
Abstract
Background
Oral anticoagulants (OACs) are commonly prescribed, have well-documented benefits for important clinical outcomes but have serious harms as well. Rates of OAC-related adverse events including thromboembolic and hemorrhagic events are especially high shortly after hospital discharge. Expert OAC management involving virtual care is a research priority given its potential to reach remote communities in a more feasible, timely, and less costly way than in-person care. Our objective is to test whether a focused, expert medication management intervention using a mix of in-person consultation and virtual care follow-up, is feasible and effective in preventing anticoagulation-related adverse events, for patients transitioning from hospital to home.
Methods and analysis
A randomized, parallel, multicenter design enrolling consenting adult patients or the caregivers of cognitively impaired patients about to be discharged from medical wards with a discharge prescription for an OAC. The interdisciplinary multimodal intervention is led by a clinical pharmacologist and includes a detailed discharge medication reconciliation and management plan focused on oral anticoagulants at hospital discharge; a circle of care handover and coordination with patient, hospital team and community providers; and early post-discharge follow-up virtual medication check-up visits at 24 h, 1 week, and 1 month. The control group will receive usual care plus encouragement to use the Thrombosis Canada website.
The primary feasibility outcomes include recruitment rate, participant retention rates, trial resources management, and the secondary clinical outcomes include adverse anticoagulant safety events composite (AASE), coordination and continuity of care, medication-related problems, quality of life, and healthcare resource utilization. Follow-up is 3 months.
Discussion
This pilot RCT tests whether there is sufficient feasibility and merit in coordinating oral anticoagulant care early post-hospital discharge to warrant a full sized RCT.
Trial registration
NCT02777047.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Strengths and limitations of this study
-
There is a lack of high-quality evidence on hospital discharge strategies which reduce oral anticoagulant (OAC)-related adverse events.
-
Our methods build on our prior research showing that early post-hospital discharge is a high-risk period for adverse events for patients, that coordination of OAC management peri-hospital discharge is hypothesized to reduce events, and that virtual care may be more cost-effective than in person care.
-
This pragmatic pilot randomized trial combines expert multidisciplinary medication management led by Clinical Pharmacology at hospital discharge with virtual care follow-up.
-
Our feasibility outcomes introduce the concept of research resource utilization and management, expressed as cost per patient completing the trial, as a key outcome.
Introduction
Background and rationale
Institute for Safe Medication Practices (ISMP) data suggest that approximately 300,000 Canadians suffer serious, disabling, or fatal medication-related harm annually [1, 2]. Anticoagulants are the most common cause of medication-related serious harm, in terms of emergency department visits, hospitalizations, and fatalities [3, 4]. Transitions in care have been identified as a particularly high-risk period for adverse events [5]. Each adverse drug event requiring a hospital visit approximately doubles the cost of care in the subsequent 6 months [1]. Indeed, our previous study of thromboembolic and hemorrhagic events after hospital discharge for patients taking oral anticoagulants (OACs) found rates to be approximately 2 to 3 times higher in the first month compared to later [6]. Root cause analyses and patient safety inquiries cite problems with recognition of individual risk factors for benefit versus harm, drug interactions and contraindications, dosing adjustments over time and around procedures, drug monitoring and reversal strategies, and with communications and poor adherence by patients [7,8,9]. Furthermore, the direct oral anticoagulants, each with different dosage regimens based on indication and no widely available test to measure the anticoagulant effect, have increased opportunities for medication errors, while greatly increasing drug costs [2, 10, 11].
Anticoagulants are a high priority drug family for improved medication management because of (a) established benefits to reduce the risk for stroke due to atrial fibrillation by approximately 70%, the risk for recurrent venous thromboembolism by more than 90%, and the risk of death by approximately 25%; (b) widespread utilization with more than 12% of people >85 years of age taking OACs and more than 7 million prescriptions dispensed annually in Canada; and (c) ongoing under-prescribing of OACs for eligible patients, over-prescribing of reduced doses off-label, and use of inferior treatments such as aspirin [12,13,14,15].
Although there are several well-developed anticoagulation management guidelines with detailed, evidence-informed recommendations, it is difficult for physicians and other health care providers to keep up with evolving evidence, and many patient situations lack high-quality evidence to support a specific approach [16,17,18]. Thrombosis Canada provides updated, user-friendly succinct clinical guidance for physicians and patients with point-of-care decision support tools; however, the value of supplemental patient education and patient decision aids to improve outcomes remains unproven [19].
Optimal medication safety requires not only best practices based on clinical evidence but also impeccable application and uptake. The latter requires coordination, communication with and education of all key providers, caregivers, and the patient, as well as frequent monitoring, handovers, and constant quality improvement [20,21,22]. All of these may be most efficiently provided on a large scale across large regions by telehealth or virtual visits [23,24,25]. However, the effectiveness and cost-effectiveness of telehealth or online interventions for medication management remain uncertain [26, 27]. In addition, methods of coordination and communication of care, including in anticoagulant management programs or clinics vary, although toolkits exist for content guidance [28,29,30]. Significantly, coordination interventions have rarely involved clinical pharmacologists—medical specialists with expertise in medication management and the ability to diagnose, prescribe, and change prescriptions.
We hypothesize that expert coordination and management of OAC therapy combined with frequent virtual visits by a pharmacist and clinical pharmacologist in the early post-hospital discharge period, plus regular communication with the patient’s circle of care, could decrease adverse anticoagulant-related events and associated healthcare resource utilization while improving patient’s health-related quality of life compared to usual care.
Objectives
The aim of this pilot randomized controlled trial (RCT) is to test whether high-quality, easily scalable, expert multidisciplinary medication management, and care coordination at hospital discharge and during short-term post-discharge virtual visits are feasible (primary) and can improve oral anticoagulant-related adverse event (thrombotic events, bleeds, and deaths) rates, medication problems, quality-of-life, cost-effectiveness, and satisfaction with care (secondary) during a high-risk transition in care period.
Methods and analysis
Trial design and setting
The protocol was developed according to the SPIRIT and TIDieR guidelines [31, 32]. This pragmatic pilot RCT is designed as a 2-arm, parallel, blinded assessment, variable block randomized trial with individual level randomization and outcomes of feasibility and clinical outcomes, during 3 months of follow-up.
Participating sites include 6 hospitals in Southwest Ontario—3 academic teaching hospitals and 3 community hospitals. The trial was originally scheduled to begin in late 2019 but was delayed initially by staff shortages, then by COVID-19 restrictions on hospital-based research [33].
Eligibility criteria
Inclusion criteria include (a) adult patients within a day of their hospital discharge from internal medicine services with a discharge prescription for an OAC intended to be taken for at least 4 weeks (including both incident and prevalent users), (b) discharge is to home or to a congregant setting such as retirement home where the patient manages their own medications, (c) English-speaking, and (d) capable of providing informed consent. To ensure that we can ethically recruit vulnerable participants including those cognitively impaired, the ability to consent will be measured by the COACHeD Capacity to Consent test, requiring a score of 14 or more (Additional file 3) [34]. If the patient does not pass, a close caregiver (defined as a family member in daily contact with the patient and involved in their medication supervision) will be invited to provide consent on the patient’s behalf by signing a caregiver consent form.
Patients will be excluded if they are less than 18 years of age and have an expected lifespan of less than 3 months and will be discharged to long-term care or other institution where medications are controlled by staff or decline informed consent.
Intervention
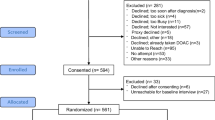
Figure 1 shows the study flow diagram including time and activities for both groups (see Additional file 1).
The Intervention Arm includes the following:
-
a.
Interdisciplinary intervention led by a clinical pharmacologist who is a leader in evidence-based prescribing—includes a detailed discharge medication reconciliation and management plan focussed on oral anticoagulants at hospital discharge; a circle of care handover and coordination with patient, hospital team, and community providers; three scheduled early post-discharge virtual medication check-up visits at 24 h, 1 week, and 1 month with triage of any problems. Medication reconciliation reviews hospital-administered medications compared to pre-admission medications [35]. Medication management is the more complex task of assessing and revising medications in light of the individual patient’s diagnoses, current symptoms and signs, risk factors, allergies and intolerances, other medications, and goals. In this study, all medications will be reviewed with a focus on OAC choice, dosage, indication, duration, potential drug interactions, patient risk factors for thromboembolism versus bleeding, drug insurance, adherence challenges, and health literacy. A study pharmacist with additional training will complete the detailed medication reconciliation then confer with the clinical pharmacologist who will lead the medication management including meeting with the patient and making prescription changes.
-
b.
Hand-overs to the community care team including the main patient caregiver (if applicable), family physician, medical specialist(s), and community pharmacist, using a templated consult summary note which includes a patient profile, details of recent hospitalization, discharge medications, an OAC monitoring checklist, circle of care and upcoming appointments, and recommendations. Figure 2 shows an example consult note (see Additional file 2). The monitoring is based on (a) best evidence (updated guidelines and dedicated evidence review using the CLOT repository of CanVECTOR and McMaster’s Health Information Research Unit), and decision aid content for patients and their families to assist in anticoagulant knowledge and adherence; (b) best practices regarding discharge medication management, virtual care, scalable coordination of care with clear accountability, communication, and teletriage where situations require medical intervention [5, 16, 19, 36,37,38,39,40]. All consult notes are reviewed in detail with the Clinical Pharmacologist.
-
c.
“Virtual visits” (secure video calls from within our electronic medical record (EMR) or phone visits where video is not possible) by the study pharmacist at three follow-up time points—24 h post-discharge to ensure the discharge prescription medications were obtained and understood, review the OAC monitoring checklist, review other medications, and solicit concerns; and at 1 week and 1 month to ensure medication adherence, review the OAC monitoring checklist and other medications, and solicit concerns. After each follow-up visit, a summary consult note will be sent to all circle of care providers, and any clinical events or serious concerns will be addressed by the Clinical Pharmacologist or directed to patient’s family physician via phone call or direct email. Each follow-up visit with intervention patients will be recorded and tracked to ensure adherence to protocols.
-
d.
Teletriage—The patients have the study pharmacist’s contact information and can phone for assistance at any time. The study pharmacist is in constant communication with a Clinical Pharmacologist investigator for guidance. An expert thrombosis specialist will be available on call as needed.
Control Arm: Patients allocated to the control group will receive usual care, plus the URL to Thrombosis Canada website. Usual care in the participating sites includes OAC management by family doctors except for new thromboembolic events which will be followed short term by thromboembolism or hematology specialists, complicated atrial fibrillation which will have cardiology involved temporarily, and a small proportion of warfarin management which is provided in an anticoagulation clinic. This choice of the control group is the most relevant for generalizability to both academic and community practices.
For both arms, there are no restrictions placed on concomitant medical intervention or treatments, as this is a pragmatic randomized trial.
Outcomes
Study outcomes, their measurement methods, timing, and analysis are presented in detail in Table 1 and Table 2 and include the following:
-
1.
Primary outcomes, which are study feasibility outcomes, include recruitment and retention rates, and estimated resources required per patient to complete the main trial. Feasibility will be achieved if we can recruit at least 30% of those eligible, retain 90% of those recruited, and spend no more than $1500 per patient running the trial. Secondary feasibility outcomes include barriers and facilitators to success of the primary outcomes, in terms of process and management issues. These will be used to determine whether a large definitive research study is likely to be feasible, taking into account the practical aspects of managing and funding the project [41, 42].
-
2.
Secondary clinical outcomes include the following:
-
a.
The Adverse Anticoagulant Safety Events composite (AASE) which is any of thromboembolic events or clinically relevant bleeding or death. Thromboembolic events include objectively verified ischemic stroke, systemic embolism, pulmonary embolism, or DVT. Clinically relevant bleeds in this study is defined as bleeding that causes death, hospitalization, or emergency department visits.
-
b.
Coordination and Continuity of Care: Adapted from Health Quality Ontario’s draft guidance and a Rand instrument, the Coordination and Continuity of Care Questionnaire is designed to measure the quality of the transitional and follow-up care [5, 43, 44].
-
c.
Patient Quality of Life: The EQ-5D-5L is the 5-level classification system of the EQ-5D, a measure of health status from the EuroQol group [45]. Using EQ-5D-5L, respondents are asked a short series of questions about mobility, self-care, usual activities, pain discomfort, and anxiety/depression, as well as a summary visual analogue scale. This scale, which provides utility measurements, has been well validated for the Canadian population [45,46,47].
-
d.
Patient Knowledge of OAC Management: Insufficient patient knowledge about OAC may predict poor medication adherence and inadequate anticoagulation control [48]. The COACHeD OAC Knowledge Questionnaire tests knowledge of the therapeutic objective, process of use, safety, and maintenance of the medications [44].
-
e.
Satisfaction with Care: Satisfaction reported by patients and by key health professionals is one of the recommended outcomes to report in medical research, as it may influence adherence [49, 50]. This outcome will be assessed by the Patient/Caregiver Study Satisfaction Survey and by the Provider Study Satisfaction Survey [44].
-
f.
Medication Problems: We will be assessing problems with appropriateness, with medication errors characterized using the National Coordinating Council for Medication Error Reporting and Prevention (NCC-MERP) scale and with medication adherence and attitudes as measured by COMPETE Medication Problems Questionnaire [51, 52].
-
g.
Resource utilization: This is a key outcome to determine cost-effectiveness and cost-utility which then determines whether health care systems might pay for this type of care [53, 54]. We will be measuring all types of health care utilization by patients [43, 46, 54].
-
a.
Sample size estimation
Pilot RCT guidance suggests that where sample size is calculated, it be based on the ability to detect a significant feasibility problem that might interfere with a subsequent full-size RCT—for example, difficulty with recruitment and retention, accuracy of outcome event capture and adjudication, frequency of events, and costing suggesting the intervention is not remotely cost-effective [55]. If a feasibility problem exists at a rate of 5% probability, it can be identified with 95% confidence with a sample of 59 participants [55].
Recruitment methods
Background work flow studies at several of the participating hospitals have confirmed that patient discharge is a complex, hurried process that is often rescheduled multiple times because of fluctuations in patient health or delays in required tests, procedures, or milestones. For feasibility reasons, we plan to use a rolling recruitment method, spending 2 weeks at each hospital on a cyclical basis until a quota is recruited (10–12 patients per site). Rounds and recruitment posters will advertise the study; in some locations, the EMR can assist with screening criteria, and in all locations, research staff are seeking eligible patients in consultation with the Most Responsible Physician team personnel.
Allocation
Participants who meet all the inclusion/exclusion criteria at screening and have completed informed consent will be enrolled in the study, complete baseline assessments, then will be randomized via a computer-generated randomization sequence stratified by site to one of the two study arms, intervention, or control.
A statistician will prepare the randomization schedule using an adaptive biased-coin strategy [56]. Randomization will be stratified by site to maintain balance and minimize the predictability of treatment assignments [57]. To restrict the treatment group imbalance, a maximal tolerable imbalance between treatment groups will be incorporated into the schedule [58]. The randomization schedule will be produced by a program written in SAS V9.4 software (SAS Institute Inc., Cary, NC, USA) and implemented in REDCap on a centralized computer where each patient’s treatment assignment will be available on-line to the research pharmacist at the time of randomization. This process ensures allocation concealment, and randomization awareness where necessary, for example, for the intervention staff.
Blinding
Since this is a pragmatic RCT of care coordination, it will not be possible to completely blind patients or their providers; however, outcome data collectors, adjudicators, and statisticians will be blinded to group allocation until analysis is completed at the end of the study.
Data collection methods
Trained research staff will conduct the interviews with the patients or caregivers, entering data electronically on study laptops directly into REDCap case report forms. The participants’ medical records will be reviewed to abstract data on baseline characteristics, medical history, and medication information. Strategies to promote participant retention and complete follow-up include reminding participants in advance of their end-of-study visit and communicating by email if email address is provided at baseline. Participants who drop out of the study will have their data to that point retained in the study, as approved by REB, to avoid bias. The reasons for study non-completion will be recorded.
Data management
REDCap (Research Electronic Data Capture) is our study software platform—secure, web-based, providing interfaces for validated data capture, role-specific access, audit trails for tracking data manipulation and exports, automated export procedures to SAS and encrypted transmissions [59, 60]. Paper study documents including signed informed consent forms will be stored in our secure research office once they are scanned into REDCap study files. Regular data quality checks, such as automatic range checks, will be performed by the study team to identify data that appear inconsistent, incomplete, or inaccurate.
Patients are not identifiable in the project results database. The identifying information required for the clinical team to deliver the intervention is kept in a separate database. Access to the final dataset will be restricted to the core research team.
Statistical analyses
The reporting of the results of this trial will follow the CONSORT extension to pilot trials [41]. We will use descriptive statistics for presentation of baseline variables and adequacy of follow-up. Feasibility analysis including recruitment rate (≥30% is considered success), participant retention rate (≥90% to end of study is considered success), study resource utilization required (CAD $1500 per patient recruited and completing the study is considered a threshold), management assessment, and scientific assessment will be descriptive.
Analysis will use intention-to-treat methods with censoring only if the patient dies or drops out of the study with refusal of negotiated further assessments. A sensitivity analysis of the subgroup of patients who received all 3 planned follow-up intervention calls and completed the end-study data collection (per protocol analysis) will be carried out. Research staff and statisticians will review outcome data and analysis blinded to group identification.
The primary clinical endpoint will be the incidence of adjudicated AASE during the follow-up period, using proportions, and chi-square testing. Secondary endpoint analyses of the coordination and continuity of care, patient quality of life, OAC management knowledge satisfaction with care (providers & patients), and resource utilization will be analyzed using t tests. The incidence of the adjudicated individual clinical events (clinically relevant bleeding events, thromboembolic events, all-cause hospitalizations, and emergency department visits) will be analyzed using the methods described above for AASE. Public unit costs from Ontario will be used to cost healthcare resource utilization collected as part of the trial. Using an area under the curve approach, quality-adjusted life years (QALYs) will be determined by weighting the EQ-5D-5L health utility scores by time spent in health state. Costs and outcomes (i.e., QALYs, AASE) between the interventions will be compared from a public payer perspective. Given the short follow-up, a low risk of the trial and pilot design, no interim analysis or imputation for missing data is planned. All analyses will be performed using SAS V9.4 software (SAS Institute Inc., Cary, NC, USA).
Data monitoring
Since this trial does not involve any investigational product or procedure, and uses the main anticoagulant safety events and medication safety as outcome events, a formal Data Safety Monitoring Board is not required. Any serious adverse event will be reviewed by our Trial Steering Committee (TSC) within a week of detection, to discern any attribution to our procedures. If found to be due to our coordination procedures, the trial steering committee will recommend whether modifications are indicated. The TSC will be composed of individuals with expertise in clinical trials, chaired by the lead statistician, include the PI, the operational statistician plus a methodologist independent of the study team. Similarly, since this is a short pilot pragmatic RCT where no harm is expected and adjustment of trial procedures may be necessary for feasibility, no formal external auditing of trial conduct is planned. There is no requirement for additional ancillary and post-trial care for those who might come to harm while in the trial, as usual medical care which covers this eventuality is already in place.
Trial management
The trial Principal Investigator will be responsible for communicating any changes to the study, new information, or unanticipated events to the REB, to the sponsor, and to Local Principal Investigators (LPI). The LPI (also called Site Investigator) is responsible for supervising any individual or party to whom they have delegated tasks at the trial site. A Trial Management Group (TMG) will be responsible for the day-to-day management of the trial and will include at a minimum the trial PI, the local PIs, and the trial Research Coordinator. The group will closely review all aspects of the conduct and progress of the trial, ensuring that there is a forum for identifying and addressing issues. Particular attention will be paid to progress towards trial milestones, adherence to the protocol and good research practices. Meetings will be minuted and retained in the study REDCap database.
Patient contributions
Patients were specifically involved in the planning of this trial in several ways. A patient representative is a co-investigator (K.A.). In preparation for this trial, we investigated barriers and facilitators to optimal OAC management, using a systematic review of the literature and then a qualitative focus group study of the opinions of 26 patients/caregivers and 16 providers [61, 62]. These influenced the intervention content and timing. In addition, our choice of outcomes was guided by our recently published systematic survey of the literature regarding patient-important outcomes in OAC trials, as advised by patient groups [63]. One of our Knowledge User—dissemination lead groups is ISMP Canada who are the national leads on medication safety with well developed 2-way communications with patients.
Ethics and dissemination
The study has been approved (study #1639) by the Hamilton Integrated Research Ethics Board, Brant Community Healthcare System Research Ethics Committee, and by the Tri-Hospital Research Ethics Board of Waterloo. Significant protocol modifications will be proactively communicated to the research ethics boards through study amendments to obtain approval prior to the changes being implemented. Each modification will be assessed to determine whether it warrants communication with trial participants.
Dissemination will be through presentations, publications, our research social media, incorporation into our knowledge partner communications to stakeholders, and possible future grant application. The results of the trial will be reported first to trial collaborators. The main report will be drafted by the trial coordinating team, and the final version will be agreed by the Trial Steering Committee before submission for publication, on behalf of the collaboration. The trial will be reported in accordance with the Consolidated Standards of Reporting Trials (CONSORT) guidelines (www.consort-statement.org). The results of the trial will be shared widely, and participants are able to request a copy of the results through contacting the local study team.
Availability of data and materials
An anonymized dataset will be shared in accordance with future requirements of our funders, the Canadian Institutes of Health Research.
Abbreviations
- AASE:
-
Adverse anticoagulant safety events
- COACHeD:
-
Coordination of oral anticoagulant care at hospital discharge
- CONSORT:
-
Consolidated standard of reporting trials
- DVT:
-
Deep vein thrombosis
- EMR:
-
Electronic medical records
- EQ-5D-5L:
-
European Quality of Life Five Dimension
- ISMP:
-
Institute for Safe Medication Practices
- LPI:
-
Local principal investigator
- NCC-MERP:
-
National Coordinating Council for Medication Error Reporting and Prevention
- OAC:
-
Oral anticoagulant
- PI:
-
Principal investigator
- QALY:
-
Quality-adjusted life years
- RCT:
-
Randomized control trial
- REB:
-
Research ethics board
- REDCap:
-
Research electronic data capture
- TMG:
-
Trial management group
- TSC:
-
Trial steering committee
References
Institute for Safe Medication Practices (ISMP). Hospital to home - facilitating medication safety at transitions: a toolkit for healthcare providers. Toronto, CA: ISMP; 2015. [Available from: www.ismp-canada.org]. Accessed 3 June 2022.
Moore TCM, Furberg C, Mattison D. QuarterWatch 2013 quarter 1: perspective on drug hypersensitivity; ISMP Canada. ResearchGate. 2014. p. 1–18. https://doi.org/10.13140/RG.2.1.5065.6168.
Bayoumi I, Dolovich L, Hutchison B, Holbrook A. Medication-related emergency department visits and hospitalizations among older adults. Can Fam Physician. 2014;60(4):e217–22.
Budnitz DS, Shehab N, Lovegrove MC, Geller AI, Lind JN, Pollock DA. US emergency department visits attributed to medication harms, 2017-2019. JAMA. 2021;326(13):1299–309.
Ontario HQ. Transitions between hospital and home: care for people of all ages: HQO; 2020 [Available from: https://www.hqontario.ca/Portals/0/documents/evidence/quality-standards/qs-transitions-between-hospital-and-home-quality-standard-en.pdf]. Accessed 10 June 2022.
Holbrook ABH, Paterson M, Martins D, Greaves S, Munil P, Gomes T. Oral anticoagulant-associated adverse event rates are high in the post-hospital discharge period. CMAJ Open. 2021;9(2):364–75.
Graves CM, Haymart B, Kline-Rogers E, Barnes GD, Perry LK, Pluhatsch D, et al. Root cause analysis of adverse events in an outpatient anticoagulation management consortium. Jt Comm J Qual Patient Saf. 2017;43(6):299–307.
Banerjee A, Benedetto V, Gichuru P, Burnell J, Antoniou S, Schilling RJ, et al. Adherence and persistence to direct oral anticoagulants in atrial fibrillation: a population-based study. Heart. 2020;106(2):119–26.
Yu A, Jeyakumar Y, Wang M, Lee J, Marcucci M, Holbrook A. How personalized are benefit and harm results of randomized trials? A systematic review. J Clin Epidemiol. 2020;126:17–25.
Majeed A, Schulman S. Bleeding and antidotes in new oral anticoagulants. Best Pract Res Clin Haematol. 2013;26(2):191–202.
Moore TCM, Furberg C. Monitoring FDA MedWatch reports: anticoagulants the leading reported drug risk in 2011. Toronto CA: ISMP Quarterwatch; 2011.
Aguilar MI, Hart R. Oral anticoagulants for preventing stroke in patients with non-valvular atrial fibrillation and no previous history of stroke or transient ischemic attacks. Cochrane Database Syst Rev. 2005;3:CD001927. https://doi.org/10.1002/14651858.CD001927.pub2.
Ben Freedman S, Gersh BJ, Lip GY. Misperceptions of aspirin efficacy and safety may perpetuate anticoagulant underutilization in atrial fibrillation. Eur Heart J. 2015;36(11):653–6.
Ogilvie IM, Newton N, Welner SA, Cowell W, Lip GY. Underuse of oral anticoagulants in atrial fibrillation: a systematic review. Am J Med. 2010;123(7):638–45 e4.
Park SKJN. Nonvalvular Atrial Fibrillation in the Era of Non-Vitamin K Antagonist Oral Anticoagulants. Int J Arrhythmia. 2022;23(1). https://doi.org/10.1186/s42444-021-00053.
Holbrook A, Schulman S, Witt DM, Vandvik PO, Fish J, Kovacs MJ, et al. Evidence-based management of anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e152S–e84S.
Witt DM, Nieuwlaat R, Clark NP, Ansell J, Holbrook A, Skov J, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: optimal management of anticoagulation therapy. Blood Adv. 2018;2(22):3257–91.
Andrade JG, Verma A, Mitchell LB, Parkash R, Leblanc K, Atzema C, et al. 2018 focused update of the Canadian Cardiovascular Society Guidelines for the management of atrial fibrillation. Can J Cardiol. 2018;34(11):1371–92.
Thrombosis Canada Clinical Guide Committee. Clinical Guides 2021: Thrombosis Canada; 2021. [Available from: https://thrombosiscanada.ca/clinicalguides/]. Accessed 23 June 2022.
De Regge MDPK, Meijboom B, Trybou J, Mortier E, Eekloo K. The role of hospitals in bridging the care continuum: a systemtic review of coordination of care and follow-up for adults with chronic conditions. BMC Health Serv Res. 2017;17(1):550. https://doi.org/10.1186/s12913-017-2500-0 PMID: 28793893; PMCID: PMC5551032.
Tricco ACAJ, Ivers NM, Ashoor HM, Khan PA, Blondal E, Ghassemi M, et al. Effectiveness of quality improvement strategies for coordination of care to reduce use of health care services: a systematic review and meta-analysis. CMAJ Open. 2014;186(15):E568–E78.
Huntley ALD, Wye L, Morris R, Checkland K, England H, Salisbury C, et al. Which feaures of primary care affect unscheduled secondary care use? A systematic review. BMJ Open. 2013;4(5):e004746. https://doi.org/10.1136/bmjopen-2013-004746 PMID: 24860000; PMCID: PMC4039790.
Sakunrag I, Danwilai K, Dilokthornsakul P, Chaiyakunapruk N, Dhippayom T. Clinical Outcomes of Telephone Service for Patients on Warfarin: A Systematic Review and Meta-Analysis. Telemed J E Health. 2020;26(12):1507–21.
Lee M, Wang M, Liu J, Holbrook A. Do telehealth interventions improve oral anticoagulation management? A systematic review and meta-analysis. J Thromb Thrombolysis. 2018;45(3):325–36.
Xia X, Wu J, Zhang J. The effect of online versus hospital warfarin management on patient outcomes: a systematic review and meta-analysis. Int J Clin Pharmacol. 2018;40(6):1420–9.
Lee PA, Greenfield G, Pappas Y. The impact of telehealth remote patient monitoring on glycemic control in type 2 diabetes: a systematic review and meta-analysis of systematic reviews of randomised controlled trials. BMC Health Serv Res. 2018;18(1):495. https://doi.org/10.1186/s12913-018-3274-8.
Ekeland AG, Bowes A, Flottorp S. Effectiveness of telemedicine: a systematic review of reviews. Int J Med Inform. 2010;79(11):736–71.
Duan-Porter W, Ullman K, Majeski B, Miake-Lye I, Diem S, Wilt TJ. Care coordination models and tools: a systematic review and key informant interviews. [Internet]. Washington (DC): Department of Veterans Affairs (US); 2020 Jun. Available from: https://www.ncbi.nlm.nih.gov/books/NBK566155/. Accessed 15 June 2022.
Michigan Anticoagulation Quality Improvement Initiative. Anticoagulation desktop reference (version 2.0.1): a consortium-developed compendium of anticoagulation information: Michigan Anticoagulation Quality Improvement Initiative (MAQI2); 2020 [updated Septmeber 2 2020. Available from: https://anticoagulationtoolkit.org/sites/default/files/toolkit_pdfs/toolkitfull.pdf. Accessed 15 June 2022.
The Joint Commission. R3 Report Issue 19: national patient safety goal for anticoagulant therapy. The Joint Commission; 2018. Available from: https://www.jointcommission.org/standards/r3-report/r3-report-issue-19-national-patient-safety-goal-for-anticoagulant-therapy/#.Ys2Vq9vMIls. Accessed 3 June 2022.
Chan AW, Tetzlaff JM, Altman DG, Dickersin K, Moher D. SPIRIT 2013: new guidance for content of clinical trial protocols. Lancet. 2013;381(9861):91–2.
Hoffmann TC, Glasziou PP, Boutron I, Milne R, Perera R, Moher D, et al. Better reporting of interventions: template for intervention description and replication (TIDieR) checklist and guide. BMJ. 2014;348:g1687. https://doi.org/10.1136/bmj.g1687.
Orkin AM, Gill PJ, Ghersi D, Campbell L, Sugarman J, Emsley R, et al. Guidelines for reporting trial protocols and completed trials modified due to the COVID-19 pandemic and other extenuating circumstances: the CONSERVE 2021 statement. JAMA. 2021;326(3):257–65.
COACHeD RCT Capacity to Consent Questionnaire. MacSphere. 2021. Available from: http://hdl.handle.net/11375/27712. Accessed 15 June 2022.
Institute for Safe Medication Practices Canada. Institute for Safe Medication Practices Canada. Medication Reconciliation (MedRec) ISMPCanada. Toronto CA: ISMPCanada; 2000. [Available from: https://www.ismp-canada.org/medrec/]. Accessed 3 June 2022.
Thrombosis Canada. Thrombosis Canada. Canada: Thrombosis Canada. [Available from: https://thrombosiscanada.ca/]. Accessed 8 June 2022.
Keshavjee K, Mirza K, Martin K. The next generation EMR. Stud Health Technol Inform. 2015;208:210–4.
Institue for Safe Medication Practices Canada. Virtual medication history interviews and discharge education. Toronto CA: ISMP; 2020. [Available from: www.ismp-canada.org]. Accessed 3 June 2022.
Core elements of anticoagulation stewardship programs 2019: anticoagulation forum; 2019 [Available from: https://acforum.org/web/downloads/ACF%20Anticoagulation%20Stewardship%20Guide.pdf]. Accessed 10 June 2022.
McMaster's Health Information Research Unit. CLOT Respository: McMaster University; [Available from: https://plus.mcmaster.ca/clotplus]. Accessed 10 June 2022.
Eldridge SM, Chan CL, Campbell MJ, Bond CM, Hopewell S, Thabane L, et al. CONSORT 2010 statement: extension to randomised pilot and feasibility trials. BMJ. 2016;355:i5239. https://doi.org/10.1136/bmj.i5239.
Thabane L, Lancaster G. A guide to the reporting of protocols of pilot and feasibility trials. Pilot Feasibility Stud. 2019;5:37. https://doi.org/10.1186/s40814-019-0423-8.
Wenger NS, Roth CP, Shekelle P, Investigators A. Introduction to the assessing care of vulnerable elders-3 quality indicator measurement set. J Am Geriatr Soc. 2007;55(Suppl 2):S247–52.
COACHeD RCT Additional Files 2021 (CCCQ, OAC Knowledge Test, Patient/Caregiver Study Satisfaction Survey). MacSphere, McMaster University. 2021. Available from: http://hdl.handle.net/11375/27213. Accessed 8 June 2022.
Euroqol. EQ-5D Instruments: Euroqol; 2018 [Available from: https://euroqol.org/eq-5d-instruments/]. Accessed 15 June 2022.
Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727–36.
Xie F, Pullenayegum E, Gaebel K, Bansback N, Bryan S, Ohinmaa A, et al. A time trade-off-derived value set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98–105.
Rolls CAOK, Chalmers L, et al. The relationship between knowledge, health literacy, and adherence among patients taking oral anticoagulants for stroke thromboprophyxlaxis in atrial fibrillation. Cardiovasc Ther. 2017;35(6). https://doi.org/10.1111/1755-5922.12304.
Williamson PR, Altman DG, Bagley H, Barnes KL, Blazeby JM, Brookes ST, et al. The COMET Handbook: version 1.0. Trials. 2017;18(Suppl 3):280. https://doi.org/10.1186/s13063-017-1978-4.
Perino AC, Shrader P, Turakhia MP, Ansell JE, Gersh BJ, Fonarow GC, et al. Comparison of patient-reported care satisfaction, quality of warfarin therapy, and outcomes of atrial fibrillation: findings from the ORBIT - AF Registry. J Am Heart Assoc. 2019;8(9):e011205. https://doi.org/10.1161/JAHA.118.011205.
National Coordinating Council for Medication Error Reporting and Prevention (NCC-MERP). About Medication Errors: MERP N; 2020 [Available from: https://www.nccmerp.org/about-medication-errors]. Accessed 15 June 2022.
Holbrook A, Pullenayegum E, Thabane L, Troyan S, Foster G, Keshavjee K, et al. Shared electronic vascular risk decision support in primary care: Computerization of Medical Practices for the Enhancement of Therapeutic Effectiveness (COMPETE III) randomized trial. Arch Intern Med. 2011;171(19):1736–44.
Carrasquillo O. In: Gellman MD, Turner JR, editors. Health Care Utilization. Encyclopedia of Behavioral Medicine. New York: Springer; 2013. p. 909–10.
Canadian Agency for Drug and Technogolies in Health (CADTH) [Internet] Canada: Canadian Journal of Health Technologies; 2021 [Available from: https://www.cadth.ca/]. Accessed 20 June 2022.
Viechtbauer W, Smits L, Kotz D, Bude L, Spigt M, Serroyen J, et al. A simple formula for the calculation of sample size in pilot studies. J Clin Epidemiol. 2015;68(11):1375–9.
Wei LJ. The adaptive biased coin design for sequential experiments. Ann Stat. 1978;6(1):92–100.
Markaaryan TRW. Exact properties of Efron's biased coin randomization procedure. Ann Statist. 2010;38(3):1546–67.
Soares JFJWC. Some restricted randomization rules in sequential designs. Commun Stat. 1983;12(17):2017–34.
Harris PA, Taylor R, Minor BL, Elliott V, Fernandez M, O'Neal L, et al. The REDCap consortium: Building an international community of software platform partners. J Biomed Inform. 2019;95:103208. https://doi.org/10.1016/j.jbi.2019.103208.
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–81.
Holbrook A, Wang M, Swinton M, Troyan S, Ho JMW, Siegal DM. Barriers and facilitators for optimizing oral anticoagulant management: perspectives of patients, caregivers, and providers. PLoS One. 2021;16(9):e0257798. https://doi.org/10.1371/journal.pone.0257798.
Wang M, Holbrook A, Lee M, Liu J, Leenus A, Chen N, et al. Barriers and facilitators to optimal oral anticoagulant management: a scoping review. J Thromb Thrombolysis. 2020;50(3):697–714.
Wang M, Chen Z, Wong M, Thabane L, Mbuagbaw L, Siegal D, et al. Are the correct outcomes being measured in studies of oral anticoagulants? A systematic survey. Thromb Res. 2021;201:30–49.
Acknowledgements
Not applicable.
Funding
This work was supported by the Canadian Institutes of Health Research grant number FRN 148803 and the Hamilton Academic Health Services Organization grant number HAH-16-06. The study funders had no role in the study design; collection, management, analysis, or interpretation of data; writing of the report; or the decision to submit the report for publication.
Author information
Authors and Affiliations
Contributions
AH is the principal investigator and conceived the study idea, wrote the protocol, wrote the grant for funding, and led the project team. SH, JD, SS, KK, JH, JET, DS, and LT were involved in the design of the study and securing funding. KA is the patient representative. AH, ST, KV, and LY drafted the protocol manuscript, applied for ethics approval, and coordinated the study. SG, BL, AA, MT, and YK are the local principal investigators. The authors have read and approved the final manuscript. AH acts as guarantor. • Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; • Drafting the work or revising it critically for important intellectual content; • Final approval of the version to be published; • Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. No professional writers were used to write this manuscript. The authors conform to the Pilot and Feasibility Studies eligibility guidelines.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study has been approved by the Hamilton, Waterloo, and Brantford Research Ethics Boards. Dissemination will include publications, presentations, follow-up larger trials, and appropriate changes in national medication safety and thromboembolism guidelines driven by our investigators and knowledge use partners.
Prior to performing any trial-specific procedure, a signed consent form will be obtained for each participant. The consent form is available in supplementary files [44]. A delegated member of the trial team will discuss the study with the patient and relevant family members/caregivers and provide them with a study consent form which will describe the purpose of the trial, the procedures to be followed, and the risk/benefits of participation. Any questions that they might have will be answered by either the trial team or the investigator. Once this is completed, we will test the patient’s capacity to consent (see questionnaire in Additional file 3) [44]. If the patient is found capable, they will be invited to sign the informed consent form. If not, a close caregiver who assists the patient with their medications and medical care, will be invited to sign. Consent will be voluntary and free from coercion. Both the person obtaining consent and the investigator will sign the informed consent form, and a copy will be given to the patient and caregiver (if applicable).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure 1
. COACHeD Flow Diagram.
Additional file 2: Figure 2
. Sample COACHeD Consult at Discharge.
Additional file 3.
COACHeD Capacity to Consent Questionnaire.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Holbrook, A.M., Vidug, K., Yoo, L. et al. Coordination of Oral Anticoagulant Care at Hospital Discharge (COACHeD): protocol for a pilot randomised controlled trial. Pilot Feasibility Stud 8, 166 (2022). https://doi.org/10.1186/s40814-022-01130-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40814-022-01130-z