
Abstract
Background
Vaccination is the one of the agendas of many countries to reduce cervical cancer caused by the Human papillomavirus. Currently, VLP-based vaccine is the most potent vaccine against HPV, which could be produced by a variety of expression systems. Our study focuses on a comparison of recombinant protein expression L1 HPV52 using two common yeasts, Pichia pastoris and Hansenula polymorpha that have been used for vaccine production on an industrial scale. We also applied bioinformatics approach using reverse vaccinology to design alternative multi-epitope vaccines in recombinant protein and mRNA types.
Results
Our study found that P. pastoris relatively provided higher level of L1 protein expression and production efficiency compared to H. polymorpha in a batch system. However, both hosts showed self-assembly VLP formation and stable integration during protein induction. The vaccine we have designed exhibited high immune activation and safe in computational prediction. It is also potentially suitable for production in a variety of expression systems.
Conclusion
By monitoring the overall optimization parameter assessment, this study can be used as the basis reference for large-scale production of the HPV52 vaccine.
Similar content being viewed by others

Background
Vaccination shows potential treatment for cancer inhibition caused by the Human papillomavirus (HPV) [1]. Vaccination using Gardasil 9 was shown to be effective to nearly 100% in preventing broad HPV-type-induced cervical, vulvar and vaginal diseases [2]. The existing strengths of local and regional communities to conduct massive production of the vaccine facilitated a relatively low-cost manufacturing process, which could cover the needs relatively faster. In addition, independency in vaccine production shows a preparedness to face the pandemic in particular for developing countries [3]. Moreover, it has been reported that HPV vaccination has become a national immunization program in more than 100 countries [4].
Recombinant protein-based vaccines are still continuously developed to fulfill the need in tackling HPV [5]. The major capsid protein L1 is the most exposed protein to the immune system that could assemble into virus-like particles (VLP), generating high immune responses [6, 7]. The main HPV vaccines that were licensed and commercially available (Gardasil and Cervarix) nowadays also utilize purified recombinant VLP-based systems [8].
HPV L1 protein could be expressed in various expression systems from prokaryotes, such as E. coli, as well as eukaryotes such as insects, plants, and yeast [9,10,11]. The yeast expression system is one of the most commonly used platforms for industrial production due to its ability to generate high protein titers [12, 13]. In addition, utilization of their post-translation modification features enhances protein solubility and folding [14].
Multi-epitope vaccine (MEV) is an alternative way to prevent and treat a pathogen infection, which has been continuously developed in a form of recombinant subunit protein or mRNA vaccines [15]. High efficacy, safe, and low-cost manufacturing are the main reasons for researchers to continue developing MEV [16]. Moreover, mRNA vaccines currently have been the breakout stars of the pandemic. Their demonstrated impressive protection has great application prospects and advantages [17, 18].
Several studies reported that L1 HPV52 (categorized as one of the high-risk HPV types) could easily be expressed in P. pastoris and H. polymorpha [19, 20]. However, the justification of which host is most likely preferable for L1 HPV52 expression is still not yet clear. Thus, this study focuses on the optimization, characterization, and comparison of codon optimized HPV52 in yeasts. Previously, L1 protein was expressed under strong methanol-inducible promoters AOX and MOX in P. pastoris GS115 and H. polymorpha NCYC495, respectively [21]. Biomass, growth rate, clone copy number, VLP formation, and integration stability of P. pastoris GS115 and H. polymorpha NCYC495 were evaluated. We also described another point of view of an alternative multi-epitope vaccine (MEV). We applied reverse vaccinology to generate a recombinant fusion protein of the top listed epitopes that we identified in our previous study. They were recognized by B and T cell epitopes, which have high-level population coverage and potentially give broad-spectrum protection against other HPV types [22]. This study could give a wide perspective on tackling the carcinogenic pathogen HVP52 through optimum vaccine production, particularly for VLP, recombinant subunit protein, and mRNA-based vaccines.
Methods
Codon and mRNA structure analysis
The global consensus sequences of L1 HPV52 [22] were used as reference sequences for codon optimization. Codon analysis was performed using available data from Kazusa (https://www.kazusa.or.jp/codon/) [23], while Codon adaptation index (CAI) was evaluated for each host using the online available CAI evaluator (http://genomes.urv.es/CAIcal/) [24]. A 50 bp mRNA structure segment of the gene of interest starting from translation T + 1 from AOX and MOX was predicted by (https://rna.urmc.rochester.edu/RNAstructureWeb/Servers/Predict1/Predict1.html) [25]. The program could predict a minimum free energy (MFE) structure that reflected the mRNA stability.
Construction and yeast transformation
To obtain an efficient translation in both P. pastoris and H. polymorpha, HPV52 L1 codon was optimized and cloned into pD902 that has AOX promoter (DNA 2.0, currently ATUM, Newark, CA). The L1 gene was subcloned into pHIPH4 that is regulated by promoter MOX and terminator tAMO. The pD902 was provided by ATUM, while pHIPH4 was kindly given by the University of Groningen. All construction was generated following basic molecular cloning [26].
The recombinant plasmid was transformed into E. coli DH5α, then isolated and linearized using NcoI and StuI, respectively, for pD902_HPV52L1 and pHIPH4_HPV52L1. As much as 5000 ng of each linearized plasmid was introduced using electro-transformation in a 2-mm cuvette after a 1.5 kV/cm, 50 μF, and 129 Ω electric field pulse (5 ms resulting pulse length) [19]. The transformants were grown on YPD (1% yeast, 2% peptone, 2% dextrose, 2% agar) agar supplemented with zeocin to select for P. pastoris containing pD902_HPV52L1 and hygromycin for H. polymorpha containing pHIPH4_HPV52L1 at 30 ℃ and 37 ℃, respectively.
Genome isolation and transformant validation
Transformants were screened using colony PCR and verified by sequencing analysis using specific primers (Table 1). The selected colonies were treated using Triton-X before being used as a PCR template [27]. Genomic DNA was isolated following a protocol described with slight modification [28]. Briefly, an overnight yeast culture was lysed with lysis buffer (50 mM Tris HCl pH7.2, 50 mM EDTA, 3% SDS, 1% ß-Mercaptoethanol). The homogenous lysate was extracted with an equal volume of PCI (1:1). Subsequently, sodium acetate buffer pH 5.2 and 0.6 V isopropanol were added to precipitate the DNA. The precipitated DNA was then washed with 70% ethanol and re-suspended in nuclease-free water (NFW) with 50 mg/ml RNase.
Protein expression, isolation, SDS PAGE, western blot (WB), and transmission electron microscopy (TEM)
The best performing colony was chosen by the expression level of each clone. The expression was performed in a shake flask system with 1:10 aeration. Yeast was grown in Buffered Glycerol Complex Medium (BMGY) to produce a high biomass yield and then transferred into Buffered Methanol-Complex Medium (BMMY) with methanol added as an inducer at the optimal concentration for each strain: 0.5% (P. pastoris), and 1% (H. polymorpha). Growth kinetics analysis was performed by measuring OD600 values every 24 h until 96 h at 22 ℃ and 30 ℃ for P. pastoris cultures, while H. polymorpha cultures were measured at 30 ℃ and 37 ℃ [29]. The analysis was performed in three biological replicates.
Protein was isolated using a glass bead added to lysis buffer (50 mM sodium phosphate, pH 7.4, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM EDTA, and 5% glycerol). The sample was separated using 12% SDS–PAGE under reducing condition, 400 mA for 90 min. The gel was transferred into the nitrocellulose membrane using the wet transfer method. The membrane was blocked using skim milk for 1 h. Next, the primary polyclonal antibody L1 HPV52 (Creative Diagnostic, CABT B8799) and anti-rabbit conjugated HRP (secondary antibody) were subsequently added at 1:20 polyclonal antibody L1 HPV52 (Creative Diagnostic, CABT B8799). Finally, the specific protein band was visualized by pouring TMB chromogenic substrate. The isolated protein was also evaluated by immunoblot.
For TEM analysis, the first step protein purification was conducted by 50% ammonium sulfate precipitation, followed by overnight dialysis. The samples were absorbed on carbon-coated copper grids and negatively stained with 2% phosphotungstic acid. The grids were air-dried before examination under a transmission electron microscope, JEOL 1010, 80 kV.
Copy number analysis and protein quantification
Copy number of integrated HPV52 L1 gene in both yeasts was determined using qPCR MyGo Pro RT-PCR with three-step amplification for 40 cycles of initiation (98 ℃, 2 min), denaturation (98 ℃ 10 s), annealing (60 ℃, 10 s), and extension (68 ℃, 30 s). Additional melting curve analysis was added in the final step. Transcription of reporter gene ARG4 and ACT in P. pastoris and H. polymorpha were respectively detected using specific primers as described in Table 1 [30, 31]. Comparison of copy number was normalized by the lowest CT value. In addition, PCR efficiency was calculated by the equation below [32]. The analysis was performed using three biological replicates.
The total amount of lysate and precipitated fraction (ammonium precipitation) were quantified by BCA Protein Assay Kit (Thermo Scientific™). The HPV52 L1 dot blot was detected using polyclonal antibody L1 HPV52 (Creative Diagnostic, CABT B8799). The concentration of L1 protein was quantified using a densitometer, compering with recombinant purified HPV52 L1 in different concentrations as a standard. ELISA was also performed to specify the amount of HPV52 L1 using a monoclonal antibody (Creative diagnostics, CABT-B8810).
Epitope-based vaccine design and validation
The top highest antigenicity level of listed epitopes was selected based on our previous study (Supplementary Table S1). The designed vaccine was connected using EAAAK that separated the entire epitope and the other supporting components. Meanwhile, AAY and GPGPG linkers were used to connect individual B and T cell epitopes, respectively. The 50S ribosomal protein L7/L12 (Locus RL7_MYCTU) derived from Mycobacterium tuberculosis was also added to boost the vaccine immunogenicity. Antigenicity of the whole construct, immune response, toxicity, antigenicity, and physicochemical were evaluated using Vaxijen [33], C-ImmSim [34, 35], Toxinpred [36], and Protparam [37], respectively.
Designed mRNA vaccine docking and immune simulation
Docking analysis was performed using the available online server The ClusPro 2.0 server [38]. The protein structure of the designed vaccine (Receptor) was predicted using I-TASSER [39] and then validated by Ramachandran (ZLab server (https://zlab.umassmed.edu/bu/rama/index.pl). The TLR4 (PDB ID: 4G8A) was applied as protein ligan, and its interaction with the receptor was identified, using pdbsum [40]. All protein structures are visualized by pymole software.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software, San Diego, CA) by applying Student’s t test to determine differences between the two-group data. The data with p < 0.05 were considered to have significant differences.
Result
Gene design, synthesis, and vector expression construction
The optimized codon was shown to be feasible for both P. pastoris and H. polymorpha with an adaptation coefficient of 0.77. The RNA structure of the optimized codon could provide a higher stability by decreasing folding free for more preferable transcription (Supplementary Figure S1) [41]. The gene encoding L1 HPV52 was inserted into the multi-cloning site between BamHI and NotI for pD902 and HindIII and SalI for pHIPH4 (Fig. 1A). Synonymous mutation of serine (TCC to AGC) was found in all positive clones from H. polymorpha (Fig. 1B), with slightly different mRNA free energy (Fig. 1C). Meanwhile, P. pastoris showed matched sequences with the designed codon in all positive colonies. The synonymous mutation found in clone H. polymorpha did not affect the reading frame of the HPVL152 protein. In addition, the shape of mRNA is relatively similar which will not interfere with the normal interaction with the ribosome which allows the production of similar protein levels.

Vector construction, synonymous mutation, and mRNA structure analysis. A Construction of gene expression vectors. PHIPH4_HPVL152 for H. polymorpha (left) and pD902_HPVL152 for P. pastoris (right). B Synonymous mutation of serine residue was found in HPV52 L1 isolated sequences from all H. polymorpha. C mRNA structure around the mutation site of HPV52 L1 was not significantly different
Growth kinetics and expression level of HPV52 L1
H. polymorpha showed a significantly different growth rate between two temperature conditions while P. pastoris did not (Fig. 2A). It correlates with the biomass production from H. polymorpha that showed significantly distinction between the two conditions while P. pastoris was observed with no change (Fig. 2B). However, they both reached a stationary phase after 72 h with the highest biomass levels observed at 30 ℃ (Fig. 2A).The optimum expression was observed at 30 ℃ with 0.5% inducer for P. pastoris and 37 ℃ with 1% inducer for H. polymorpha (Fig. 3A, B).

Biomass analysis of HPV52 L1 expression. A Growth curve after induction. B Differences of final OD between two hosts. n = 3, p < 0.05 were considered to have significant differences

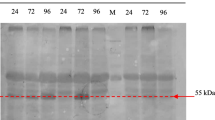
Expression profile of HPV52 L1 induced by different amounts of methanol. A WB profile of HPV52 L1 expression in P. pastoris. B WB profile of HPV52 L1 expression in H. polymorpha. C Immunoblot of HPV52 L1 expression using polyclonal antibody. H-5 indicated H. polymorpha colony 5, while P-1 represents P. pastoris colony 1, the protein was isolated from 72 h culture in the optimum condition
WB and immunoblot analyses showed the expression level of ~ 59 kDa HPV52 L1 in P. pastoris was ~ 1.5 times higher than H. polymorpha (detected by ELISA) (Fig. 3C). In addition, the percentage protein recovery of P. pastoris also exhibited ~ 1.4 times higher than H. polymorpha even though the protein recovery from ammonium precipitation were similar in both yeasts (Table 2).
Copy number and stability during methanol induction
The isolated genome of each clone was serially diluted and set as a template for copy number analysis. The r2 regression value of ACT and ARG showed a good correlation with r2 > 0.9 (Fig. 4A, B). The melting curve also showed specific amplification, described by a single peak from all samples (data not shown). The best performing colony stably expressed the protein during methanol induction (Fig. 4C). The fact that both hosts have a stable copy number during induction indicated a stable integration leading to a stable expression [42].

Standard curve and stability analysis. A Standard curve of the reporter gene, ARG4 in P. pastoris. B Standard curve of the reporter gene, ACT in H. polymorpha. C Stability of HPV52 L1 copy number in each host during methanol induction. The protein expression was induced using an optimal amount of inducer
TEM analysis
The benefit of using a yeast expression system is the ability to form self-assembly VLP. VLP formation was observed by TEM with approximately 50 nm size-like nature virions (Fig. 5). The unsynchronized form was commonly found at the protein expression without further purification, which can be caused by differential expression and VLP assembly periods. To increase homogeneity and stability of VLP disassembly and reassembly VLP is still required.

TEM images showing self-assembled VLP of HPV52 L1. A VLP formation of HPV52 L1 was obtained from P. pastoris dialyzed crude. B VLP formation of HPV52 L1 was obtained from H. polymorpha dialyzed crude. View examples of VLP are indicated by arrows
Vaccine design and validation
The predicted protein structure of vaccine design showed 85% Highly Preferred Conformations (Supplementary Figure S2). It also showed a stable expression, high solubility, and nontoxic features (Supplementary Table S2). The probable antigenicity of the designed vaccine exhibited slightly higher than the capsid protein itself in VaxiJen simulation (0.50 > 0.48). It covered B and T cell epitopes (Fig. 6A), with charge distribution profile (Fig. 6B) and B cell surface recognition site distributed across the whole structure (Fig. 6C). It was reported that this epitope potentially gives a cross-protection profile across other HPV types. Moreover, the vaccine is also considered can cover a wide region of the population, nontoxic, stable, and non-allergenic as well [22].

The designed vaccine and epitope mapping and interaction. A Scheme of mRNA vaccine design. B Surface and charge structure of multi-epitope recombinant vaccine HPV 52, the color is according to the vaccine scheme. C Linear (upper, indicated in yellow) and conformational B (lower, indicated in yellow) cells epitope mapping showed the overlapping region with T cells epitope. D Docking analysis of TLR4 and recombinant vaccine. E Interaction mapping residues of TLR4 and a recombinant vaccine
Docking analysis designed mRNA vaccine to TLR
No less than 29 complexes formation was generated by Claspro with the lowest energy of – 1283 selected as the best complex (Fig. 6D). The complex formation between the designed vaccine and TLR4 is stabilized by 1 salt bridge and 6 hydrogen bonds in chain D of TLR4 and 5 salt bridges and 20 hydrogen bonds in chain B (Fig. 6E). Their interaction is facilitated by the net charge of the contacting area between the ligand-receptor.
Immune simulation
Immune response was increased following antigen exposures, it showed that secondary and tertiary responses were higher than primary induction (Fig. 7A). Immunoglobulin response of IgM showed a higher level than IgG in the primary induction. In the second and third doses, the IgG1 + IgG2, as well as IgG1, exhibited a higher level than IgM along with antigen reduction. The increasing level of B, Th (helper), Tc (cytotoxic), and NK (Natural Killer) cells were also observed for a long period which indicated memory formation (Fig. 7B–E). In addition, the IFN gamma showed robust activation during vaccination with a variety of immune responses indicated by a lower Simpson index (Fig. 7F). The data suggested that the designed vaccine has full filled good vaccine indicators and can be considered for further in vitro and in vivo analysis.

Vaccine immune simulation using C-ImmSim. A Immunoglobulin production in response to antigen exposures, B B cell population, C T helper cell population, D Total production of T-cytotoxic cells, E natural killer cells production, and F cytokine level profile after the injections
Discussion
Recombinant protein expression has been used extensively to produce vaccines for a long time. This also applies to the vaccine manufacture against HPV which mostly use major capsid protein L1. It is the most exposed protein that can stimulate immune response similar to the native virion [7]. Yeast is the one of the best platforms for producing viral-like particle for HPV. In addition to the self-assembly HPV VLPs, the ability of yeasts to produce large amounts of protein has always been advantage for the industry [43].
In this study, we highlighted two different approaches to produce HPV vaccine. First, we optimized recombinant expression L1 HPV52 using two different yeast (P. pastoris and H. polymorpha) and took advantage from their self-assembly VLP system. We also compared their profile in the optimized condition which include growth kinetics, biomass, temperature, inducer amount, copy number stability, and VLP formation. The second approach is to use the potential antigenic peptide from our study to generate peptide-based vaccine in the form of recombinant or mRNA vaccines.
Selecting the right host strain is the first step in opening the bottleneck for protein production. We used yeast expression system to produce heterologous protein that have been used for industrial and biopharmaceutical in the large amount for many years. Yeast is easy to be manipulated genetically, short generation times, large scalable using fermentation, less expensive, and suitable for various proteins that needs post translation modification [44].
We optimized codon preference for yeast to enhance the protein production. By replacing rare codons to match with natural host codons leads to proper protein folding by preserving slow translation regions [45]. We also used AOX and MOX which are categorized as strong methanol inducible promoters to give a higher level of heterologous protein expression [45].
Increasing cell biomass as a strategy to elevate protein yield has been extensively studied by manipulating the host and its environments such as medium or feeding strategy [46, 47]. Our research using a shake flask system showed that H. polymorpha gave a higher biomass compared to P. pastoris. Based on our data, biomass has not significant effect on the L1 HPV52 expression. It was also suggested that accumulation of biomass does not necessarily linear with protein production [47]. Because our study used shake flask system which has limitations in controlling methanol uptake, we used the same aeration and methanol concentration throughout the production. It decreased methanol and oxygen shortage that required by higher cell [48]. This may result differently once the feed-batch strategy is implemented at the fermenter scale.
High copy number integrant in yeast could enhance protein titers. However, as with biomass, copy number does not always have a directly proportional correlation to the protein titer. An inverse correlation was demonstrated in our study by P. pastoris having higher expression level of L1 HPV52 in a relatively low copy number. This is in line with the number of inducers indicating that the lowest inducer gave a higher expression level. It might be that P. pastoris requires a slow production of L1HPV52 to reduce metabolic stress. In contrast for H. polymorpha was relatively more resistance to foreign gene (in this context L1 HPV52), causing its expression to be higher at a relatively higher copy number. However, both yeasts showed stabile integration during protein production.
The overexpression of heterologous protein may cause stress on a secretory pathway, enhancing the unfolding protein response that leads to protein degradation and other cellular stress responses [49]. Moreover, the combination of transcription and translation levels, as well as protein folding play a role in the protein features like solubility and stability that affect the protein function [50, 51].
VLP formation correlates to protein folding and its environmental properties to minimize free energy in higher structures [52]. Nevertheless, this study showed reproducible data with previous reports that confirmed the self-assembly of the VLPs. For medical application purposes, further purification steps and subsequent VLPs reassembly are required to obtain the correct particle size at the appropriate amount of yield to induce an adequate immune response [3, 53,54,55].
Recombinant antigen protein purification for biopharmaceutical product should not have any additional component that may affect the biological system (safety issue). We used non-tagging protein purification strategy which not required additional tagging cleavage step that can increase purification efficiency. It can use size exclusion chromatography tandem with ion exchange or hydrophobic interaction chromatography. In addition, we did ammonium precipitation as initial step before further purifications. Immune response validation should also be tested in animal models, this may help in assessing which host can produce better L1 HPV52 folding as well as VLP maturation.
Until now the best, licensed HPV vaccine with high efficacy uses a VLP-based platform. It utilizes capsid protein L1 as the main component, inhibiting viral replication using the neutralizing mechanism. VLP gives a high titer of antibody production because it mimics a viral nature form that leads to undistinguished by the immune system [56].
Peptide based vaccine via recombinant protein or mRNA platform can be used as another option to produce potential vaccine. Moreover currently mRNA vaccine was become new star in infectious diseases prevention such as in tackling COVID19. We used computational approach using in silico protein modelling from our potential peptide we tested in previous study to get initial insight for the real biological event.
In general vaccine could be recognized by pathogen recognition receptors (PRRs), mainly expressed by cells of the innate immune system. VLP internalization prompts cell maturation and epitope presentation through major histocompatibility complex (MHC) class I or class II molecules [57]. MHC I bind to CD8+ T cells responsible for the cytotoxicity activity, while MCH II binds to CD4+ T cells bridging antibodies production by B cells.
The mRNA normally produced by in vitro transcription and then delivered by vector-mediated internalization using a lipid, nanoparticle, or polymer-based delivery system into the body [58]. mRNA is translated by cellular machinery in the cytoplasm and then undergoes posttranslational modification to stabilize the tertiary structure to have fully functional protein properties. MHC I-targeting domain (MITD) which is present in the construct responsible for peptide secretion through transporting the peptide to particular compartments in the endoplasmic reticulum and Golgi body, then MHC-I and MHC-II could present them on the surface of cells [59].
The right linker should be added to avoid inter-domain interaction leading to impaired bioactivity [60]. A balance between flexibility and rigidity that maintain a stable conformation when expressed is required [61]. mRNA ORF required an extra region for the polymerase to stay and start the transcription. We used a common Xenopus beta globulin in 5′ and alpha 3′ UTR that flank the mRNA ORF which was reported to significantly enhance the stability of mRNA as well as protein translation [62]. The open reading frame from the first epitope to the end with linker in between can be used for recombinant subunit vaccine using common protein expression. In addition, adjuvant is also a key point that could enhance the immune response by several mechanisms such as depot formation, recruitment of immune cells, induction of cytokines and chemokines, enhancement of antigen uptake, presentation, and transport to draining lymph nodes [63]. All the vaccine mechanism of action is summarized in Fig. 8.

Purposed mechanism of action of VLP, mRNA, and multi epitope protein-based vaccines. The schema was created using Biorender (https://biorender.com/)
In general, our construct was computationally predicted have a good vaccine properties. It has a good solubility index and potentially induce immune response, however in vitro and in vivo validation is still needed.
Conclusion
Our study showed that the expression system of P. pastoris GS115 and H. polymorpha NCYC495 allowed the self-assembly of VLPs into a correct human papilloma pseudovirion structure. P. pastoris tend to give a higher level of L1 protein than H. polymorpha in a batch system; however, both hosts gave a stable integration that keeps the protein expression stable during production. Our purposed vaccine predicted has a high immune activation, is safe and is easy to produce in various expression systems. However, the designed vaccine needs to validate further in vitro and in vivo assay. In the end, a combination of different parameters in characterizing protein expression is a powerful way of exploring protein expression profiles that could be helpful for massive production industrial-scale production.
Availability of data and materials
All data generated or analyzed during this activity are included in this published article.
Abbreviations
- HPV:
-
Human papilloma virus
- VLP:
-
Virus-like particles
- MEV:
-
Multi epitope vaccine
- CAI:
-
Codon adaptation index
- MFE:
-
Minimum free energy
- PCR:
-
Polymerase chain reaction
- NFW:
-
Nuclease-free water
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- WB:
-
Western blot
- TEM:
-
Transmission electron microscope
- BMGY:
-
Buffered glycerol complex medium
- BMMY:
-
Buffered methanol-complex medium
- PMSF:
-
Phenylmethylsulfonyl fluoride
- BCA:
-
Bicinchoninic acid
- EDTA:
-
Ethylenediaminetetraacetic acid
- TMB:
-
3,3′,5,5′-Tetramethylbenzidine
- PRRs:
-
Pathogen recognition receptors
- MHC:
-
Major histocompatibility complex
- MITD:
-
MHC I-targeting domain
- TLR:
-
Toll-like receptor
References
Lei J, Ploner A, Elfström KM, Wang J, Roth A, Fang F, Sundström K (2020) HPV vaccination and the risk of invasive cervical cancer. N Engl J Med 383:1340–1348
Chatterjee A (2014) The next generation of HPV vaccines: nonavalent vaccine V503 on the horizon. Expert Rev Vaccines 13:1279–1290
Fuenmayor J, Gòdia F, Cervera L (2017) Production of virus-like particles for vaccines. New Biotechnol 39:174–180
Markowitz LE, Schiller JT (2021) Human Papillomavirus Vaccines. J Infect Dis 224:S367-s378
Nascimento IP, Leite LCC (2012) Recombinant vaccines and the development of new vaccine strategies. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas 45:1102–1111
Saylor K, Gillam F, Lohneis T, Zhang C (2020) Designs of Antigen Structure and Composition for Improved Protein-Based Vaccine Efficacy. Front Immunol 11:283–283
Syomin BV, Ilyin YV (2019) Virus-like particles as an instrument of vaccine production. Mol Biol 53:323–334
Wang JW, Roden RBS (2013) Virus-like particles for the prevention of human papillomavirus-associated malignancies. Expert Rev Vaccines 12:129–141
Bazan SB, de Alencar Muniz A, Chaves KA, Aires AM, Cianciarullo RL, Garcea, and P. L. Ho, (2009) Expression and characterization of HPV-16 L1 capsid protein in Pichia pastoris. Adv Virol 154:1609
Hitzeroth II, Chabeda A, Whitehead MP, Graf M, Rybicki EP (2018) Optimizing a human papillomavirus type 16 L1-Based Chimaeric Gene for Expression in Plants. Front Bioeng Biotechno 6.
Carter JJ, Yaegashi N, Jenison SA, Galloway DA (1991) Expression of human papillomavirus proteins in yeast Saccharomyces cerevisiae. Virology 182:513–521
Karbalaei M, Rezaee SA, Farsiani H (2020) Pichia pastoris: A highly successful expression system for optimal synthesis of heterologous proteins. J Cell Physiol 235:5867–5881
Hollenberg CP, Gellissen G (1997) Production of recombinant proteins by methylotrophic yeasts. Curr Opin Biotechnol 8:554–560
Tokmakov AA, Kurotani A, Takagi T, Toyama M, Shirouzu M, Fukami Y, Yokoyama S (2012) Multiple post-translational modifications affect heterologous protein synthesis. J Biol Chem 287:27106–27116
Cai X, Li JJ, Liu T, Brian O, Li J (2021) Infectious disease mRNA vaccines and a review on epitope prediction for vaccine design. Brief Funct Genomics 20:289–303
Niu Y, Liu Y, Yang L, Qu H, Zhao J, Hu R, Li J, Liu W (2016) Immunogenicity of multi-epitope-based vaccine candidates administered with the adjuvant Gp96 against rabies. Virologica Sinica 31:168–175
Barbier AJ, Jiang AY, Zhang P, Wooster R, Anderson DG (2022) The clinical progress of mRNA vaccines and immunotherapies. Nat Biotechnol 40:840–854
Weng Y, Huang Y (2021) Advances of mRNA vaccines for COVID-19: A new prophylactic revolution begins. Asian J Pharm Sci 16:263–264
Liu C, Yao Y, Yang X, Bai H, Huang W, Xia Y, Ma Y (2015) Production of Recombinant Human Papillomavirus Type 52 L1 Protein in Hansenula polymorpha Formed Virus-Like Particles. J Microbiol Biotechnol 25:936–940
Dewi KS, Chairunnisa S, Swasthikawati S, Yuliawati D, Agustiyanti F, Mustopa AZ, Kusharyoto W, Ningrum RA (2022) Production of codon-optimized Human papillomavirus type 52 L1 virus-like particles in Pichia pastoris BG10 expression system. Prep Biochem Biotechnol 1–9
Hartner FS, Glieder A (2006) Regulation of methanol utilisation pathway genes in yeasts. Microb Cell Fact 5:39–39
Firdaus MER, Mustopa AZ, Triratna L, Syahputra G, Nurfatwa M (2022) Dissection of capsid protein HPV 52 to rationalize vaccine designs using computational approaches immunoinformatics and molecular docking. Asian Pac J Cancer Prev 23:2243–2253
Nakamura Y, Gojobori T, Ikemura T (2000) Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res 28:292
Puigbò P, Bravo IG, Garcia-Vallvé S (2008) E-CAI: a novel server to estimate an expected value of Codon Adaptation Index (eCAI). BMC Bioinformatics 9:65–65
Bellaousov S, Reuter JS, Seetin MG, Mathews DH (2013) RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res 41:W471-474
Sambrook, J. (2001) Molecular cloning : a laboratory manual. Third edition. Cold Spring Harbor, N.Y. : Cold Spring Harbor Laboratory Press, [2001] ©2001.
Akada, R., T. Murakane, and Y. Nishizawa (2000) DNA extraction method for screening yeast clones by PCR. Biotechniques. 28: 668–670, 672, 674.
Tapia-Tussell R, Lappe P, Ulloa M, Quijano-Ramayo A, Cáceres-Farfán M, Larqué-Saavedra A, Perez-Brito D (2006) A rapid and simple method for DNA extraction from yeasts and fungi isolated from Agave fourcroydes. Mol Biotechnol 33:67–70
Cunbao L, Yufeng Y, Xu Y, Hongmei B, Weiwei H, Ye X (2015) Production of Recombinant Human Papillomavirus Type 52 L1 Protein in Hansenula polymorpha Formed Virus-Like Particles. J Microbiol Biotechnol 25:936–940
Kim H, Thak EJ, Yeon JY, Sohn MJ, Choo JH, Kim JY, Kang HA (2018) Functional analysis of Mpk1-mediated cell wall integrity signaling pathway in the thermotolerant methylotrophic yeast Hansenula polymorpha. J Microbiol 56:72–82
Abad S, Kitz K, Hörmann A, Schreiner U, Hartner FS, Glieder A (2010) Real-time PCR-based determination of gene copy numbers in Pichia pastoris. Biotechnol J 5:413–420
Shirvani R, Barshan-tashnizi M, Shahali M (2020) An investigation into gene copy number determination in transgenic yeast; The importance of selecting a reliable real-time PCR standard. Biologicals 65:10–17
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8:4
Rapin N, Lund O, Bernaschi M, Castiglione F (2010) Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 5:e9862
Khan K, Khan SA, Jalal K, Ul-Haq Z, Uddin R (2022) Immunoinformatic approach for the construction of multi-epitopes vaccine against omicron COVID-19 variant. Virology 572:28–43
Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP (2013) In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 8:e73957
Gasteiger, E., C. Hoogland, A. Gattiker, S. e. Duvaud, M. R. Wilkins, R. D. Appel, and A. Bairoch (2005) Protein identification and analysis tools on the ExPASy server. pp. 571–607In: J. M. Walker (ed.). The Proteomics Protocols Handbook. Humana Press, City.
Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S (2017) The ClusPro web server for protein–protein docking. Nat Protoc 12:255–278
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I-TASSER Suite: protein structure and function prediction. Nat Methods 12:7–8
Laskowski RA, Jabłońska J, Pravda L, Vařeková RS, Thornton JM (2018) PDBsum: Structural summaries of PDB entries. Protein Sci 27:129–134
Reuter JS, Mathews DH (2010) RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11:129
Patricia Gita, N., N. Neni, R. Elvi, N. Dessy, R. Debbie Soefie, N. Wardono, and G.-R. Ernawati Arifin (2020) Integration stability of sHBsAg-multi expression cassettes in Pichia pastoris GS115 during methanol induction. HAYATI Journal of Biosciences. 283.
Kim H, Yoo SJ, Kang HA (2015) Yeast synthetic biology for the production of recombinant therapeutic proteins. FEMS Yeast Res 15:1–16
Nielsen KH (2014) Protein expression-yeast. Methods Enzymol 536:133–147
Fu H, Liang Y, Zhong X, Pan Z, Huang L, Zhang H, Xu Y, Zhou W, Liu Z (2020) Codon optimization with deep learning to enhance protein expression. Sci Rep 10:17617
Vieira É, D., G. Andrietta Mda, and S. R. Andrietta, (2013) Yeast biomass production: a new approach in glucose-limited feeding strategy. Braz J Microbiol 44:551–558
Ferndahl C, Bonander N, Logez C, Wagner R, Gustafsson L, Larsson C, Hedfalk K, Darby RAJ, Bill RM (2010) Increasing cell biomass in Saccharomyces cerevisiae increases recombinant protein yield: the use of a respiratory strain as a microbial cell factory. Microb Cell Fact 9:47
Macauley-Patrick S, Fazenda ML, McNeil B, Harvey LM (2005) Heterologous protein production using the Pichia pastoris expression system. Yeast 22:249–270
Aw R, Polizzi KM (2013) Can too many copies spoil the broth? Microb Cell Fact 12:128–128
Nie L, Wu G, Zhang W (2006) Correlation of mRNA expression and protein abundance affected by multiple sequence features related to translational efficiency in Desulfovibrio vulgaris: a quantitative analysis. Genetics 174:2229–2243
Liu Y (2020) A code within the genetic code: codon usage regulates co-translational protein folding. Cell Communication and Signaling 18:145
Le DT, Müller KM (2021) In vitro assembly of virus-like particles and their applications. Life (Basel, Switzerland) 11:334
Zhao H, Li HY, Han JF, Deng YQ, Li YX, Zhu SY, He YL, Qin ED, Chen R, Qin CF (2013) Virus-like particles produced in Saccharomyces cerevisiae elicit protective immunity against Coxsackievirus A16 in mice. Appl Microbiol Biotechnol 97:10445–10452
Scotti N, Rybicki EP (2013) Virus-like particles produced in plants as potential vaccines. Expert Rev Vaccines 12:211–224
Gopal R, Schneemann A (2018) Production and application of insect virus-based VLPs. Methods Mol Biol 1776:125–141
Mariani L, Venuti A (2010) HPV vaccine: an overview of immune response, clinical protection, and new approaches for the future. J Transl Med 8:105
Zepeda-Cervantes, J., J. O. Ramírez-Jarquín, and L. Vaca (2020) Interaction Between Virus-Like Particles (VLPs) and Pattern Recognition Receptors (PRRs) From Dendritic Cells (DCs): Toward Better Engineering of VLPs. Frontiers in Immunology. 11.
Liang, Y., L. Huang, and T. Liu (2021) Development and delivery systems of mRNA vaccines. Frontiers in Bioengineering and Biotechnology. 9.
Ahammad I, Lira SS (2020) Designing a novel mRNA vaccine against SARS-CoV-2: an immunoinformatics approach. Int J Biol Macromol 162:820–837
Chen X, Zaro JL, Shen WC (2013) Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev 65:1357–1369
Huang, Z., C. Zhang, and X.-H. Xing (2021) Chapter Two - Design and construction of chimeric linker library with controllable flexibilities for precision protein engineering. pp. 23–49In: M. Merkx (ed.). Methods in enzymology. Academic Press, City.
Schlake T, Thess A, Fotin-Mleczek M, Kallen K-J (2012) Developing mRNA-vaccine technologies. RNA Biol 9:1319–1330
Awate S, Babiuk LA, Mutwiri G (2013) Mechanisms of action of adjuvants. Front Immunol 4:114
Acknowledgements
We would like to thank A.M. (Arjen) Krikken, University of Groningen, for providing an H. polymorpha NCYC495 and pHIPH4 plasmid, and Dr. Linda Juniar, Department of Cell and Molecular Biology, Uppsala University, Sweden, for protein structure analysis.
Funding
This study was supported by a grant from Lembaga Pengelola Dana Pendidikan (LPDP) No. 105/E1/PRN/2020 and DIPA PN Rumah Program Hasil Pengungkapan dan Pemanfaatan Biodiversitas Nusantara OR Hayati dan Lingkungan BRIN 2022 (No. RP1WBS3-064) and 2023 (No RP1Tema3-45). The funder had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
MERF and AZM designed the study. MERF, AZM, SC, RKA, SI, and AP conducted laboratory work and data analysis. MERF, AH, NE writing, reviewing, and editing. AK and MN supported materials and data analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Listed epitopes included in the construct. Figure S1. mRNA structure of HPV52 L1 starting from predicted transcription initiation complex. A mRNA structure of the native codon P. pastoris. B mRNA structure of the optimized codon P. pastoris. C mRNA structure of the native codon H. polymorpha, D mRNA structure of the optimized codon H. polymorpha. Figure S2. The Ramachandran plot of predicted vaccine structure using ZLab server; green: highly preferred conformations, delta ≥ −2; brown: preferred conformations, −2 > delta ≥ −4; and red: questionable conformations, delta < −4.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Firdaus, M.E.R., Mustopa, A.Z., Ekawati, N. et al. Optimization, characterization, comparison of self-assembly VLP of capsid protein L1 in yeast and reverse vaccinology design against human papillomavirus type 52. J Genet Eng Biotechnol 21, 68 (2023). https://doi.org/10.1186/s43141-023-00514-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-023-00514-9