine lungs to describe the details of lung development at critical stages of alveolarization and improve data for LungMap. A recent study applied in situ hybridization analysis to determine the origin of clonogenic mesenchymal cells in the human lung, especially from an adventitial fibroblast subset. The spatial heterogeneity of mesenchymal cells and their potential characteristics have also been described [70].
Cellular composition and spatial atlas of lung
The Human Cell Atlas (HCA) aims to establish atlas data of different organs and tissues in healthy individuals at single-cell resolution [71, 72], including the Human Cell Lung Atlas (HCLA), which focuses on the respiratory system. In respiratory research, the cell atlas identifies previously undefined cell catalogues and their phenotypes and interactions, enhancing our understanding of respiratory and lung diseases [73, 74]. Combined with scRNA-seq, many studies have generated lung molecular cell atlases of health and disease, such as LungMAP [75], discovAIR [76], etc. [4, 77, 78]. Researchers have defined 58 different cell types and gene expression profiles in the human lung using droplet- and plate-based scRNA-seq [79]. However, these data are inaccurate due to the lack of spatial context and resolution required to describe the extreme cellular heterogeneity of lungs’ anatomical features. Indeed, a recent study used ST to distinguish 80 cell types and states, including 11 cell populations that had not been annotated in previous lung atlas studies, and define a gland-associated immune niche [80]. Combined with the gene expression profile of scRNA-seq and the spatially resolved transcriptomics on the complete tissue section, Sountoulidis et al. [81] constructed a comprehensive topographic atlas of the early development of human lung, describing the development track leading to significant heterogeneity of lung cell. We believe that the spatial diversity of the lung at the mRNA level is associated with the proteome and, further, with physiological functions. In addition, several in situ hybridization techniques, such as proximity ligation in situ hybridization technology (PLISH) [82] and SCRINSHOT [83] have been developed and used for identifying and localizing lung and airway cell types in mice, including the recently discovered ionocytes. In the distal airways and alveoli, 15 markers were robustly used to identify macrophages and epithelial cells, such as AT1 and AT2 cells, club cells, and neuroendocrine cells.
Lung cancer
In the past few centuries, tumors have been considered a highly organized “organ” instead of a simple aggregation of abnormally proliferating cells [84]. Lung cancer is the main cause of death from tumor diseases [85]. A significant challenge for medical research is to identify normal cellular trajectory points at the start and progression of lung pathologies and analyse the cellular responses after treatment [86].
Lung cancer microenvironment
Immune checkpoint (ICP)-targeted therapy has shown considerable success in lung cancer, including non-small cell lung cancer (NSCLC), lung adenocarcinoma (LUAD), squamous and non-squamous carcinoma. However, studies have shown that the rate of positive responses in patients receiving targeted drugs remains low, with a possibly high degree of immune-related adverse events [87]. In addition, clinical benefits are often prevented by resistance to ICP-targeted drugs in the primary tumor [63]. Increasing evidence suggests that TME is strongly related to tumor development, metastasis, and recurrence and is more critical than ICPs in immune evasion [88, 89]. Therefore, it is urgent to improve the understanding regarding TME to better classify patients and determine new treatment targets to improve prognosis. The cell catalogue transcriptome of the TME in lung tumors is available at single-cell resolution, and its phenotypes and concerted behavior have been described [90]. Spatial transcription analysis can reveal the spatial distribution preference of stromal cells in the lung TME. For example, 10 × Chromium and in situ imaging were used to explore the loss of Tgfbr2 leading to TME remodeling and promoting immune exclusion in a lung cancer mouse model [91]. Tumor-associated macrophages (TAMs), one of the most abundant immune cells in the TME [92], are essential regulators of anti-tumor immunity. However, the mechanisms regulating their abundance in the TME remain to be explored. Larroquette et al. [93] analysed preconditioned tumor samples from advanced NSCLC patients undergoing ICP blocker therapy, reporting that tumor compartment enrichment in TAMs was associated with immunotherapy resistance. A spatial analysis of 78 in situ transcripts from 16 tumor specimens was performed using NanoString GeoMx. The results revealed that the prognostic effect of TAMs in NSCLC was directly related to the distance from tumor cells, and the three significantly up-regulated genes CD27, CCL5, and ITGAM in tumors with high-level TAM infiltration might be potential targets for immunotherapy. Furthermore, applying multiplex immunohistochemistry (mIHC) and digital spatial profiler (DSP) has been observed for TME analysis in samples of NSCLC patients following immune checkpoint inhibitor (ICI) treatment [94].
Heterogeneity of lung cancer
Many studies based on scRNA-seq have revealed the heterogeneity of lung cancer cells [95,96,97]. With advances in spatial transcription analysis and integration of scRNA-seq, researchers have combined specific molecular phenotypes with unknown cell interaction patterns or clinical manifestations to classify tumor cell subgroups. For example, Sinjab et al. [64] identified cell lineages, states, and transcriptomic features that evolved geospatially from normal tissue regions to LUAD, where significant expression of CD24 in epithelial cells drives primary tumor features. Their data provided a spatial atlas of LUAD to identify potential targets for early interception. Zhang et al. [98] used the Visium platform to identify spatial location-specific subclones in the lung squamous cell carcinoma (LUSC). The results showed that the immune cell composition of the tumor subclones was significantly different from the tumor proportion, and the tumor purity was contrary to the trend of tumor epithelial-mesenchymal transition (EMT). The effect of high intratumoral heterogeneity (ITH) on therapy efficacy in advanced lung cancer has recently been reported [99]. The innovative use of DSP has provided compelling evidence for improved prediction of therapeutic outcomes in dual-specificity antibody therapy by integrating genetic information from the stromal region.
Development and metastasis of lung cancer
Cancer development involves tumor cells’ adaptation to the environment and is the inevitable and continuous result of life [14]. To date, tracking cancer evolution in humans has focused on DNA mutations. However, genotypes are not necessarily phenotypes [100], and cancer cell populations within a tumor often exhibit significant differences and transcriptional diversity [101, 102]. In this line, lung cancers exhibit more complex molecular and morphological heterogeneity and different combinations of subclone mutations, reducing the reproducibility of lung cancer research and posing a challenge for effective treatment [103]. Zhu et al. [65] integrated RNA-seq and ST techniques and constructed a single-cell spatiotemporal multi-omics atlas of LUAD to explore the dynamic evolution trajectory of early LUAD. The results suggested that LUAD might originate from Clara and AT2 cells, eventually evolving into the UBE2C+ cancer cell subpopulation, in which UBE2C mediates the proliferation and metastasis of tumor cells. As LUAD progresses from adenocarcinoma in situ (AIS) to invasive adenocarcinoma (IAC), the spatial distribution of cancer cells may be more important than their type. This finding compensates for the absence of spatial information for immunotyping LUAD by scRNA-seq. Furthermore, studies have reported the mechanisms of brain metastasis (BrMs) in NSCLC based on DSP. These findings highlight the highly immunosuppressive microenvironment associated with BrMs lesions, as compared to primary tumors, characterized by the reduced abundance of B and T cells and increased infiltration of neutrophils, providing a framework for the spatial heterogeneity of BMS in lung cancer and identifying the characteristic genes of metastasis to predict patients’ prognosis [104].
Intratumoral microbiota
Lung cancers have a unique microbial composition [105]. Several studies have shown that bacterial populations within tumors are tumor type-specific and might directly regulate cancer initiation and progression [106]. Wong-Rolle et al. [66] obtained spatial macro transcriptome information from 12 lung cancer patients to understand the spatial distribution of the microbiota in lung cancers and its effect on host cell heterogeneity, reporting that bacteria were significantly concentrated in tumor cells, and the content increased from normal tissues and tertiary lymphoid structures (TLS) to tumor cells and peaked in the airway, suggesting that the bacteria in lung cancer may be derived from the airway rather than the intestinal flora. Further gene expression correlation analysis showed that bacterial content positively correlated with the CTNNB, HIF1A, and VEGFA genes. In addition, several signaling pathways related to tumorigenesis also positively correlated with bacterial content in lung cancer. We believe that spatial transcriptome and local interaction group analysis can predict individual tumor behavior and provide useful resources for understanding and reversing lung cancer progression.
Pulmonary fibrosis
Pulmonary fibrosis is a highly heterogeneous end-stage pathological change of the lung, whose pathogenesis has not been fully elucidated [107, 108]. The first published study of pulmonary fibrosis using ST focused on epithelial cells [68], demonstrating the unique molecular signature of epithelial cell/fibroblast foci sandwiches from typical interstitial pneumonia/idiopathic pulmonary fibrosis (IPF) patients. The pathogenesis of IPF is associated with epithelial dysfunction. Another study used the GeoMx DSP platform to explore the transcriptional differences between fibroblastic foci and fibrous and normal areas in IPF cases and identified new fibrogenic biomarkers expressed in fibroblastic foci [109]. The spatiotemporal analysis brings hope for the development of pulmonary fibrosis. For example, Shi et al. [110] explored the spatiotemporal distribution of heterogeneous fibroblasts in the progression of secondary pulmonary fibrosis due to silica inhalation. The results showed that GREM1, as a driving factor causing inflammation, is involved in changes in this pulmonary condition and may be a potential target for the early treatment of silicosis.
Pneumonia
The corona virus disease 2019 (COVID-19) pandemic, induced by severe acute respiratory syndrome coronavirus 2 (SARS-CoV2), has caused millions of cases of severe acute respiratory illness worldwide [111, 112], the pathogenesis of which is not fully understood, and the host response to its infection should be better defined. ST provides new insights into identifying cell types and elucidating heterogeneity after SARS-CoV2 infection, as shown in Table 1 [69, 113,114,115,116,117,118,119,120,121,122,123,124]. The Broad Institute, in collaboration with Harvard Medical School and others, created the post-infection lung spatial transcriptome atlas [69], revealing extensive remodelling of the lung epithelial, immune, and stromal compartments and mapping cell types and genes associated with disease severity. Analysis of COVID-19-infected patients highlighted two stages of the disease that lead to death in patients. Desai et al. [113] showed that RNA levels were associated with disease duration, reporting significant spatiotemporal heterogeneity in viral load and immune response. It is estimated that COVID-19 infection will lead to various complications and post-acute sequelae of SARS-CoV2 (PASC). Dinnon et al. [114] constructed a mouse model and identified the transcriptional profiles of acute and chronic disease stages using RNA-ISH and GeoMx DSP, providing strategies for testing and improving the “long COVID”. Moreover, DSP data obtained from lung tissues in areas affected by acute respiratory distress syndrome (ARDS) induced by SARS-CoV2 and H1N1 indicate unique transcriptional signatures, thus identifying novel therapeutic targets [115].
Other respiratory diseases
In situ sequencing and capture techniques have been applied to the mechanistic study and therapeutic target discovery of tuberculosis [125,126,127] and influenza infections [128]. For example, a study used mice infected with H1N1 as the model for studying ARDS [129], demonstrating that over-activated fibroblasts could produce extracellular matrix remodelling enzymes, thereby promoting the infiltration of immune cells and leading to compromised lung function. Many studies have evaluated other respiratory diseases, such as pulmonary arterial hypertension [130], asthma [131], and chronic obstructive pulmonary disease (COPD) [132], using scRNA-seq. The introduction of space omics technology is expected to deepen our understanding of respiratory diseases.
Challenges and perspectives
Although ST and related frontier bioinformatics analysis have entirely changed the research on complex organs and tissues and brought great hope for developing systems and disease mechanisms, several challenges should still be addressed to develop its potential beyond these (Fig. 4).

The challenges and perspectives of spatial transcriptomics (ST). ST moves toward high-throughput and full-length transcriptome, in vivo analysis of living cells, and integrates multi-omics and spatiotemporal omics, enabling the construction of 3D spatial atlas with the help of sample processing and bioinformatics tools and providing powerful new techniques for interrogating tissue structure and function
Preparation and treatment of samples
The stability and availability of pre-sequencing samples might be a major obstacle limiting ST. Any intact tissue containing mRNA is suitable for the spatial transcriptome [10]; however, the sample preparation regimen should be further optimized according to the characteristics of different tissues. Morphological differences between tissue types should also be considered. For example, single cell/tissue spots significantly affect transcription levels, and hyperpigmentation in skin cell samples can negatively affect image acquisition due to light absorption [133]. Similarly, lung tissues with alveolar sacs must be handled carefully when frozen [134]. In addition, the sample collection, storage, and processing methods have measurement deviations from the original transcriptional data. One study explored the temporal and spatial gene expression map of the developing human heart [135]. Heart tissue was chopped up and cultured in a suspension using trypsin and collagenase to produce individual cells. Some studies have applied exogenous reagents for isolation [136, 137]; however, they have hardly mentioned whether physicochemical-based isolation and unnatural processing would affect the cells’ transcription levels, reducing the reliability of the generated scRNA-seq data [138]. Concerning spatial transcriptome technologies, in situ capture and sequencing-based strategies [51, 139, 140] have achieved tissue dissection-free sequencing, significantly reducing the risk of sample preparation. However, the effect of tissue slice preparation on the transcriptome should also be considered from a macroscopic perspective. Therefore, further innovations are necessary in sample preparation. Ideally, the upstream processing of tissue sections for sequencing is automated and accessible to nonprofessional users.
A growing body of evidence suggests that ST can potentially aid in clinical diagnosis, understanding human disease, and making proper decisions concerning medicines [141]. Formalin-fixed paraffin-embedded (FFPE) tissue blocks are the gold standard of human tissue preservation methods for clinical diagnosis. The rapid development of space transcriptomics is compatible with FFPE blocks, a breakthrough in the in-depth pathological analysis of more than one billion FFPE sections in the sample library [142, 143]. Unfortunately, RNA molecules in samples are often seriously degraded; therefore, a platform that can analyze FFPE samples is urgently needed. The 10 × Genomics Visium and NanoString DSP have demonstrated their compatibility with FFPE blocks [144, 145]. The Visium Cytassist, launched in late 2022 by 10 × Genomics, enables automated transfer analysis of tissue sections in 30 min, with simultaneous transcriptome and protein analysis of one FFPE sample [146]. Nevertheless, since FFPE tissue blocks are usually stored in a fixing solution, and the time from biopsy to fixation may differ between samples, the integrity of RNA will also be much lower than that of fresh frozen tissue. In addition, since the quality of data may depend on the specific sample, its reliability and robustness should be improved before clinical applications.
High-throughput full-length transcriptome
The ST has solved the problem of losing spatial location information by cell dissociation in the scRNA-seq method. However, apart from spatial addressing, the transcription coverage and depth of these methods are still in their early development stages. Most ST methods can only retrieve single-ended transcripts rather than full-length ones [147]. The residual sequence in the polyadenylated RNA molecule and the spectrum of non-polyadenylated transcripts have not been detected, hindering the study of the immune cell receptor spectrum and alternative splicing (AS). Microdissection-based methods exhibit ideal coverage but suboptimal throughput and spatial resolution of transcription, with a trade-off between the coverage of tissue sections and the detection sensitivity of transcripts. ISH-based methods exhibit excellent spatial resolution and detection efficiency; however, they are difficult to use for large tissue sections and high throughput. ISS has a high resolution but sacrifices the transcript capture depth. Recently, a method of vast transcriptome analysis of single cells by dA-tailing was proposed [148], namely, VASA-seq, which is the only single-cell sequencing technology to combine high sensitivity, full-length transcriptome coverage, and high throughput. However, VASA-seq cannot obtain the spatial location information of cells. In addition, although combining high-throughput microfluidics and barcode index improves the throughput, the quality of RNA might decrease due to cell fixation and permeability treatment. In addition, the cell/bead co-encapsulation efficiency should be improved. Therefore, techniques with higher gene coverage, lower detection deviation, and higher spatial resolution down to the single-cell level are highly anticipated [55].
3D spatial atlas
Currently, most advanced space technologies are limited to displaying cell organization and gene expression on a 2D pattern rather than an actual 3D spatial atlas, which cannot summarize the highly complex spatial cell environment. Tomo-seq performs RNA-seq on different tissue slices to obtain spatial information [149]. Similarly, Geo-seq is combined with the LCM technique to study the transcriptome of small samples with geographical positions. Peng et al. [150] used it to construct a 3D transcriptome atlas of murine embryos and accurately map single ectodermal cells back to their in vivo locations. However, the Geo-seq operation is complicated and requires continuous tissue sections, partitioning by LCM technology, and the sequencing process of database construction [39]. Recently, NanoString’s GeoMx DSP and STRP-seq have been developed to combine bioinformatics tools to reconstruct 3D structural maps such as murine brains [151], and human hearts [152, 153]. However, these technologies are limited by spatial resolution and large sample sizes. We believe that high-precision reconstruction and characterization of 3D structures will further provide a solid foundation for clinical application and research.
Living cells/tissue in vivo
The current ST methods cannot be applied to in vivo histological research on living cells because they investigate only cell snapshots at a certain time in fixed samples. The observed gene expression profile may only be the product of expression heterogeneity [154]. These technologies limit the cell to the active transcriptional state at a single time point, while other cells with similar functions may be dormant. Therefore, their ability to correctly infer cells at the individual gene level and time scale is still controversial. Some research groups have tried to study the transcriptome of living cells in vitro. For example, TIVA has pioneered the capture of mRNA in living cells for transcriptional analysis [42]. However, this method is currently not applicable to the analysis of many cells. ZipSeq marks DNA codes (Zipcodes) on living cells of intact tissue using photocaged oligonucleotides and specific patterned illumination to explore its spatial heterogeneity [43]. However, its low spatial resolution limits its application scope. Similarly, Live-seq is the first to realize continuous observation of whole gene expression in living cells [155]. However, some problems remain, such as inapplicability in vivo and the limitation of multiple sampling. Therefore, the next research direction is how to infer the future or correlate past events with current gene expression, realize the exploration of spatial omics in living cells or tissues in vivo, and track mRNA dynamics in real time with minimal cell disturbance.
SM-omics and spatiotemporal omics
ST is progressing rapidly towards multi-omics data and achieving single-cell resolution. This advancement enables the retrieval of comprehensive information encompassing splicing variation, genetic and epigenetic changes, proteomics, and time-dimension data, all within a single experimental setup [15]. This will assist in understanding cell–cell interactions and the overall cell phenotype/state to solve the tissue function from multiple spatial scales [156]. As mentioned previously, the most robust strategy to obtain a more comprehensive multi-omics profile is to process continuous tissue sections, in which each section is queried by different omics. However, continuous tissue section sampling might give rise to sample heterogeneity deviation.
Researchers have significantly advanced in developing of various single-cell multi-omics technologies, as exemplified in Table 2 [157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181]. Furthermore, multiple databases have been specifically curated to cater to human single-cell omics, providing comprehensive analytical tools for data analysis and interpretation [182, 183]. On this basis, SM-omics technologies, including multi omic single-scan assay with integrated combinatorial analysis (MOSAICA) [184], SM-omics [185], spatial molecular imaging (SMI) [186], DBiT-seq [56] and spatial protein and transcriptome sequencing (SPOTS) [187], which retain spatial information coordinates, have been developed. Developing new technologies such as mIHC [188] and cytometry by time of flight (CyTOF) [189], also promotes joint analysis with ST. At present, SM-omics is on Nature’s annual technologies list in 2022. Nevertheless, SM-omics sequencing is still in its infancy. Splicing variants have traditionally been difficult to detect at the RNA level because most techniques are based on single-ended transcription analysis. At the protein level, the analysis of targeted proteins is limited by the number of available variants, such as fluorescent dyes, barcodes, or antibodies, and is limited to the analysis of targeted proteins. In addition, histone modification, chromatin accessibility, and metabolomics research still lack spatial counterparts. The integrated approach requires high-quality acquisition of multiple parameters, such as gene coverage, throughput, accuracy, and sequencing depth. Finally, how to achieve actual single-cell resolution should also be considered.
On the other hand, gene expression in biological systems is highly dynamic [190], and analysing cellular interactions and regulatory mechanisms from temporal and spatial dimensions assists in understanding the rules in complex systems, namely, spatiotemporal omics. RNA metabolic labelling strategies were introduced in RNA-seq to distinguish new mRNA from old ones, such as 4-thio-uridine (4sU) [191] and 5-ethynyl-uridine (5-EU) [192]. In addition, RNA timestamps bind RNA-seq to infer the “age” of RNA in hours [193]. However, these methods are only suitable for describing short time scales or points in time characteristics of cells. Therefore, continuous analysis and spatial localization of the same cell make spatiotemporal omics possible, such as “high-throughput Patch-seq” [194] and “Live-seq with spatial context” [155].
Bioinformatics support
Increased spatial dimensionality, data volume, and complexity present formidable challenges during the data analysis phase. Numerous bioinformatics tools employed for scRNA-seq analysis can be readily adapted for spatial analysis, encompassing deconvolution, clustering, cell type annotation [195, 196], and other essential processing steps [197]. However, it ignores spatial location and structural characteristics; therefore, the current bioinformatics pipeline must be improved to analyze the unique properties of ST. Bioinformatics analysis software for ST is emerging endlessly. Trendseek [198], SpatialDE [199], graph laplacian-based integrative single-cell spatial analysis (GLISS), and SpaGCN [200] have been developed to analyze the relationships between spatial location and gene expression. Methods for studying the interactions between cells include Graph Convolutional Neural networks for Genes (GCNG), spatial variance component analysis (SVCA) [201], novaSpaRc [202], and SpaOTsc [203]. Methods like stLearn, BayesSpace [204], spatial clustering using the hidden Markov random field based on empirical bayes (SC-MEB) [205] and MULTILAYER [206] are used for spatial clustering. It is anticipated that advancements in spatial omics technologies will result in the proliferation of analytical computing tools, facilitating the harmonization of data across diverse platforms and fostering the integration of information tools such as machine learning and image segmentation. This integration is crucial for a deeper understanding of intricate spatial structures and expanding availability to a wide array of available data sources [207].
With the exception of commercial platforms like 10 × Visium, Stereo-seq, and GeoMx DSP, the majority of the aforementioned ST techniques are generally confined to laboratory settings. This limitation is expected, given the recent publication of these methods and the high costs associated with translating experiments into diagnostic applications. Standardizing experimental procedures and data analysis pipelines is anticipated to facilitate the commercialization and widespread accessibility of spatial omics analysis techniques. Moreover, ongoing efforts to integrate automated sample processing, 3D structure, deep section scanning, and time series data will further advance this field by revealing new cell structures and expanding our understanding of biological processes. Given that the future progress of ST necessitates the convergence of multiple disciplines, close collaboration is imperative between researchers in bioinformatics analytics, automation devices, clinical translational research, and biomedical fields [208].
Conclusions
This review presented cutting-edge technologies in ST and their applications to organ/tissue physiological and pathological processes. As a newer iteration of scRNA-seq, the field of ST is expanding rapidly, significantly improving our understanding of developmental biology and pathogenesis and transforming our ability to diagnose, understand, and treat diseases. With this growing panoply and collaborative efforts of bioinformatics, engineering, and SM-omics, we expect to obtain high-throughput molecular information concerning full-length transcriptomes when the cost significantly decreases. These data will provide a more comprehensive view for clarifying interactions between cell biological mechanisms within tissue ecosystems.
Availability of data and materials
Data availability is not applicable to this article as no new data were created or analyzed in this study.
Abbreviations
- ACE2:
-
Angiotensin converting enzyme 2
- AIS:
-
Adenocarcinoma in situ
- Apt-seq:
-
Aptamers and single cell sequencing
- ARDS:
-
Acute respiratory distress syndrome
- AS:
-
Alternative splicing
- ATP:
-
Adenosine triphosphate
- BOLORAMIS:
-
Barcoded oligonucleotides ligated on RNA amplified for multiplexed and parallel in situ analyses
- BrMs:
-
Brain metastasis
- CBSST-seq:
-
Cross-amplified barcodes on slides for spatial transcriptomics sequencing
- CID:
-
Coordinate identity
- CITE-seq:
-
Cellular indexing of transcriptomes and epitopes by sequencing
- COPD:
-
Chronic obstructive pulmonary diseases
- COVID-19:
-
Corona virus disease 2019
- CyTOF:
-
Cytometry by time of flight
- DAD:
-
Diffuse alveolar damage
- DBiT-seq:
-
Deterministic barcoding in tissue for spatial omics sequencing
- DMF-DR-seq:
-
Digital microfluidics gDNA-mRNA sequencing
- DNA:
-
Deoxyribonucleic acid
- DR-seq:
-
GDNA-mRNA sequencing
- DSP:
-
Digital spatial profiler
- EASI-FISH:
-
Expansion-assisted iterative fluorescence in situ hybridization
- ECCITE-seq:
-
Expanded CRISPR-compatible cellular indexing of transcriptomes and epitopes by sequencing
- EEL FISH:
-
Enhanced electric FISH
- EMT:
-
Epithelial-mesenchymal transition
- exFISH:
-
Expansion FISH
- ExSeq:
-
Expansion sequencing
- FACS:
-
Fluorescence activated cell sorting
- FFPE:
-
Formalin-fixed paraffin-embedded
- FISSEQ:
-
Fluorescent in situ sequencing
- GCNG:
-
Graph Convolutional Neural networks for Genes
- Geo-seq:
-
Geographical position sequencing
- GLISS:
-
Graph laplacian-based integrative single-cell spatial analysis
- G&T-seq:
-
Genome and transcriptome sequencing
- HCA:
-
Human Cell Atlas
- HCLA:
-
Human Cell Lung Atlas
- HCR:
-
Hybridization chain reaction
- HDST:
-
High-definition ST
- HybISS:
-
Hybridization-based ISS
- IAC:
-
Invasive adenocarcinoma
- ICI:
-
Immune checkpoint inhibitor
- ICP:
-
Immune checkpoint
- IISS:
-
Improved ISS
- inCITE-seq:
-
Intranuclear cellular indexing of transcriptomes and epitopes
- INs-seq:
-
Intracellular staining and sequencing
- IPF:
-
Idiopathic pulmonary fibrosis
- ISH:
-
In situ hybridization
- ISS:
-
In situ sequencing
- ITH:
-
Intratumoral heterogeneity
- LCM:
-
Laser capture microdissection
- lncRNA:
-
Long noncoding RNA
- LUAD:
-
Lung adenocarcinoma
- LUSC:
-
Lung squamous cell carcinoma
- MERFISH:
-
Multiplexed error-robust FISH
- MID:
-
Molecular identifiers
- mIHC:
-
Multiplex immunohistochemistry
- miRNA:
-
MicroRNA
- MISAR-seq:
-
Microfluidic indexing based spatial ATAC and RNA sequencing
- MOSAICA:
-
Multi omic single-scan assay with integrated combinatorial analysis
- mRNA:
-
Messenger RNA
- NGS:
-
Next-generation sequencing
- NK cell:
-
Natural killer cell
- NSCLC:
-
Non-small cell lung cancer
- osmFISH:
-
Ouroboros smFISH
- Paired-seq:
-
Parallel analysis of individual cells for RNA expression and DNA accessibility by sequencing
- PASC:
-
Post-acute sequelae of SARS-CoV2
- PEA/STA:
-
Proximity extension assays/specific (RNA) target amplification
- Pixel-seq:
-
Poly-indexed library-sequencing
- PLAYR:
-
Proximity ligation assay for RNA
- PLISH:
-
Proximity ligation in situ hybridization technology
- RCA:
-
Rolling circle amplification
- REAP-seq:
-
RNA expression and protein sequencing assay
- RNA-ISH:
-
RNA-in situ hybridization
- ROI:
-
Regions of interest
- SABER:
-
Signal amplification by exchange reaction
- scCAT-seq:
-
Single-cell chromatin accessibility and transcriptome sequencing
- sc-GEM:
-
Single-cell analysis of genotype, expression and methylation
- sciCAR:
-
Single-cell combinatorial indexing of chromatin accessibility and mRNA
- SCITO-seq:
-
Single-cell combinatorial indexed cytometry sequencing
- SC-MEB:
-
Spatial clustering using the hidden Markov random field based on empirical bayes
- scMT-seq:
-
Single-cell methylome and transcriptome sequencing
- scM&T-seq:
-
Single-cell genome-wide methylome and transcriptome sequencing
- scNMT-seq:
-
Single-cell nucleosome, methylation and transcription sequencing
- scRNA-seq:
-
Single-cell RNA sequencing
- scTHS-seq:
-
Single-cell transposome hypersensitive site sequencing
- scTrio-seq:
-
Single-cell triple omics sequencing
- SEDAL:
-
Sequencing with error-reduction by dynamic annealing and ligation
- seqFISH:
-
Sequential FISH
- SIDR:
-
Simultaneous isolation of genomic DNA and total RNA
- smFISH:
-
Single-molecule fluorescence in situ hybridization
- smHCR:
-
Single-molecule hybridization chain reaction
- SMI:
-
Spatial molecular imaging
- SM-omics:
-
Spatial multi-omics
- SNARE-seq:
-
Single-nucleus chromatin accessibility and mRNA expression sequencing
- snDrop-seq:
-
Single-nucleus droplet-based sequencing
- snoRNA:
-
Small nucleolar RNA
- SNVs:
-
Single nucleotide variants
- spatial-CITE-seq:
-
Spatial co-indexing of transcriptomes and epitopes
- SARS-CoV2:
-
Severe acute respiratory syndrome coronavirus 2
- SPOTS:
-
Spatial protein and transcriptome sequencing
- ST:
-
Spatial transcriptomics
- STARmap:
-
Spatially-resolved transcript amplicon readout mapping
- Stereo-seq:
-
Spatial enhanced resolution omics sequencing
- STRS:
-
Spatial total RNA sequencing
- SVCA:
-
Spatial variance component analysis
- TAMs:
-
Tumor-associated macrophages
- TIVA:
-
Transcriptome in vivo analysis
- TLS:
-
Tertiary lymphoid structures
- TME:
-
Tumor microenvironment
- tRNA:
-
Transfer RNA
- UMI:
-
Unique molecular identifier
- xDBiT:
-
Multiplexed deterministic barcoding in tissue
- yPAP:
-
Yeast poly(A) polymerase
- 3D:
-
Three-dimensional
- 4sU:
-
4-Thio-uridine
- 5-EU:
-
5-Ethynyl-uridine
References
Zepp JA, Morrisey EE. Cellular crosstalk in the development and regeneration of the respiratory system. Nat Rev Mol Cell Biol. 2019;20(9):551–66.
Kiley JP. Advancing respiratory research. Chest. 2011;140(2):497–501.
Goldstraw P, Ball D, Jett JR, Le Chevalier T, Lim E, Nicholson AG, et al. Non-small-cell lung cancer. Lancet. 2011;378(9804):1727–40.
Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med. 2019;25(7):1153–63.
Crosetto N, Bienko M, van Oudenaarden A. Spatially resolved transcriptomics and beyond. Nat Rev Genet. 2015;16(1):57–66.
Li X, Wang CY. From bulk, single-cell to spatial RNA sequencing. Int J Oral Sci. 2021;13(1):36.
Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–82.
Wang Y, Wang JY, Schnieke A, Fischer K. Advances in single-cell sequencing: insights from organ transplantation. Mil Med Res. 2021;8(1):45.
Tian L, Chen F, Macosko EZ. The expanding vistas of spatial transcriptomics. Nat Biotechnol. 2022;41(6):773–82.
Williams CG, Lee HJ, Asatsuma T, Vento-Tormo R, Haque A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022;14(1):68.
Thommen DS, Schumacher TN. T cell dysfunction in cancer. Cancer Cell. 2018;33(4):547–62.
Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353(6294):78–82.
Close JL, Long BR, Zeng H. Spatially resolved transcriptomics in neuroscience. Nat Methods. 2021;18(1):23–5.
Seferbekova Z, Lomakin A, Yates LR, Gerstung M. Spatial biology of cancer evolution. Nat Rev Genet. 2022;24(5):295–313.
Vandereyken K, Sifrim A, Thienpont B, Voet T. Methods and applications for single-cell and spatial multi-omics. Nat Rev Genet. 2023. https://doi.org/10.1038/s41576-023-00580-2.
Park HE, Jo SH, Lee RH, Macks CP, Ku T, Park J, et al. Spatial transcriptomics: technical aspects of recent developments and their applications in neuroscience and cancer research. Adv Sci (Weinh). 2023;10(16):e2206939.
Moffitt JR, Lundberg E, Heyn H. The emerging landscape of spatial profiling technologies. Nat Rev Genet. 2022;23(12):741–59.
Levsky JM, Singer RH. Fluorescence in situ hybridization: past, present and future. J Cell Sci. 2003;116(Pt 14):2833–8.
Liu S, Punthambaker S, Iyer EPR, Ferrante T, Goodwin D, Fürth D, et al. Barcoded oligonucleotides ligated on RNA amplified for multiplexed and parallel in situ analyses. Nucleic Acids Res. 2021;49(10):e58.
Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single-cell in situ RNA profiling by sequential hybridization. Nat Methods. 2014;11(4):360–1.
Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348(6233):aaa6090.
Shah S, Lubeck E, Schwarzkopf M, He TF, Greenbaum A, Sohn CH, et al. Single-molecule RNA detection at depth by hybridization chain reaction and tissue hydrogel embedding and clearing. Development. 2016;143(15):2862–7.
Eng CHL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. 2019;568(7751):235–9.
Codeluppi S, Borm LE, Zeisel A, La Manno G, Van Lunteren JA, Svensson CI, et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods. 2018;15(11):932–5.
Kishi JY, Lapan SW, Beliveau BJ, West ER, Zhu A, Sasaki HM, et al. SABER amplifies FISH: enhanced multiplexed imaging of RNA and DNA in cells and tissues. Nat Methods. 2019;16(6):533–44.
Borm LE, Mossi Albiach A, Mannens CCA, Janusauskas J, Özgün C, Fernández-García D, et al. Scalable in situ single-cell profiling by electrophoretic capture of mRNA using EEL FISH. Nat Biotechnol. 2023;41(2):222–31.
Chen F, Wassie AT, Cote AJ, Sinha A, Alon S, Asano S, et al. Nanoscale imaging of RNA with expansion microscopy. Nat Methods. 2016;13(8):679–84.
Wang Y, Eddison M, Fleishman G, Weigert M, Xu S, Wang T, et al. EASI-FISH for thick tissue defines lateral hypothalamus spatio-molecular organization. Cell. 2021;184(26):6361-77.e24.
Ke R, Mignardi M, Pacureanu A, Svedlund J, Botling J, Wählby C, et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat Methods. 2013;10(9):857–60.
Chen X, Sun YC, Church GM, Lee JH, Zador AM. Efficient in situ barcode sequencing using padlock probe-based BaristaSeq. Nucleic Acids Res. 2018;46(4):e22.
Tang X, Chen J, Zhang X, Liu X, Xie Z, Wei K, et al. Improved in situ sequencing for high-resolution targeted spatial transcriptomic analysis in tissue sections. J Genet Genomics. 2023. https://doi.org/10.1016/j.jgg.2023.02.004.
Li Q, Lin Z, Liu R, Tang X, Huang J, He Y, et al. Multimodal charting of molecular and functional cell states via in situ electro-sequencing. Cell. 2023;186(9):2002-17.e21.
Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018;361(6400):eaat5691.
Zeng H, Huang J, Zhou H, Meilandt WJ, Dejanovic B, Zhou Y, et al. Integrative in situ mapping of single-cell transcriptional states and tissue histopathology in a mouse model of Alzheimer’s disease. Nat Neurosci. 2023;26(3):430–46.
Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343(6177):1360–3.
Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Ferrante TC, Terry R, et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat Protoc. 2015;10(3):442–58.
Alon S, Goodwin DR, Sinha A, Wassie AT, Chen F, Daugharthy ER, et al. Expansion sequencing: spatially precise in situ transcriptomics in intact biological systems. Science. 2021;371(6528):eaax2656.
Wu CC, Kruse F, Vasudevarao MD, Junker JP, Zebrowski DC, Fischer K, et al. Spatially resolved genome-wide transcriptional profiling identifies BMP signaling as essential regulator of zebrafish cardiomyocyte regeneration. Dev Cell. 2016;36(1):36–49.
Chen J, Suo S, Tam PP, Han JDJ, Peng G, Jing N. Spatial transcriptomic analysis of cryosectioned tissue samples with Geo-seq. Nat Protoc. 2017;12(3):566–80.
Boisset JC, Vivié J, Grün D, Muraro MJ, Lyubimova A, Van Oudenaarden A. Mapping the physical network of cellular interactions. Nat Methods. 2018;15(7):547–53.
Medaglia C, Giladi A, Stoler-Barak L, De Giovanni M, Salame TM, Biram A, et al. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science. 2017;358(6370):1622–6.
Lovatt D, Ruble BK, Lee J, Dueck H, Kim TK, Fisher S, et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods. 2014;11(2):190–6.
Hu KH, Eichorst JP, McGinnis CS, Patterson DM, Chow ED, Kersten K, et al. ZipSeq: barcoding for real-time mapping of single cell transcriptomes. Nat Methods. 2020;17(8):833–43.
Merritt CR, Ong GT, Church SE, Barker K, Danaher P, Geiss G, et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat Biotechnol. 2020;38(5):586–99.
Haase C, Gustafsson K, Mei S, Yeh SC, Richter D, Milosevic J, et al. Image-seq: spatially resolved single-cell sequencing guided by in situ and in vivo imaging. Nat Methods. 2022;19(12):1622–33.
Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363(6434):1463–7.
Vickovic S, Eraslan G, Salmén F, Klughammer J, Stenbeck L, Schapiro D, et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat Methods. 2019;16(10):987–90.
Stickels RR, Murray E, Kumar P, Li J, Marshall JL, Di Bella DJ, et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat Biotechnol. 2021;39(3):313–9.
Wei X, Fu S, Li H, Liu Y, Wang S, Feng W, et al. Single-cell Stereo-seq reveals induced progenitor cells involved in axolotl brain regeneration. Science. 2022;377(6610):eabp9444.
Chen A, Liao S, Cheng M, Ma K, Wu L, Lai Y, et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell. 2022;185(10):1777-92.e21.
Srivatsan SR, Regier MC, Barkan E, Franks JM, Packer JS, Grosjean P, et al. Embryo-scale, single-cell spatial transcriptomics. Science. 2021;373(6550):111–7.
Lee Y, Bogdanoff D, Wang Y, Hartoularos GC, Woo JM, Mowery CT, et al. XYZeq: spatially resolved single-cell RNA sequencing reveals expression heterogeneity in the tumor microenvironment. Sci Adv. 2021;7(17):eabg4755.
Cho CS, Xi J, Si Y, Park SR, Hsu JE, Kim M, et al. Microscopic examination of spatial transcriptome using Seq-Scope. Cell. 2021;184(13):3559-72.e22.
Fu X, Sun L, Dong R, Chen JY, Silakit R, Condon LF, et al. Polony gels enable amplifiable DNA stamping and spatial transcriptomics of chronic pain. Cell. 2022;185(24):4621-33.e17.
Cao J, Chen X, Huang S, Shi W, Fan Q, Gong Y, et al. Microfluidics-based single cell analysis: from transcriptomics to spatiotemporal multi-omics. Trac Trends Anal Chem. 2023;158:116868.
Liu Y, Yang M, Deng Y, Su G, Enninful A, Guo CC, et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell. 2020;183(6):1665-81.e18.
Wirth J, Huber N, Yin K, Brood S, Chang S, Martinez-Jimenez CP, et al. Spatial transcriptomics using multiplexed deterministic barcoding in tissue. Nat Commun. 2023;14(1):1523.
Jin Z, Yu N, Bai J, Liu Z, Li H, Zhang J, et al. Cross-amplified barcodes on slides for spatial transcriptomics sequencing. bioRxiv. 2022. https://doi.org/10.1101/2022.08.25.504658.
Zhao H, Tian G, Hu A. Matrix-seq: an adjustable-resolution spatial transcriptomics via microfluidic matrix-based barcoding. bioRxiv. 2022. https://doi.org/10.1101/2022.08.05.502952.
Jiang F, Zhou X, Qian Y, Zhu M, Wang L, Li Z, et al. Simultaneous profiling of spatial gene expression and chromatin accessibility for mouse brain development. bioRxiv. 2023. https://doi.org/10.1101/2022.03.22.485333.
Liu Y, DiStasio M, Su G, Asashima H, Enninful A, Qin X, et al. Spatial-CITE-seq: spatially resolved high-plex protein and whole transcriptome co-mapping. Res Sq. 2022. https://doi.org/10.21203/rs.3.rs-1499315/v1.
McKellar DW, Mantri M, Hinchman MM, Parker JSL, Sethupathy P, Cosgrove BD, et al. Spatial mapping of the total transcriptome by in situ polyadenylation. Nat Biotechnol. 2023;41(4):513–20.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–23.
Sinjab A, Han G, Treekitkarnmongkol W, Hara K, Brennan PM, Dang M, et al. Resolving the spatial and cellular architecture of lung adenocarcinoma by multiregion single-cell sequencing. Cancer Discov. 2021;11(10):2506–23.
Zhu J, Fan Y, Xiong Y, Wang W, Chen J, Xia Y, et al. Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single-cell RNA sequencing and spatial transcriptomics. Exp Mol Med. 2022;54(11):2060–76.
Wong-Rolle A, Dong Q, Zhu Y, Divakar P, Hor JL, Kedei N, et al. Spatial meta-transcriptomics reveal associations of intratumor bacteria burden with lung cancer cells showing a distinct oncogenic signature. J Immunother Cancer. 2022;10(7):e004698.
Ljungberg MC, Sadi M, Wang Y, Aronow BJ, Xu Y, Kao RJ, et al. Spatial distribution of marker gene activity in the mouse lung during alveolarization. Data Brief. 2019;22:365–72.
Calabrese F, Lunardi F, Tauro V, Pezzuto F, Fortarezza F, Vedovelli L, et al. RNA sequencing of epithelial cell/fibroblastic foci sandwich in idiopathic pulmonary fibrosis: new insights on the signaling pathway. Int J Mol Sci. 2022;23(6):3323.
Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe Å, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. 2021;595(7865):107–13.
Kadefors M, Rolandsson Enes S, Åhrman E, Michaliková B, Löfdahl A, Dellgren G, et al. CD105+CD90+CD13+ identifies a clonogenic subset of adventitial lung fibroblasts. Sci Rep. 2021;11(1):24417.
Rozenblatt-Rosen O, Stubbington MJT, Regev A, Teichmann SA. The Human Cell Atlas: from vision to reality. Nature. 2017;550(7677):451–3.
Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, et al. The human cell atlas. Elife. 2017;6:e27041.
Schupp JC, Adams TS, Cosme C Jr, Raredon MSB, Yuan Y, Omote N, et al. Integrated single-cell atlas of endothelial cells of the human lung. Circulation. 2021;144(4):286–302.
Deprez M, Zaragosi LE, Truchi M, Becavin C, Ruiz García S, Arguel MJ, et al. A single-cell atlas of the human healthy airways. Am J Respir Crit Care Med. 2020;202(12):1636–45.
Ardini-Poleske ME, Clark RF, Ansong C, Carson JP, Corley RA, Deutsch GH, et al. LungMAP: the molecular atlas of lung development program. Am J Physiol Lung Cell Mol Physiol. 2017;313(5):L733–40.
Luecken MD, Zaragosi LE, Madissoon E, Sikkema L, Firsova AB, De Domenico E, et al. The discovAIR project: a roadmap towards the Human Lung Cell Atlas. Eur Respir J. 2022;60(2):2102057.
Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature. 2018;560(7718):377–81.
Goldfarbmuren KC, Jackson ND, Sajuthi SP, Dyjack N, Li KS, Rios CL, et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat Commun. 2020;11(1):2485.
Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature. 2020;587(7835):619–25.
Madissoon E, Oliver AJ, Kleshchevnikov V, Wilbrey-Clark A, Polanski K, Richoz N, et al. A spatially resolved atlas of the human lung characterizes a gland-associated immune niche. Nat Genet. 2023;55(1):66–77.
Sountoulidis A, Marco Salas S, Braun E, Avenel C, Bergenstråhle J, Theelke J, et al. A topographic atlas defines developmental origins of cell heterogeneity in the human embryonic lung. Nat Cell Biol. 2023;25(2):351–65.
Nagendran M, Riordan DP, Harbury PB, Desai TJ. Automated cell-type classification in intact tissues by single-cell molecular profiling. Elife. 2018;7:e30510.
Sountoulidis A, Liontos A, Nguyen HP, Firsova AB, Fysikopoulos A, Qian X, et al. SCRINSHOT enables spatial mapping of cell states in tissue sections with single-cell resolution. PLoS Biol. 2020;18(11):e3000675.
Fu T, Dai LJ, Wu SY, Xiao Y, Ma D, Jiang YZ, et al. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J Hematol Oncol. 2021;14(1):98.
Gibson GJ, Loddenkemper R, Lundbäck B, Sibille Y. Respiratory health and disease in Europe: the new European Lung White Book. Eur Respir J. 2013;42(3):559–63.
Rajewsky N, Almouzni G, Gorski SA, Aerts S, Amit I, Bertero MG, et al. Publisher correction: LifeTime and improving European healthcare through cell-based interceptive medicine. Nature. 2021;592(7852):E8.
Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017;355(6322):eaaf8399.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–50.
Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–72.
Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24(8):1277–89.
Dhainaut M, Rose SA, Akturk G, Wroblewska A, Nielsen SR, Park ES, et al. Spatial CRISPR genomics identifies regulators of the tumor microenvironment. Cell. 2022;185(7):1223-39.e20.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–45.
Larroquette M, Guegan JP, Besse B, Cousin S, Brunet M, Le Moulec S, et al. Spatial transcriptomics of macrophage infiltration in non-small cell lung cancer reveals determinants of sensitivity and resistance to anti-PD1/PD-L1 antibodies. J Immunother Cancer. 2022;10(5):e003890.
Monkman J, Kim H, Mayer A, Mehdi A, Matigian N, Cumberbatch M, et al. Multi-omic and spatial dissection of immunotherapy response groups in non-small cell lung cancer. Immunology. 2023. https://doi.org/10.1111/imm.13646.
Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 2017;17(9):557–69.
Ji JJ, Fan J. Discovering myeloid cell heterogeneity in the lung by means of next generation sequencing. Mil Med Res. 2019;6(1):33.
Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity. 2022;55(9):1564–80.
Zhang L, Mao S, Yao M, Chao N, Yang Y, Ni Y, et al. Spatial transcriptome sequencing revealed spatial trajectory in the non-small cell lung carcinoma. bioRxiv. 2021. https://doi.org/10.1101/2021.04.26.441394.
Song X, Xiong A, Wu F, Li X, Wang J, Jiang T, et al. Spatial multi-omics revealed the impact of tumor ecosystem heterogeneity on immunotherapy efficacy in patients with advanced non-small cell lung cancer treated with bispecific antibody. J Immunother Cancer. 2023;11(2):e00623.
Ciriello G, Magnani L. The many faces of cancer evolution. iScience. 2021;24(5):102403.
Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science. 2018;360(6386):331–5.
Gaiti F, Chaligne R, Gu H, Brand RM, Kothen-Hill S, Schulman RC, et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature. 2019;569(7757):576–80.
Tavernari D, Battistello E, Dheilly E, Petruzzella AS, Mina M, Sordet-Dessimoz J, et al. Nongenetic evolution drives lung adenocarcinoma spatial heterogeneity and progression. Cancer Discov. 2021;11(6):1490–507.
Zhang Q, Abdo R, Iosef C, Kaneko T, Cecchini M, Han VK, et al. The spatial transcriptomic landscape of non-small cell lung cancer brain metastasis. Nat Commun. 2022;13(1):5983.
Wong-Rolle A, Wei HK, Zhao C, Jin C. Unexpected guests in the tumor microenvironment: microbiome in cancer. Protein Cell. 2021;12(5):426–35.
Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. 2020;368(6494):973–80.
Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941–52.
Wijsenbeek M, Cottin V. Spectrum of fibrotic lung diseases. N Engl J Med. 2020;383(10):958–68.
Keow J, Cecchini MJ, Jayawardena N, Zompatori M, Joseph MG, Mura M. Digital quantification of p16-positive foci in fibrotic interstitial lung disease is associated with a phenotype of idiopathic pulmonary fibrosis with reduced survival. Respir Res. 2022;23(1):147.
Shi X, Wang J, Zhang X, Yang S, Luo W, Wang S, et al. GREM1/PPP2R3A expression in heterogeneous fibroblasts initiates pulmonary fibrosis. Cell Biosci. 2022;12(1):123.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3.
Whitworth J. COVID-19: a fast evolving pandemic. Trans R Soc Trop Med Hyg. 2020;114(4):241–8.
Desai N, Neyaz A, Szabolcs A, Shih AR, Chen JH, Thapar V, et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat Commun. 2020;11(1):6319.
Dinnon KH 3rd, Leist SR, Okuda K, Dang H, Fritch EJ, Gully KL, et al. SARS-CoV-2 infection produces chronic pulmonary epithelial and immune cell dysfunction with fibrosis in mice. Sci Transl Med. 2022;14(664):eabo5070.
Margaroli C, Benson P, Sharma NS, Madison MC, Robison SW, Arora N, et al. Spatial mapping of SARS-CoV-2 and H1N1 lung injury identifies differential transcriptional signatures. Cell Rep Med. 2021;2(4):100242.
Butler D, Mozsary C, Meydan C, Foox J, Rosiene J, Shaiber A, et al. Shotgun transcriptome, spatial omics, and isothermal profiling of SARS-CoV-2 infection reveals unique host responses, viral diversification, and drug interactions. Nat Commun. 2021;12(1):1660.
Downes DJ, Cross AR, Hua P, Roberts N, Schwessinger R, Cutler AJ, et al. Identification of LZTFL1 as a candidate effector gene at a COVID-19 risk locus. Nat Genet. 2021;53(11):1606–15.
Kulasinghe A, Tan CW, Ribeiro Dos Santos Miggiolaro AF, Monkman J, Sadeghirad H, Bhuva DD, et al. Profiling of lung SARS-CoV-2 and influenza virus infection dissects virus-specific host responses and gene signatures. Eur Respir J. 2022;59(6):2101881.
Yang CX, Tomchaney M, Landecho MF, Zamacona BR, Marin Oto M, Zulueta J, et al. Lung spatial profiling reveals a T cell signature in COPD patients with fatal SARS-CoV-2 infection. Cells. 2022;11(12):1864.
Erjefält JS, de Souza Xavier Costa N, Jönsson J, Cozzolino O, Dantas KC, Clausson CM, et al. Diffuse alveolar damage patterns reflect the immunological and molecular heterogeneity in fatal COVID-19. EBioMedicine. 2022;83:104229.
Xu Z, Wang X, Fan L, Wang F, Lin B, Wang J, et al. Integrative analysis of spatial transcriptome with single-cell transcriptome and single-cell epigenome in mouse lungs after immunization. iScience. 2022;25(9):104900.
Cross AR, de Andrea CE, Villalba-Esparza M, Landecho MF, Cerundolo L, Weeratunga P, et al. Spatial transcriptomic characterization of COVID-19 pneumonitis identifies immune circuits related to tissue injury. JCI Insight. 2023;8(2):e157837.
Mothes R, Pascual-Reguant A, Koehler R, Liebeskind J, Liebheit A, Bauherr S, et al. Distinct tissue niches direct lung immunopathology via CCL18 and CCL21 in severe COVID-19. Nat Commun. 2023;14(1):791.
Tan X, Grice LF, Tran M, Mulay O, Monkman J, Blick T, et al. A robust platform for integrative spatial multi-omics analysis to map immune responses to SARS-CoV-2 infection in lung tissues. bioRxiv. 2023. https://doi.org/10.1101/2023.02.19.529128.
Carow B, Hauling T, Qian X, Kramnik I, Nilsson M, Rottenberg ME. Spatial and temporal localization of immune transcripts defines hallmarks and diversity in the tuberculosis granuloma. Nat Commun. 2019;10(1):1823.
Magoulopoulou A, Qian X, Pediatama Setiabudiawan T, Marco Salas S, Yokota C, Rottenberg ME, et al. Spatial resolution of Mycobacterium tuberculosis bacteria and their surrounding immune environments based on selected key transcripts in mouse lungs. Front Immunol. 2022;13:876321.
Dutt TS, Karger BR, Fox A, Youssef N, Dadhwal R, Ali MZ, et al. Mucosal exposure to non-tuberculous mycobacteria elicits B cell-mediated immunity against pulmonary tuberculosis. Cell Rep. 2022;41(11): 111783.
Beppu AK, Zhao J, Yao C, Carraro G, Israely E, Coelho AL, et al. Epithelial plasticity and innate immune activation promote lung tissue remodeling following respiratory viral infection. bioRxiv. 2022. https://doi.org/10.1101/2021.09.22.461381.
Boyd DF, Allen EK, Randolph AG, Guo XZJ, Weng Y, Sanders CJ, et al. Exuberant fibroblast activity compromises lung function via ADAMTS4. Nature. 2020;587(7834):466–71.
Rodor J, Chen SH, Scanlon JP, Monteiro JP, Caudrillier A, Sweta S, et al. Single-cell RNA sequencing profiling of mouse endothelial cells in response to pulmonary arterial hypertension. Cardiovasc Res. 2022;118(11):2519–34.
Tang W, Li M, Teng F, Cui J, Dong J, Wang W. Single-cell RNA-sequencing in asthma research. Front Immunol. 2022;13:988573.
Sauler M, McDonough JE, Adams TS, Kothapalli N, Barnthaler T, Werder RB, et al. Characterization of the COPD alveolar niche using single-cell RNA sequencing. Nat Commun. 2022;13(1):494.
Saiselet M, Rodrigues-Vitória J, Tourneur A, Craciun L, Spinette A, Larsimont D, et al. Transcriptional output, cell-type densities, and normalization in spatial transcriptomics. J Mol Cell Biol. 2020;12(11):906–8.
Yagi Y, Aly RG, Tabata K, Barlas A, Rekhtman N, Eguchi T, et al. Three-dimensional histologic, immunohistochemical, and multiplex immunofluorescence analyses of dynamic vessel co-option of spread through air spaces in lung adenocarcinoma. J Thorac Oncol. 2020;15(4):589–600.
Asp M, Giacomello S, Larsson L, Wu C, Fürth D, Qian X, et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. 2019;179(7):1647-60.e19.
Jansova D, Tetkova A, Koncicka M, Kubelka M, Susor A. Localization of RNA and translation in the mammalian oocyte and embryo. PLoS ONE. 2018;13(3):e0192544.
Zhao L, Song W, Chen YG. Mesenchymal-epithelial interaction regulates gastrointestinal tract development in mouse embryos. Cell Rep. 2022;40(2):111053.
Choe K, Pak U, Pang Y, Hao W, Yang X. Advances and challenges in spatial transcriptomics for developmental biology. Biomolecules. 2023;13(1):156.
Stenbeck L, Taborsak-Lines F, Giacomello S. Enabling automated and reproducible spatially resolved transcriptomics at scale. Heliyon. 2022;8(6):e09651.
Fazal FM, Han S, Parker KR, Kaewsapsak P, Xu J, Boettiger AN, et al. Atlas of subcellular RNA localization revealed by APEX-Seq. Cell. 2019;178(2):473-90.e26.
Rao A, Barkley D, França GS, Yanai I. Exploring tissue architecture using spatial transcriptomics. Nature. 2021;596(7871):211–20.
Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell. 2020;182(4):976-91.e19.
Nagarajan MB, Tentori AM, Zhang WC, Slack FJ, Doyle PS. Spatially resolved and multiplexed microRNA quantification from tissue using nanoliter well arrays. Microsyst Nanoeng. 2020;6:51.
Meylan M, Petitprez F, Becht E, Bougoüin A, Pupier G, Calvez A, et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity. 2022;55(3):527-41.e5.
McCart Reed AE, Bennett J, Kutasovic JR, Kalaw E, Ferguson K, Yeong J, et al. Digital spatial profiling application in breast cancer: a user’s perspective. Virchows Arch. 2020;477(6):885–90.
Janesick A, Shelansky R, Gottscho AD, Wagner F, Rouault M, Beliakoff G, et al. High resolution mapping of the breast cancer tumor microenvironment using integrated single cell, spatial and in situ analysis of FFPE tissue. bioRxiv. 2022. https://doi.org/10.1101/2022.10.06.510405.
Liao J, Lu X, Shao X, Zhu L, Fan X. Uncovering an organ’s molecular architecture at single-cell resolution by spatially resolved transcriptomics. Trends Biotechnol. 2021;39(1):43–58.
Salmen F, De Jonghe J, Kaminski TS, Alemany A, Parada GE, Verity-Legg J, et al. High-throughput total RNA sequencing in single cells using VASA-seq. Nat Biotechnol. 2022;40(12):1780–93.
Kruse F, Junker JP, van Oudenaarden A, Bakkers J. Tomo-seq: a method to obtain genome-wide expression data with spatial resolution. Methods Cell Biol. 2016;135:299–307.
Peng G, Suo S, Chen J, Chen W, Liu C, Yu F, et al. Spatial transcriptome for the molecular annotation of lineage fates and cell identity in mid-gastrula mouse embryo. Dev Cell. 2016;36(6):681–97.
Dong K, Zhang S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat Commun. 2022;13(1):1739.
Sun D, Liu Z, Li T, Wu Q, Wang C. STRIDE: accurately decomposing and integrating spatial transcriptomics using single-cell RNA sequencing. Nucleic Acids Res. 2022;50(7):e42.
He Y, Tang X, Huang J, Ren J, Zhou H, Chen K, et al. ClusterMap for multi-scale clustering analysis of spatial gene expression. Nat Commun. 2021;12(1):5909.
Patange S, Girvan M, Larson DR. Single-cell systems biology: probing the basic unit of information flow. Curr Opin Syst Biol. 2018;8:7–15.
Chen W, Guillaume-Gentil O, Rainer PY, Gäbelein CG, Saelens W, Gardeux V, et al. Live-seq enables temporal transcriptomic recording of single cells. Nature. 2022;608(7924):733–40.
Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5(10):877–9.
MacAulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods. 2015;12(6):519–22.
Dey SS, Kester L, Spanjaard B, Bienko M, van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol. 2015;33(3):285–9.
Han KY, Kim KT, Joung JG, Son DS, Kim YJ, Jo A, et al. SIDR: simultaneous isolation and parallel sequencing of genomic DNA and total RNA from single cells. Genome Res. 2018;28(1):75–87.
Rodriguez-Meira A, Buck G, Clark SA, Povinelli BJ, Alcolea V, Louka E, et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel RNA sequencing. Mol Cell. 2019;73(6):1292-305.e8.
Xu X, Lin L, Yang J, Qian WZ, Su R, Guo XX, et al. Simultaneous single-cell genome and transcriptome sequencing in nanoliter droplet with digital microfluidics identifying essential driving genes. Nano Today. 2022;46:101596.
Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, Hu TX, et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods. 2016;13(3):229–32.
Hu Y, Huang K, An Q, Du G, Hu G, Xue J, et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol. 2016;17:88.
Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016;26(3):304–19.
Cheow LF, Courtois ET, Tan Y, Viswanathan R, Xing Q, Tan RZ, et al. Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat Methods. 2016;13(10):833–6.
Clark SJ, Argelaguet R, Kapourani CA, Stubbs TM, Lee HJ, Alda-Catalinas C, et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun. 2018;9(1):781.
Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol. 2018;36(1):70–80.
Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361(6409):1380–5.
Zhu C, Yu M, Huang H, Juric I, Abnousi A, Hu R, et al. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat Struct Mol Biol. 2019;26(11):1063–70.
Chen S, Lake BB, Zhang K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat Biotechnol. 2019;37(12):1452–7.
Liu L, Liu C, Quintero A, Wu L, Yuan Y, Wang M, et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat Commun. 2019;10(1):470.
Frei AP, Bava FA, Zunder ER, Hsieh EWY, Chen SY, Nolan GP, et al. Highly multiplexed simultaneous detection of RNAs and proteins in single cells. Nat Methods. 2016;13(3):269–75.
Genshaft AS, Li S, Gallant CJ, Darmanis S, Prakadan SM, Ziegler CGK, et al. Multiplexed, targeted profiling of single-cell proteomes and transcriptomes in a single reaction. Genome Biol. 2016;17(1):188.
Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–8.
Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol. 2017;35(10):936–9.
Delley CL, Liu L, Sarhan MF, Abate AR. Combined aptamer and transcriptome sequencing of single cells. Sci Rep. 2018;8(1):2919.
Mimitou EP, Cheng A, Montalbano A, Hao S, Stoeckius M, Legut M, et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat Methods. 2019;16(5):409–12.
Katzenelenbogen Y, Sheban F, Yalin A, Yofe I, Svetlichnyy D, Jaitin DA, et al. Coupled scRNA-Seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell. 2020;182(4):872-85.e19.
Hwang B, Lee DS, Tamaki W, Sun Y, Ogorodnikov A, Hartoularos GC, et al. SCITO-seq: single-cell combinatorial indexed cytometry sequencing. Nat Methods. 2021;18(8):903–11.
Chung H, Parkhurst CN, Magee EM, Phillips D, Habibi E, Chen F, et al. Joint single-cell measurements of nuclear proteins and RNA in vivo. Nat Methods. 2021;18(10):1204–12.
Xu X, Zhang M, Zhang X, Liu Y, Cai L, Zhang Q, et al. Decoding expression dynamics of protein and transcriptome at the single-cell level in paired picoliter chambers. Anal Chem. 2022;94(23):8164–73.
Pan L, Shan S, Tremmel R, Li W, Liao Z, Shi H, et al. HTCA: a database with an in-depth characterization of the single-cell human transcriptome. Nucleic Acids Res. 2023;51(D1):D1019–28.
Shi X, Yu Z, Ren P, Dong X, Ding X, Song J, et al. HUSCH: an integrated single-cell transcriptome atlas for human tissue gene expression visualization and analyses. Nucleic Acids Res. 2023;51(D1):D1029–37.
Vu T, Vallmitjana A, Gu J, La K, Xu Q, Flores J, et al. Spatial transcriptomics using combinatorial fluorescence spectral and lifetime encoding, imaging and analysis. Nat Commun. 2022;13(1):169.
Vickovic S, Lötstedt B, Klughammer J, Mages S, Segerstolpe Å, Rozenblatt-Rosen O, et al. SM-omics is an automated platform for high-throughput spatial multi-omics. Nat Commun. 2022;13(1):795.
He S, Bhatt R, Brown C, Brown EA, Buhr DL, Chantranuvatana K, et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat Biotechnol. 2022;40(12):1794–806.
Ben-Chetrit N, Niu X, Swett AD, Sotelo J, Jiao MS, Stewart CM, et al. Integration of whole transcriptome spatial profiling with protein markers. Nat Biotechnol. 2023;41(6):788–93.
Govek KW, Troisi EC, Miao Z, Aubin RG, Woodhouse S, Camara PG. Single-cell transcriptomic analysis of mIHC images via antigen mapping. Sci Adv. 2021;7(10):eabc5464.
Hartmann FJ, Mrdjen D, McCaffrey E, Glass DR, Greenwald NF, Bharadwaj A, et al. Single-cell metabolic profiling of human cytotoxic T cells. Nat Biotechnol. 2021;39(2):186–97.
Rabani M, Raychowdhury R, Jovanovic M, Rooney M, Stumpo DJ, Pauli A, et al. High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell. 2014;159(7):1698–710.
Ding J, Sharon N, Bar-Joseph Z. Temporal modelling using single-cell transcriptomics. Nat Rev Genet. 2022;23(6):355–68.
Battich N, Beumer J, de Barbanson B, Krenning L, Baron CS, Tanenbaum ME, et al. Sequencing metabolically labeled transcripts in single cells reveals mRNA turnover strategies. Science. 2020;367(6482):1151–6.
Rodriques SG, Chen LM, Liu S, Zhong ED, Scherrer JR, Boyden ES, et al. RNA timestamps identify the age of single molecules in RNA sequencing. Nat Biotechnol. 2021;39(3):320–5.
Cadwell CR, Palasantza A, Jiang X, Berens P, Deng Q, Yilmaz M, et al. Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq. Nat Biotechnol. 2016;34(2):199–203.
Li PH, Kong XY, He YZ, Liu Y, Peng X, Li ZH, et al. Recent developments in application of single-cell RNA sequencing in the tumour immune microenvironment and cancer therapy. Mil Med Res. 2022;9(1):52.
Su M, Pan T, Chen QZ, Zhou WW, Gong Y, Xu G, et al. Data analysis guidelines for single-cell RNA-seq in biomedical studies and clinical applications. Mil Med Res. 2022;9(1):68.
Miao Z, Humphreys BD, McMahon AP, Kim J. Multi-omics integration in the age of million single-cell data. Nat Rev Nephrol. 2021;17(11):710–24.
Edsgärd D, Johnsson P, Sandberg R. Identification of spatial expression trends in single-cell gene expression data. Nat Methods. 2018;15(5):339–42.
Svensson V, Teichmann SA, Stegle O. SpatialDE: identification of spatially variable genes. Nat Methods. 2018;15(5):343–6.
Hu J, Li X, Coleman K, Schroeder A, Ma N, Irwin DJ, et al. SpaGCN: integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat Methods. 2021;18(11):1342–51.
Arnol D, Schapiro D, Bodenmiller B, Saez-Rodriguez J, Stegle O. Modeling cell-cell interactions from spatial molecular data with spatial variance component analysis. Cell Rep. 2019;29(1):202-11.e6.
Nitzan M, Karaiskos N, Friedman N, Rajewsky N. Gene expression cartography. Nature. 2019;576(7785):132–7.
Cang Z, Nie Q. Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat Commun. 2020;11(1):2084.
Zhao E, Stone MR, Ren X, Guenthoer J, Smythe KS, Pulliam T, et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat Biotechnol. 2021;39(11):1375–84.
Yang Y, Shi X, Liu W, Zhou Q, Chan Lau M, Chun Tatt Lim J, et al. SC-MEB: spatial clustering with hidden Markov random field using empirical Bayes. Brief Bioinform. 2022;23(1):bbab466.
Moehlin J, Mollet B, Colombo BM, Mendoza-Parra MA. Inferring biologically relevant molecular tissue substructures by agglomerative clustering of digitized spatial transcriptomes with multilayer. Cell Syst. 2021;12(7):694-705.e3.
Park J, Kim J, Lewy T, Rice CM, Elemento O, Rendeiro AF, et al. Spatial omics technologies at multimodal and single cell/subcellular level. Genome Biol. 2022;23(1):256.
Li X. Harnessing the potential of spatial multiomics: a timely opportunity. Signal Transduct Target Ther. 2023;8(1):234.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82271629), the Central Funds Guiding the Local Science and Technology Development of Shenzhen (2021Szvup024), the Jiangsu Provincial Key Research and Development Program (BE2021664), and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX23_0312). The images were created by Biorender.
Author information
Authors and Affiliations
Contributions
WJW and LXC collected the related papers and wrote the manuscript. LYH and MJZ investigated and prepared the figure and the tables. KTD and CG helped to revise the manuscript. QYG, ZGW and XWZ made important suggestions to the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, WJ., Chu, LX., He, LY. et al. Spatial transcriptomics: recent developments and insights in respiratory research. Military Med Res 10, 38 (2023). https://doi.org/10.1186/s40779-023-00471-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40779-023-00471-x