
Abstract
The 5-carbon positions on cytosine nucleotides preceding guanines in genomic DNA (CpG) are common targets for DNA methylation (5mC). DNA methylation removal can occur through both active and passive mechanisms. Ten-eleven translocation enzymes (TETs) oxidize 5mC in a stepwise manner to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). 5mC can also be removed passively through sequential cell divisions in the absence of DNA methylation maintenance. In this chapter, we describe approaches that couple TET-assisted bisulfite (TAB) and oxidative bisulfite (OxBS) conversion to the Illumina MethylationEPIC BeadChIP (EPIC array) and show how these technologies can be used to distinguish active versus passive DNA demethylation. We also describe integrative bioinformatics pipelines to facilitate this analysis.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
- DNA methylation
- DNA hydroxymethylation
- Active demethylation
- Passive demethylation
- Illumina MethylationEPIC BeadChip
- Tet-assisted bisulfite (TAB)
- Oxidative bisulfite (OxBS)
- TETs
- DNMTs
- Epigenetics
1 Introduction
Methylation on the 5-carbon of cytosine nucleotides in genomic DNA of eukaryotes is the most extensively studied epigenetic modification. To date, over 70,000 research papers, methods chapters, and review articles have been dedicated to the study of DNA methylation (5mC). 5mC provides diverse functionality in the regulation of gene expression, genome stability, chromatin compaction, and developmental timing [1]. Indeed, DNA methylation is largely regarded as one of the most stable epigenetic modifications, as its inheritance to daughter cells following cell division is faithfully copied during DNA replication by the maintenance methylation machinery, DNA methyltransferase 1 (DNMT1) and Ubiquitin-like, containing PHD and RING finger domains, 1 (UHRF1) [2,3,4,5]. 5mC patterning is conserved across most somatic tissues, with the most dynamics occurring at enhancers and other distal regulatory regions of the genome that influence gene expression [6, 7]. Additional dynamic changes in 5mC are observed in disease transformation and in early mammalian development, as further described below.
DNA methylation can be passively removed in dividing cells that lack DNA methylation maintenance activity. An active mechanism for 5mC removal remained elusive until 2009, when the existence of an oxidized form of DNA methylation, DNA hydroxymethylation (5hmC) was thrust into the spotlight with the discovery of its abundance in neuronal tissue and the identification of an enzyme that could oxidize 5mC to 5hmC, Ten-eleven translocation 1 (TET1) [8,9,10]. Subsequently, two additional TET enzymes, TET2 and TET3, also demonstrated the ability to oxidize 5mC in a stepwise manner to 5hmC, 5-formylcytosine (5fC), and 5-carboxylcytoine (5caC) [11, 12]. Oxidation of 5mC to 5fC and 5caC allows for base-excision repair of the oxidized nucleotide by thymidine deglycosylase (TDG) and replacement by unmodified cytosine (5C) [13,14,15]. Combined, these discoveries laid the foundation for what is now widely accepted as the active DNA demethylation pathway.
While recent evidence suggests that the oxidized forms of 5mC can act in a regulatory manner through the recruitment of reader proteins [16, 17], perhaps the most well-studied roles for the active DNA demethylation pathway are in the early stages of mammalian development [18]. Following fertilization, both the paternal and maternal genomes undergo massive changes in DNA methylation patterning that occurs through both active and passive DNA demethylation, respectively [19,20,21,22,23]. Primordial germ cells (PGCs) also undergo a dramatic loss of DNA methylation that can be attributed to both passive and active DNA demethylation mechanisms [24, 25]. Embryonic stem cells (ESCs) also rely on TET proteins to maintain self-renewal properties as well as to direct lineage specification upon induction of differentiation [11, 26].
Given the importance of 5mC for maintaining proper control of chromatin structure and function, aberrant patterning of 5mC has been widely studied in the context of aging, psychiatric and developmental disorders, and cancer [27,28,29,30]. As hypermethylation of tumor suppressor genes is a hallmark of cancer, significant effort has been devoted to developing therapies that induce DNA demethylation of these genes in order to restore their expression and function in cancer cells [27]. Accordingly, both passive and active DNA demethylation mechanisms are now being targeted for combination cancer therapies with DNMT inhibitors like 5-aza-2′-deoxycytidine (DAC) and with l-ascorbic acid (Vitamin C, VitC), a co-factor for TET dioxygenase activity [31,32,33].
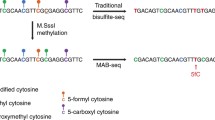
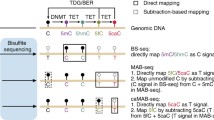
In this chapter, we use DAC and VitC to induce active and passive DNA demethylation in the human germ cell tumor-derived cell line NCCIT, known to express TET enzymes [34, 35]. To distinguish between active and passive DNA demethylation at base-resolution, we coupled Tet-assisted bisulfite (TAB) and oxidative bisulfite (OxBS) conversion chemistries to DNA methylation analysis with the Illumina MethylationEPIC BeadChIP (EPIC array) [36,37,38,39,40]. The EPIC array is a high-throughput platform that interrogates the DNA methylation status of approximately 850,000 individual CpG dinucleotides at base-resolution across multiple features of the genome (e.g., CpG islands, promoters, enhancers). Using bisulfite-converted genomic DNA (gDNA) as an input, single-stranded DNA probes hybridize to the bisulfite-converted gDNA, and single base-pair extensions with fluorescently labeled nucleotides reveal the underlying modification status of the gDNA (Fig. 1a). For example, if a cytosine nucleotide is unmodified in the gDNA, bisulfite conversion will deaminate the cytosine to uracil, which will then be read as thymine following whole-genome amplification. Once the probe for this specific CpG hybridizes to the bisulfite-converted gDNA, an adenine nucleotide will be incorporated and give off a fluorescent signal to indicate that the cytosine was unmethylated (Fig. 1a). Vice versa, cytosine nucleotides that are modified (5mC/5hmC ) are protected from bisulfite conversion and will remain cytosines [41]. Following bisulfite conversion, whole-genome amplification, and hybridization, a fluorescently labeled guanine nucleotide will be incorporated, informing that the underlying cytosine was methylated (Fig. 1a).

Coupling TAB and OxBS to EPIC array analysis to enable distinction of 5mC and 5hmC. (a) Sodium bisulfite (BS) conversion of genomic DNA (gDNA) deaminates unmodified cytosine (5C) residues to uracil (U). 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) are protected from deamination. Bisulfite-converted gDNA is PCR amplified and hybridized to the Illumina Infinium MethylationEPIC BeadChip (EPIC array) for analysis. Both 5mC and 5hmC are read as C, while 5C residues are read as T on this platform. (b) TET-assisted bisulfite conversion (TAB) incorporates transfer of a sugar moiety (gluc) to 5hmC by β-glucosyltransferase (β-GT). Prior to bisulfite conversion, β-GT-modified gDNA is reacted with the catalytic domain of TET2 (TET2-CD), which catalyzes stepwise oxidation of 5mC to 5hmC, 5-formylcytosine (5fC), and 5-carboxycytosine (5caC). Gluc-5hmC is protected from further oxidation and is resistant to bisulfite conversion. 5fC, 5caC, and 5C are converted as described above. (c) With Oxidative Bisulfite conversion (OxBS ), gDNA is treated with potassium perruthenate (KRuO4) to oxidize 5hmC to 5fC prior to reaction with sodium bisulfite. Image of EPIC array modified from Illumina
TAB conversion is an upstream modification to the standard bisulfite conversion method that allows for only 5hmC nucleotides to be read as modified cytosines [38, 42]. 5hmC nucleotides in gDNA are first protected from downstream steps by addition of a glucose moiety mediated by T4 β-glucosyltransferase (β-GT) (Fig. 1b). 5mC nucleotides are targeted for TET-mediated stepwise oxidation to 5hmC, 5fC, and 5caC by incubating β-GT-treated gDNA with the recombinant catalytic domain of TET2 and its required co-factors. Following bisulfite conversion and amplification, only 5hmC nucleotides will be read as cytosine by the EPIC array, and 5mC/5C nucleotides are read as thymine (Fig. 1b). With OxBS conversion, 5hmC nucleotides in gDNA are oxidized by potassium perruthenate (KRuO4) to 5fC prior to bisulfite conversion (Fig. 1c) [43]. Following EPIC array processing, 5mC will be read as cytosine while all oxidized cytosines and 5C will be thymine (Fig. 1c).
In this chapter, we demonstrate the utility of EPIC arrays for determining active versus passive DNA demethylation using the techniques shown in Fig. 1. We provide bioinformatic pipelines that can be used to analyze the 5mC and 5hmC signals from TAB and OxBS arrays. Additionally, we detail assays that can be used to determine relative global change in 5mC and 5hmC across gDNA samples, which we use to check samples prior to EPIC array analysis. Finally, we provide a comparison of the TAB array and OxBS array approaches and discuss how to determine which platform is best suited for different experiments.
2 Materials
2.1 Benchtop Assays to Detect DNA Modification Change
2.1.1 Locus-Specific High-Resolution Melt (HRM) Analysis
2.1.1.1 Equipment and Reagents
-
1.
NanoDrop spectrophotometer.
-
2.
ZYMO EZ DNA Methylation Kit.
-
3.
Bio-Rad Precision Melt Supermix.
-
4.
Heat block, water-bath or thermocycler capable of holding temp at 37 and 50 °C.
-
5.
Nuclease-free water.
-
6.
Real-Time PCR instrument with SYBR detection capabilities.
-
7.
Compatible Real-Time PCR plates (96-well).
-
8.
Compatible Real-Time PCR plate seals.
2.1.2 Global Quantification of 5hmC
2.1.2.1 ELISA-Based Assay
2.1.2.1.1 Equipment and Reagents
-
1.
EpiGentek MethylFlash Global DNA Hydroxymethylation (5-hmC) ELISA Easy Kit (Colorimetric).
-
2.
8-channel pipette.
-
3.
Aerosol resistant pipette tips.
-
4.
Incubator at 37 °C.
-
5.
Microplate reader capable of reading absorbance at 450 nm.
2.1.2.2 DNA Dot Blot
2.1.2.2.1 Equipment and Reagents
-
1.
1 M NaOH.
-
2.
10 M ammonium acetate.
-
3.
20× SSC buffer: 3 M NaCl, 300 mM sodium citrate.
-
4.
1× TE buffer: 10 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0.
-
5.
1× PBST: 2.68 mM KCl, 1.47 mM KH2PO4, 136.9 mM NaCl, 9.5 mM Na2PO4, 1% Tween-20.
-
6.
Stripping buffer: 5% acetic acid, 500 mM NaCl.
-
7.
5% methylene blue stain.
-
8.
Thermo Scientific Superblock T20 blocking buffer.
-
9.
Nitrocellulose membrane and two pieces of filter paper cut to 4.5″ × 3.1″.
-
10.
NanoDrop spectrophotometer.
-
11.
Stratagene UV Stratalinker 2400.
-
12.
Hybridization oven.
-
13.
Bio-rad Bio-Dot apparatus.
-
14.
12-channel pipette.
-
15.
Multi-channel filtered pipette tips.
-
16.
96-well plate with concave bottom wells.
-
17.
Active Motif anti-rabbit 5hmC antibody (pAb: 39791).
-
18.
Film developer.
2.2 Modifications to Bisulfite Conversion Chemistry to Distinguish 5mC from 5hmC
2.2.1 TET-Assisted Bisulfite (TAB) Array
2.2.1.1 Equipment and Reagents
-
1.
Covaris E220 evolution sonicator.
-
2.
Covaris microtube (130 μL volume).
-
3.
Thermocycler.
-
4.
PCR-tube strips (200 μL).
-
5.
Heat block or incubator to 37 °C.
-
6.
DynaMag magnet.
-
7.
Invitrogen Qubit fluorometer.
-
8.
Invitrogen Qubit assay tubes.
-
9.
Transilluminator (312 nm).
-
10.
Agarose gel electrophoresis apparatus.
-
11.
QUMA analysis software [44].
-
12.
3 M Sodium Acetate pH 4.8.
-
13.
100% Ethanol.
-
14.
Nuclease-free water.
-
15.
Invitrogen Qubit dsDNA HS assay kit.
-
16.
T4-Phage β-glucosyltransferase (T4-βGT).
-
17.
ZYMO 5-Methylcytosine and 5-Hydroxymethylcytosine DNA Standard Set.
-
18.
KAPA Biosystems KAPA Pure beads.
-
19.
Tet oxidation reagent #1: 1.5 mM Fe(NH4)2(SO4)2.
-
20.
Tet oxidation reagent #2: 83.3 mM NaCl, 167 mM HEPES pH 8.0, 4 mM ATP, 8.3 mM DTT, 3.33 mM α-ketoglutaric acid, 6.7 mM l-ascorbic acid.
-
21.
TET2 catalytic domain (TET2-CD) 2.0 mg/mL.
-
22.
ZYMO EZ DNA Methylation Kit.
-
23.
Taq polymerase.
-
24.
Agarose.
-
25.
DNA Gel Extraction Kit.
-
26.
Promega pGEM-T Vector System I.
-
27.
DH5a high-efficiency competent cells.
-
28.
X-gal.
-
29.
Ampicillin agar bacterial plates.
-
30.
illustra TempliPhi DNA Sequencing Template Amplification Kit.
2.2.2 Oxidative Bisulfite (OxBS) Array
2.2.2.1 Equipment and Reagents
-
1.
NuGEN TrueMethyl oxBS Module, Tecan Genomics, Inc. (Catalog #: 0414-32).
3 Methods
3.1 Benchtop Assays to Detect DNA Modification Change
We treated NCCIT cells (biological duplicate) with PBS (NoTx), 1 μM DAC to induce passive DNA demethylation, and 1 μM DAC with 57 μM VitC (DAC + VitC) to induce both passive and active DNA demethylation (Fig. 2a). In order to conduct TAB array or OxBS array, each gDNA sample must undergo two different treatments: (1) bisulfite conversion and (2) TAB/OxBS conversion. Both treatments of an individual gDNA sample are then submitted for processing on the EPIC array, meaning that the user cost is doubled for analysis of each sample. Depending on the nature of the experiment, querying both 5mC and 5hmC on the EPIC array can become quite expensive. In this section, we describe quick, low-cost benchtop assays commonly used in our laboratory to detect locus-specific and global changes in 5mC and 5hmC across gDNA samples of interest prior to submission on the EPIC array.

Assays to measure DNA methylation changes in isolated gDNA samples prior to EPIC array analysis. (a) Drug treatment paradigm in NCCIT cells. Cells were treated with PBS (NoTx), 1 μM DAC (DAC), or 1 μM DAC and 57 μM Vitamin C (DAC + VitC) for 72 h as shown. (b) High-resolution melt (HRM) curve analysis of candidate modified (exons of PPP1R18 and DAXX) and unmodified (promoter of RPL30) regions in NCCIT cells. An average of technical duplicates is shown. gDNA from HCT116 cells with knockout of DNMT1 and DNMT3B alleles (DKO1) is included as a positive control for DNA demethylation. (c) Global quantification of 5hmC in NCCIT cells (treated as described in a) with the ELISA-based MethylFlash Hydroxymethylated DNA 5-hmC Quantification Kit (EpiGentek). Error bars represent SEM among biological duplicates and technical duplicates. Unpaired t-tests were conducted to determine p-values. (d) DNA dot blot analysis for 5hmC in NCCIT cells (treated as described in a). 800 ng of DNA was loaded on the top row followed by serial twofold dilutions in the subsequent rows. Methylene blue staining is a loading control for total DNA
3.1.1 Locus-Specific High-Resolution Melt (HRM) Analysis
High-resolution melt (HRM) analysis is a quantitative, real-time PCR-based method that allows the user to determine the relative nucleotide composition of a region of double-stranded DNA by analyzing the melting curve of a PCR amplicon [45]. Initially designed to identify mutations and polymorphisms in a gDNA sample, HRM has been adapted for use in epigenetics research to determine the relative amount of DNA modifications at a given locus [46, 47]. Following DNA isolation, a sodium bisulfite conversion step is performed to make single nucleotide polymorphisms (SNPs) to the DNA that indicate if a cytosine nucleotide was modified. If the cytosine is modified (either 5mC or 5hmC), the C will stay a C, but if the cytosine is unmodified, it will be deaminated to uracil (and converted into thymine during PCR amplification). HRM takes advantage of these methylation-specific SNPs. After bisulfite conversion, regions of interest in the genome are amplified by real-time qPCR, and then a high-resolution melt step, in which the temperature is raised in very small increments and fluorescence is detected after each increment, is conducted to determine the melting temperature of the amplicon. The more Ts (unmodified cytosines) in the amplicon, the lower the melting temperature; the more Cs (modified cytosines) in the amplicon, the higher the melting temperature. By using differences in melting temperature of an amplicon across samples, the relative DNA modification state of a sample can be determined.
In our treatment paradigm (Fig. 2a), we measured a decrease in the peak melting temperature for DAC and DAC + VitC samples relative to the NoTx group, indicating that the genomic loci being queried (PPP1R18, DAXX) have less modified cytosines than the NoTx group (Fig. 2b). The RPL30 promoter served as a negative control for cytosine modifications, as it is completely unmodified in NCCIT (Fig. 2b). RPL30 also served as a positive control for bisulfite conversion, as amplification of this region could not occur without high conversion efficiency. Collectively, these results demonstrate that the DAC treatment was effective in reducing the overall modification level of cytosines at known regions of modification in the NoTx sample, indicating that these samples and treatment paradigms were good candidates for EPIC array analysis.
3.1.1.1 General Procedure
-
1.
Design bisulfite qPCR primers for regions of interest, including a known fully modified region and a known fully unmodified region, using MethPrimer [48] with the following specifications:
-
(a)
Primer Length: between 20 and 30 bp.
-
(b)
Amplicon Length: between 100 and 150 bp.
-
(c)
Tm of primer set: between 58 and 61 °C.
-
(d)
May allow one CpG in the first 1/3 of the primer.
-
(e)
Aim to have at least three CpGs in the amplicon so that melting temperatures will be noticeably divergent.
-
(a)
-
2.
Using the ZYMO EZ DNA Methylation kit, bisulfite convert 500 ng of sample gDNA as described in the kit protocol. Elute in 10 μL of nuclease-free water, then dilute sample with 42 μL of nuclease-free water (see Notes 1–3).
-
3.
Optimize primers by real-time qPCR on bisulfite-converted gDNA. Ensure that only one amplicon (one peak in the melting curve) is produced.
-
4.
Set up each PCR reaction as follows:
Reagent
Amount (μL)
10× Bio-Rad Precision Supermix
10
Bisulfite primers (2 μM)
2
Nuclease-free water
3
Bisulfite-converted gDNA
5
-
5.
Set up the PCR protocol as follows:
Step 1
95 °C
2 min
Step 2
95 °C
10 s
Step 3
Annealing Temp
30 s
Repeat Steps 2 and 3 39×.
Step 4
95 °C
30 s
Step 5
60 °C
1 min
Step 6
Melt Curve
65–95 °C—10 s/step—increase temperature by 0.1 °C each step, and capture fluorescence at end of each step.
-
6.
The CFX manager software will automatically calculate the melting temperature of each sample. All fluorescence data can also be exported for each individual temperature measurement (“Melt Curve Derivative Results.xlsx”) to build the plots as shown in Fig. 2b (see Note 4).
3.1.2 Global Quantification of 5hmC
As 5mC is substantially more abundant in the genome than 5hmC (approximately 14-fold higher), quantification by HRM for passive loss of DNA methylation is sufficient. However, detecting global changes in 5hmC and active DNA demethylation is challenging due to the low level of this modification on cytosine nucleotides. In this section, we discuss two approaches to determine the global level of 5hmC across samples: (1) ELISA-based quantification and (2) 5hmC DNA Dot Blot.
3.1.2.1 ELISA-Based Assay
HRM analysis of DAC and DAC + VitC treated samples suggested substantial loss in cytosine modifications relative to NoTx (Fig. 2b). As 5mC is the most abundant cytosine modification, detection of changes in 5hmC are likely masked by 5mC changes in the HRM assay. Using the EpiGentek MethylFlash Global DNA Hydroxymethylation (5-hmC) ELISA Easy Kit (Colorimetric), we profiled global 5hmC levels in our gDNA samples to determine if treatment with DAC or DAC + VitC induced changes in 5hmC. Indeed, while DAC treatment did not significantly affect global 5hmC levels relative to NoTx, the addition of VitC to the DAC treatment lead to a significant increase in 5hmC detectable by this assay (Fig. 2c). Taken together with our HRM results of these gDNA samples, we concluded that our treatment conditions induced changes to both 5mC and 5hmC.
3.1.2.1.1 General Procedure
-
1.
Prepare gDNA samples in a 96-well plate at a concentration of 25 ng/μL. A total of 100 ng gDNA is added to the assay wells.
-
2.
Follow all assay instructions in the EpiGentek MethylFlash Global DNA Hydroxymethylation (5-hmC) ELISA Easy Kit (Colorimetric) manual (see Notes 5 and 6).
-
3.
Follow analysis instructions as outlined in the EpiGentek MethylFlash Global DNA Hydroxymethylation (5-hmC) ELISA Easy Kit (Colorimetric) manual (see Note 7). Analyze all biological and technical duplicates separately.
3.1.2.2 DNA Dot Blot
For additional confirmation that our treatments sufficiently promoted changes in 5hmC levels, we performed gDNA dot blot analysis. Briefly, gDNA is denatured and immobilized on a nitrocellulose membrane prior to being probed with a 5hmC antibody. With this assay, changes in global 5hmC were detected in samples treated with DAC and DAC + VitC relative to NoTx (Fig. 2d). Complimenting the results of our HRM analysis and ELISA-based assays, we further concluded that both active and passive DNA demethylation would be observed in our samples after application to TAB array and OxBS array.
3.1.2.2.1 General Procedure
-
1.
Pre-chill 10 M ammonium acetate on ice.
-
2.
Use a NanoDrop spectrophotometer to measure gDNA sample concentration.
-
3.
For each sample, prepare 2 μg gDNA in 225 μL 1× TE buffer (see Note 8).
-
4.
Denature samples in 0.1 M NaOH at 95 °C for 10 min.
-
5.
Neutralize samples with 1 M ammonium acetate on ice. Incubate sample on ice for 10 min.
-
6.
Load 240 μL of each sample into the top row of a 96-well plate. Load 120 μL 1× TE buffer in each sequential row. Using a multichannel pipette, ensure the samples in the top row containing gDNA are thoroughly mixed and transfer 120 μL to the row below. Repeat this process working down the rows to achieve twofold serial dilutions.
-
7.
Equilibrate nitrocellulose membrane and two sheets of filter paper in 6× SSC buffer.
-
8.
Secure membrane on top of filter papers in the dot blot apparatus. Tighten knobs as much as possible, apply vacuum, and re-tighten knobs.
-
9.
Wash wells with 200 μL 1× TE buffer (see Note 9).
-
10.
Using a multichannel pipette, apply 109 μL of each sample to the membrane. Final amount of gDNA is 800 ng followed by twofold serial dilutions. Allow samples to sit on membrane 2–5 min before applying vacuum (see Note 9).
-
11.
Apply vacuum to pull samples through the manifold. Once each well has cleared, wash wells in 200 μL 2× SSC buffer.
-
12.
Remove membrane from apparatus, mark corners with a pencil to maintain orientation, place in a covered container (we use pipette tip box lids), and dry at 80 °C for 45 min in a hybridization oven.
-
13.
UV-crosslink gDNA to membrane at 120,000 μJ.
-
14.
Block for 1 h in Superblock at room temperature.
-
15.
Incubate blot overnight at 4 °C in Active Motif anti-rabbit 5hmC antibody (pAb: 39791) diluted 1:5000 in Superblock.
-
16.
Wash blot 3 × 5 min in 1× PBST buffer (see Note 10).
-
17.
Incubate blot in rabbit secondary antibody diluted 1:5000 in Superblock at room temperature for 1 h.
-
18.
Wash blot 3 × 5 min in 1× PBST buffer (see Note 10).
-
19.
Use chemiluminescence to visualize blot.
-
20.
For verification of gDNA loading, incubate blot in stripping buffer for 20–30 min. Rinse with distilled water and incubate in 5% methylene blue stain for 15–20 min. Rinse with distilled water and place between plastic to scan image.
3.2 Modifications to Bisulfite Conversion Chemistry to Distinguish 5mC from 5hmC
3.2.1 Tet-Assisted Bisulfite (TAB) Array
3.2.1.1 General Procedure
3.2.1.1.1 Preparation of gDNA
-
1.
Quantify gDNA by Invitrogen Qubit fluorometer dsDNA HS assay and dilute 5 μg gDNA in nuclease-free water to a final volume of 130 μL.
-
2.
Transfer prepared gDNA to a Covaris microtube, and shear sample with Covaris E220 sonicator to a final size of <10,000 bp using the following parameters:
Peak incident power (W)
140
Duty factor
2%
Cycles per burst
200
Treatment time
10 s
-
3.
Transfer sheared gDNA from the Covaris microTUBE to a 1.5 mL microcentrifuge tube.
-
4.
Precipitate the sheared gDNA by adding 13 μL 3 M Sodium Acetate (1/10 volume) and 325 μL 100% ethanol to each sample. Store samples at −20 °C for 30 min to overnight.
-
5.
Centrifuge samples at 17,090 RCF for 30 min at 4 °C to pellet precipitated gDNA.
-
6.
Wash samples once with 70% ethanol, and centrifuge at 17,090 RCF for 10 min at room temperature.
-
7.
Air-dry pelleted gDNA upside-down over a KimWipe for approximately 8–10 min at room temperature.
-
8.
Resuspended gDNA in 30 μL nuclease-free water.
-
9.
Quantify gDNA using the Invitrogen Qubit dsDNA HS assay.
3.2.1.1.2 T4-β-glucosyltransferase (T4-βGT) Reaction
-
1.
In a PCR-tube strip, combine the following reagents from the NEB T4-βGT kit:
Reagent
Amount
10× CutSmart Buffer
2 μL
UDP-Glucose (2 mM)
0.6 μL
T4-βGT (10 U/mL)
1 μL
Sheared gDNA
1 μg (as measured by Qubit)
ZYMO 5mC/or 5hmC standard
5 ng
Nuclease-free water
up to 20 μL
-
2.
In a thermocycler, incubate reaction overnight at 37 °C.
-
3.
Add 80 μL of nuclease-free water to 20 μL of reaction. Transfer total volume to 1.5 mL tube.
-
4.
Add an additional 100 μL of nuclease-free water to bring the total volume to 200 μL.
-
5.
Add KAPA Pure Beads to sample at a 1:1 ratio. In this case, add 200 μL of beads (see Note 11).
-
6.
Mix sample and beads well by flicking the tube multiple times.
-
7.
Incubate at room temperature for 10 min.
-
8.
Place samples on DynaMag magnetic rack and let beads move to the back of the tube. Usually this step takes about 10 min for the supernatant to become completely clear.
-
9.
Remove the supernatant (see Note 12).
-
10.
With the beads still on the rack, wash beads with 500 μL of 80% ethanol.
-
11.
Let wash sit on beads for 30 s and then remove.
-
12.
Repeat wash.
-
13.
Remove the wash, and let beads air-dry for 4 min.
-
14.
Remove the beads from the magnetic rack and resuspend in 30 μL of nuclease-free water to elute gDNA.
-
15.
Incubate beads at room temperature for 10 min.
-
16.
Place beads back on the magnetic rack and allow beads to move to the back of the tube.
-
17.
Carefully withdraw elution and save in 1.5 mL tube.
-
18.
Quantify β-GT treated DNA by Invitrogen Qubit dsDNA HS assay.
3.2.1.1.3 TET Oxidation Treatments
-
1.
Add components in the following order and amounts (see Notes 13–15):
Reagent
Amount
Water
Up to 50 μL
β-GT treated DNA
500 ng (as measured by Qubit)
Tet oxidation reagent #2
15 μL
Tet oxidation reagent #1
3.5 μL
TET2-CD (2.0 mg/mL)
8 μL
-
2.
Incubate samples in the dark at 37 °C for 2 h.
-
3.
Re-add Tet oxidation reagent #2, Tet oxidation reagent #1, and TET2-CD enzyme in same amounts as listed above.
-
4.
Bring final volume up to 100 μL with nuclease-free water.
-
5.
Incubate at 37 °C for 2 h in the dark.
-
6.
Add an additional 100 μL of nuclease-free water to bring the total volume to 200 μL.
-
7.
Add KAPA Pure Beads to sample at a 1:1 ratio. In this case, add 200 μL of beads.
-
8.
Repeat steps 6 through 17 from β-GT bead gDNA purification clean-up. Elute in 33 μL nuclease-free water. Save first elution in 1.5 mL microcentrifuge tube.
-
9.
Add an additional 20 μL of nuclease-free water to the KAPA Pure beads following removal of the TAB-treated gDNA elution.
-
10.
Incubate beads at room temperature for 10 min.
-
11.
Place beads back on the magnetic rack and allow beads to move to the back of the tube.
-
12.
Carefully withdraw elution and save in a different 1.5 mL microcentrifuge tube than the first elution. This elution will be used to process the 5mC/5hmC standards described below.
-
13.
Quantify TAB-treated gDNA from the first elution using Invitrogen Qubit dsDNA HS assay. TAB-treated gDNA may be stored at −20 °C for up to 2 weeks.
-
14.
Submit TAB-treated gDNA and non-treated gDNA from the same sample to a genomics core that processes EPIC arrays (see Note 16).
3.2.1.1.4 Bisulfite Sanger Sequencing of 5mC/5hmC Standards
-
1.
Using 10 μL of the second elution of the TAB-treated gDNA recovered from step 12 above, perform bisulfite conversion overnight with the ZYMO DNA EZ Methylation kit per the manufacturer’s instructions (see Note 2). Elute in 10 μL nuclease-free water.
-
2.
Set up PCR reaction mixture to amplify the 5mC/or 5hmC spike-in standard from step 1 of β-GT reaction (see Note 17):
Reagent
Amount
2× MyTaq
10 μL
Primers (5 μM F + 5 μM R)
1 μL
DNA
5 μL
Nuclease-free water
4 μL
-
3.
Amplify the 5mC/5hmC spike-in standard with the following PCR protocol:
Step 1
95 °C
30 s
Step 2
95 °C
30 s
Step 3
59 °C
45 s
Step 4
72 °C
45 s
Repeat Steps 2–4 44×.
Step 5
72 °C
5 min
Step 6
4 °C
Hold
-
4.
Run amplification products on a 1.5% agarose gel at 100 V for 30 min.
-
5.
Excise amplification product from the agarose gel and purify using NEB Monarch Gel Extraction Kit following all manufacturer’s instructions. Elute PCR product in 6 μL nuclease-free water.
-
6.
Ligate PCR product into Promega pGEM-T vector overnight at room temperature using the following reaction mixture:
Reagent
Amount
2× Rapid Ligation Buffer
5 μL
pGEM-T Vector
0.6 μL
DNA
4 μL
T4 DNA Ligase
1 μL
-
7.
Thaw NEB DH5α competent cells on ice for 10 min.
-
8.
Aliquot 50 μL of competent cells per ligation product into a new tube.
-
9.
Add 5 μL of ligation product to cells, gently flick the tube a few times, and incubate the cells on ice for 30 min.
-
10.
Heat shock cells for exactly 30 s at 42 °C.
-
11.
Place the cells on ice for 5 min.
-
12.
Add 450 μL of SOC media to the cells and incubate with shaking at 37 °C for 1 h.
-
13.
Split the cells onto two different ampicillin bacterial agar plates that have been coated with 80 μL of 80 mg/mL X-gal and spread until mostly dry.
-
14.
Incubate agar plates overnight at 37 °C.
-
15.
The next day, make a master ampicillin agar plate for each PCR product and pick at least 30 white clones to grow up individually on the plate.
-
16.
Incubate the master agar plates overnight at 37 °C.
-
17.
Perform colony PCR using the reaction conditions from steps 2 and 3 on at least 20 clones to verify successful insertion of the product.
-
18.
Using illustra TempliPhi DNA Sequencing Template Amplification Kit, prepare a 96-well plate of clones to be sequenced by adding 5 μL of Denature Buffer to each well and a small amount of a positive colony (see Note 18).
-
19.
Denature the samples at 95 °C for 3 min and let the samples cool to 4 °C.
-
20.
Add 5 μL of the Premix buffer to the cooled samples, seal the plate, and submit for Sanger sequencing.
-
21.
Analyze sequences using QUMA online software with all parameters set to account for CpH methylation [44].
3.2.2 Oxidative Bisulfite (OxBS) Method
3.2.2.1 General Procedure
-
1.
Follow all manufacturer instructions exactly. gDNA samples should be in water rather than TE buffer. gDNA input is 500 ng for both the sample that will be treated with oxidant and the sample without oxidant.
3.3 Bioinformatic Pipelines for EPIC Array Analysis
To model the utility of BS array (Fig. 1a), TAB array (Fig. 1b), and OxBS array (Fig. 1c) for detecting active and passive DNA demethylation, we treated NCCIT embryonal carcinoma cells with compounds to inhibit the DNMTs (DAC) and enhance TET activity (VitC). Notably, NCCIT cells are derived from a germ cell tumor, giving them pluripotent properties and the ability to differentiate upon treatment with retinoic acid [49]. Given these properties, NCCIT cells serve as an excellent model to study active and passive DNA demethylation, as cytosine modification patterning by both DNMTs and TETs is dynamic [35, 50]. To specifically inhibit the catalytically active DNMTs, of which all are highly expressed in NCCIT, we treated cells with 1 μM DAC for 24 h, and then refreshed cells with media lacking DAC for the remainder of the growth period (Fig. 2a). To both inhibit DNMTs and enhance TET activity, we treated cells with a 24-h pulse of 1 μM DAC and then added VitC at a physiologic concentration (57 μM) every 24 h until collection. Cells treated with PBS (NoTx) served as our control (Fig. 2a). All treatments were done in biological duplicate over 72 h, and differences in population doublings across treatments were insignificant (data not shown), indicating that all treatment groups went through DNA replication roughly an equivalent number of times. As discussed, we performed benchtop assays to determine the effectiveness of our drug treatments for 5mC loss (Fig. 2b) and induction of 5mC conversion to 5hmC (Fig. 2c, d) prior to submission on the EPIC array. All EPIC array analysis is conducted in the R statistical software environment (Version 3.6.1) (R Core Team).
3.3.1 TAB Array Processing
We validated the efficiency of TAB oxidation reactions by standard bisulfite Sanger sequencing (detailed in Subheading 3.1) of fully modified 5mC and 5hmC spike-in standards (Fig. 3a). Following validation of the reaction, BS and TAB array were completed for both biological duplicates of NoTx, DAC, and DAC + VitC samples to measure the levels of 5mC/5hmC and 5hmC, respectively. While BS array samples demonstrated a high retention rate of probes following SeSAMe processing with default settings, TAB array samples were more likely to fail array QC standards due to a high detection p-value (≥0.05). As the intensity values from the unmethylated and methylated fluorescent channels are used to determine the quality of probe detection, we hypothesized that SeSAMe was overestimating our failure rate due to the low signal from the methylated fluorescent channel [51]. In an effort to retain more probes in the TAB-treated samples that were biologically meaningful, we relaxed the detection p-value threshold to include all probes with a detection p-value ≤0.15. At this threshold, we were able to retain almost 70,000 more probes in our analysis without compromising our biological conclusions. For this analysis, we included all probes that had a detection p-value ≤0.15 across all samples queried on BS and TAB arrays (12 samples, n = 466,341 probes).

TAB array data analysis. (a) Bisulfite genomic sequencing (BGS) of bacterial 5mC (left) or 5hmC (right) DNA controls (ZYMO) spiked in with NCCIT gDNA prior to β-GT reaction. White circles represent unmodified cytosines (5C), black circles represent modified cytosines (5mC/5hmC ), and X represents mismatches. (b) Bean plots of β-value distributions for BS and TAB EPIC array CpG probes passing QC (n = 457,338 CpGs). A β-value of 0 represents fully unmodified cytosines and a β-value of 1 represents fully modified cytosines. (c) β-Value density plots for BS array (left), TAB array (middle), and calculated 5mC (right), derived by subtracting TAB array β-values from BS array β-values
DNA modifications across a sample’s population of DNA molecules are quantified on the EPIC array by the β-value in which a β-value of 1 indicates the cytosine is fully modified (5mC/5hmC ) in the population and a β-value of 0 indicates the cytosine is completely unmodified (5C) in the population. For initial sample characterization, we profiled the density distribution of β-values for cytosine modifications (BS array) and 5hmC alone (TAB array) (Fig. 3b, c). BS array analysis demonstrated a bimodal distribution of β-values for the NoTx samples in which the majority of cytosines were either fully modified or fully unmodified (Fig. 3b (top), c (left panel)). For both DAC and DAC + VitC samples, a leftward shift in β-value distributions was observed, consistent with loss of DNA modifications. The DAC + VitC samples also appeared to lose slightly more cytosine modifications relative to the DAC samples, although the difference between the median losses was not as pronounced as compared to the NoTx samples (Fig. 3b, top). Unlike BS array, TAB array samples yielded a unimodal distribution of β-values closer to 0, as the level of 5hmC in a sample population is typically very low (Fig. 3b, bottom; c, middle). While it was difficult to determine whether DAC induced increases in 5hmC distributions relative to NoTx, DAC + VitC samples demonstrated a clear leftward shift in β-value distributions (Fig. 3b, bottom; Figure 3c, middle), indicating that treatment of NCCIT cells with Vitamin C effectively enhanced TET activity and the conversion of 5mC to 5hmC.
As BS array β-values are a summation of 5mC and 5hmC signal, 5mC signal alone can be calculated by subtraction of TAB array β-values from BS array β-values from the same sample. In principle, this subtraction works well and yields 5mC β-values that are interpretable. As previously reported, this subtraction occasionally results in negative β-values, typically when the cytosine nucleotide is primarily modified by 5hmC in the population with little to no detectable 5mC [38, 52]. To account for negative β-values, a correction was applied that discarded all probes that yield a β-value for 5mC that was <−0.05. Calculated 5mC β-values that fell between −0.05 and 0 were adjusted to have a β-value of 0.001 [38]. Performing this correction on our dataset resulted in a loss of 8953 probes from our analysis. Distribution of 5mC β-values among all samples revealed that the DAC + VitC samples demonstrated a more significant leftward β-value shift than DAC and NoTx, indicating that DAC + VitC treatment induced more DNA demethylation than DAC (Fig. 3c, right panel). In Subheading 3.3.4, the calculated β-values for 5mC and TAB array β-values for 5hmC from this processing pipeline were used to determine the significance of modification changes across all samples.
3.3.1.1 General Pipeline
-
1.
Load necessary R packages for analysis.
library(sesame) library(colorRamps) library(ggplot2) library(data.table) library(dplyr) library(gplots) library(grDevices) library(reshape2) library(tidyverse) library(minfi) library(RColorBrewer) library(limma)
-
2.
Move all IDAT files for analysis to the same directory, and then set the working directory to the location of the IDAT files.
setwd("~/rothbart_secondary/Rochelle/ROTS_20191216_EPICoxBS/TAB/")
-
3.
Make a signal summary dataset for all the IDAT files and run SeSAMe to generate and normalize β-values for each sample [51]. Relax the pval.threshold to 0.15 to include more probes in the analysis. Name your samples as needed. Make sure the sample order is the same as the order of EPIC array number and position.
ssets <- lapply(searchIDATprefixes("~/rothbart_secondary/Rochelle/ROTS_20191216_EPICoxBS/TAB/"),readIDATpair) TABbetas <- openSesame(ssets, pval.threshold = 0.15) colnames(TABbetas) <- c("NoTx1_TAB","DAC1_TAB","DAC2_BS","DAC2_aa_BS","DAC1_aa_TAB", "NoTx2_TAB","DAC2_TAB", "DAC2_aa_TAB","NoTx1_BS","DAC1_BS","DAC1_aa_BS","NoTx2_BS") head(TABbetas,2) ## NoTx1_TAB DAC1_TAB DAC2_BS DAC2_aa_BS DAC1_aa_TAB NoTx2_TAB ## cg00000029 0.0899248 0.06510993 0.1414132 0.1678307 NA 0.06671769 ## cg00000103 NA NA NA NA NA NA ## DAC2_TAB DAC2_aa_TAB NoTx1_BS DAC1_BS DAC1_aa_BS NoTx2_BS ## cg00000029 0.1138816 0.07507755 0.1499086 0.148832 0.1138976 0.1693033 ## cg00000103 NA NA NA NA NA NA
-
4.
To make a bean plot of the β-value distributions as shown in Figs. 3b, 4a, and 4d, use the following command from minfi:
densityBeanPlot(TABbetas, main = "Beta Values")
-
5.
Transform the β-value data matrix into a data frame and remove all probes that do not have a β-value for all samples queried. Plot the density of β-value distributions across samples.
TABbetas_df <- data.frame(TABbetas) TABbetas_df <- TABbetas_df[complete.cases(TABbetas_df),] plot(density(TABbetas_df$NoTx1_BS), col = "#190B28", lty = 1, lwd = 2, xlim = c(0,1), ylim = c(0,6)) lines(density(TABbetas_df$NoTx2_BS), col = "#190B28", lty = 2, lwd = 2) lines(density(TABbetas_df$DAC1_BS), col = "#EF3E36", lty = 1, lwd = 2) lines(density(TABbetas_df$DAC2_BS), col = "#EF3E36", lty = 2, lwd = 2) lines(density(TABbetas_df$DAC1_aa_BS), col = "#17BEBB", lty = 1, lwd = 2) lines(density(TABbetas_df$DAC2_aa_BS), col = "#17BEBB", lty = 2, lwd = 2)
-
6.
Calculate true 5mC β-values by subtracting the TAB array β-values from the BS array β-values for each individual sample.
TABbetas_df <- mutate(TABbetas_df, NoTx1_5mC = NoTx1_BS - NoTx1_TAB) TABbetas_df <- mutate(TABbetas_df, DAC1_5mC = DAC1_BS - DAC1_TAB) TABbetas_df <- mutate(TABbetas_df, DAC1_aa_5mC = DAC1_aa_BS - DAC1_aa_TAB) TABbetas_df <- mutate(TABbetas_df, NoTx2_5mC = NoTx2_BS - NoTx2_TAB) TABbetas_df <- mutate(TABbetas_df, DAC2_5mC = DAC2_BS - DAC2_TAB) TABbetas_df <- mutate(TABbetas_df, DAC2_aa_5mC = DAC2_aa_BS - DAC2_aa_TAB)
-
7.
To correct for negative 5mC β-values, write an if-else statement such that any β-value that is less than −0.05 will be given the new value “10,” and any β-value between −0.05 and 0 will be corrected to 0.001. If the β-value does not meet either of these criteria, it will remain as it was originally calculated from the code above. Finally, remove all 5mC β-values that were transformed into “10” as they will not remain in the analysis.
TABbetas_df <- mutate(TABbetas_df, NoTx1_5mC = ifelse(NoTx1_5mC < -0.05, 10, ifelse(NoTx1_5mC < 0 & NoTx1_5mC >= -0.05, 0.001, NoTx1_5mC))) TABbetas_df <- mutate(TABbetas_df, NoTx2_5mC = ifelse(NoTx2_5mC < -0.05, 10, ifelse(NoTx2_5mC < 0 & NoTx2_5mC >= -0.05, 0.001, NoTx2_5mC))) TABbetas_df <- mutate(TABbetas_df, DAC1_5mC = ifelse(DAC1_5mC < -0.05, 10, ifelse(DAC1_5mC < 0 & DAC1_5mC >= -0.05, 0.001, DAC1_5mC))) TABbetas_df <- mutate(TABbetas_df, DAC2_5mC = ifelse(DAC2_5mC < -0.05, 10, ifelse(DAC2_5mC < 0 & DAC2_5mC >= -0.05, 0.001, DAC2_5mC))) TABbetas_df <- mutate(TABbetas_df, DAC1_aa_5mC = ifelse(DAC1_aa_5mC < -0.05, 10, ifelse(DAC1_aa_5mC < 0 & DAC1_aa_5mC >= -0.05, 0.001, DAC1_aa_5mC))) TABbetas_df <- mutate(TABbetas_df, DAC2_aa_5mC = ifelse(DAC2_aa_5mC < -0.05, 10, ifelse(DAC2_aa_5mC < 0 & DAC2_aa_5mC >= -0.05, 0.001, DAC2_aa_5mC))) TABbetas_df <- subset(TABbetas_df, NoTx1_5mC != 10 & NoTx2_5mC != 10 & DAC1_5mC != 10 & DAC2_5mC != 10 & DAC1_aa_5mC != 10 & DAC2_aa_5mC != 10)

OxBS array analysis. (a) Bean plots of β-value distributions for BS and OxBS array CpG probes passing QC (n = 563,208 CpGs). (b) β-Value density plots for BS array (solid line), OxBS array (dotted line), and calculated 5hmC values derived by subtracting OxBS array β-values from EPIC array β-values. (c) Number of CpG probes with a calculated 5hmCβ-value (BS array–OxBS array) either above or below 0. (d) Bean plots of 5mC and 5hmCβ-value distributions computed from the Bioconductor package ENmix for all CpG probes. (e) β-Value density plots of 5mC (solid line) and 5hmC (dashed line) computed from the Bioconductor package ENmix for all CpG probes. (f) Histogram of β-values for 5hmC (left) and 5mC (right) for DAC-treated samples (both with and without Vitamin C) relative to PBS-treated samples (NoTx). Negative β-values indicate loss of the modification relative to NoTx while positive β-values indicate a gain in the modification
3.3.2 OxBS Array Processing
BS and OxBS array were performed on an individual set of drug treatments (NoTx1, DAC1, DAC1 + VitC) to determine levels of 5hmC/5mC and 5mC, respectively. Distribution of β-values for NoTx and DAC treatments were similar to those observed on the BS array conducted alongside the TAB array, where a bimodal distribution was observed for highly modified and completely unmodified cytosines (Fig. 4a, top). Similar to the BS array results in Subheading 5.1, we observed a leftward β-value shift in DAC treated samples, indicating a loss of DNA modifications. Importantly, the difference in this leftward shift between DAC1 and DAC1 + VitC was minimal in the BS array β-value distribution (Fig. 4a, top). While the NoTx1 and DAC1 β-value distribution from the OxBS array were similar to the BS array pattern, the DAC1 + VitC distribution of β-values on OxBS array demonstrated a greater degree of a leftward shift relative to both NoTx1 and DAC1 than observed by BS array, suggesting that 5hmC patterning was also changing in this sample (Fig. 4a, bottom).
To determine the β-values for 5hmC in drug treatments, we performed the same calculation as described for TAB array, except subtraction of OxBS array (5mC) from BS array (5mC/5hmC ) yielded 5hmCβ-values rather than 5mC. Next, we plotted the density distributions of β-values from the BS array, OxBS array, and calculated 5hmC values. Unlike the results from TAB array, we noticed that a large fraction of calculated 5hmCβ-values fell below zero, particularly for NoTx1 and DAC1 treated samples (Fig. 4b). We quantified the number of CpG probes with a 5hmCβ-value below and above zero among all samples and determined that while NoTx1 5hmCβ-values were evenly split, more probes fell above zero in DAC1 than NoTx1. DAC1 + VitC 5hmCβ-values were almost all above zero (Fig. 4c). Taken together with the results from ELISA-based assays (Fig. 2c) and TAB array 5hmCβ-value distributions (Fig. 3b, bottom; c middle), we believe that the overall abundance of 5hmC in a sample population can predict the ability of OxBS array to quantify 5hmCβ-values via the subtraction method, a perspective that will be further discussed in Subheading 6.
To correct for the subtraction method disparity, we employed a Bioconductor package specifically designed to correct for this problem in OxBS array data, OxBS-MLE [53]. OxBS-MLE uses the paired CpG probe intensity values from the BS array and OxBS array to calculate maximum likelihood estimates of 5mC and 5hmCβ-values within a sample. OxBS-MLE correction produced β-value distributions (Fig. 4d, e) for 5mC (top) and 5hmC (bottom) that closely resembled results obtained by BS array and TAB array (Fig. 3b, c), where DAC + VitC samples demonstrated the greatest loss of 5mC and the greatest increase in 5hmC relative to both NoTx and DAC. Finally, we calculated β-values for 5mC and 5hmC of DAC and DAC + VitC relative to the NoTx sample to quantify changes in 5mC and 5hmC (Fig. 4f). As we only performed OxBS array on a single drug treatment set, we used β-values to determine significance of these changes in Subheading 5.3.
3.3.2.1 General Pipeline
-
1.
Load necessary R packages for analysis.
library(sesame) library(colorRamps) library(ggplot2) library(data.table) library(dplyr) library(gplots) library(grDevices) library(reshape2) library(tidyverse) library(minfi) library(RColorBrewer) library(ENmix)
-
2.
Move all IDAT files for analysis to the same directory, and then set the working directory to the location of the IDAT files.
setwd("~/rothbart_secondary/Rochelle/ROTS_20191216_EPICoxBS /OxBS /")
-
3.
Make a signal summary dataset for all the IDAT files and run SeSAMe to generate and normalize β-values for each sample. Name your samples as needed. Make sure the sample order is the same as the order of EPIC array number and position.
ssets <- lapply(searchIDATprefixes("~/rothbart_secondary/Rochelle/ROTS_20191216_EPICoxBS /OxBS /"),readIDATpair) OxBSbetas <- openSesame(ssets) colnames(OxBSbetas) <- c("NoTx1_BS","DAC1_BS","DAC1_aa_BS","NoTx1_Ox","DAC1_Ox", "DAC1_aa_Ox") head(OxBSbetas,2) ## NoTx1_BS DAC1_BS DAC1_aa_BS NoTx1_Ox DAC1_Ox DAC1_aa_Ox ## cg00000029 NA NA 0.1144161 NA NA NA ## cg00000103 NA NA NA NA NA NA
-
4.
Transform the β-value data matrix into a data frame and remove all probes that do not have a β-value for all samples queried.
OxBSbetas_df <- data.frame(OxBSbetas) OxBSbetas_df <- OxBSbetas_df[complete.cases(OxBSbetas_df),]
-
5.
Calculate true 5hmCβ-values by subtracting the OxBS array β-values from the BS array β-values for each individual sample.
OxBSbetas_df <- mutate(OxBSbetas_df, NoTx_5hmC = NoTx1_BS - NoTx1_Ox) OxBSbetas_df <- mutate(OxBSbetas_df, DAC1_5hmC = DAC1_BS - DAC1_Ox) OxBSbetas_df <- mutate(OxBSbetas_df, DAC1_aa_5hmC = DAC1_aa_BS - DAC1_aa_Ox)
-
6.
Determine the number of CpG probes with a 5hmCβ-values above and below 0.
NoTx_5hmC _above <- subset(OxBSbetas_df, NoTx_5hmC> 0) #298901 CpGs NoTx_5hmC _below <- subset(OxBSbetas_df, NoTx_5hmC< 0) #264307 CpGs DAC1_5hmC _above <- subset(OxBSbetas_df, DAC1_5hmC> 0) #365646 CpGs DAC1_5hmC _below <- subset(OxBSbetas_df, DAC1_5hmC< 0) #197562 CpGs DAC1_aa_5hmC _above <- subset(OxBSbetas_df, DAC1_aa_5hmC> 0) #484142 CpGs DAC1_aa_5hmC _below <- subset(OxBSbetas_df, DAC1_aa_5hmC< 0) #76066 CpGs
-
7.
To correct for the number of 5hmCβ-values below 0, use the OxBS-MLE command from ENmix [53]. First, isolate the β-values for BS array and then isolate the β-values for OxBS array.
colnames(OxBSbetas) <- c("NoTx","DAC","DAC.aa","NoTx","DAC","DAC.aa") beta.BS <- OxBSbetas[,c(1:3)] beta.oxBS <- OxBSbetas[,c(4:6)]
-
8.
Next, isolate the intensity values independently for both BS array and the OxBS array. A critical note is that all samples must remain in the same order and be named the same thing between BS array and OxBS array.
NoTx <- totalIntensities(ssets$`203855160107_R03C01`) DAC <- totalIntensities(ssets$`203855160107_R04C01`) DAC.aa <- totalIntensities(ssets$`203855160107_R05C01`) N.BS <- cbind(NoTx,DAC, DAC.aa) N.BS <- N.BS[order(row.names(N.BS)),] NoTx <- totalIntensities(ssets$`203855160107_R06C01`) DAC <- totalIntensities(ssets$`203855160107_R07C01`) DAC.aa <- totalIntensities(ssets$`203855160107_R08C01`) N.oxBS <- cbind(NoTx,DAC, DAC.aa) N.oxBS <- N.oxBS [order(row.names(N.oxBS )),]
-
9.
Using the isolated β-values and intensity values from above, run OxBS-MLE to recalculate 5mC and 5hmCβ-values.
OxBS .EN <- oxBS .MLE(beta.BS, beta.oxBS, N.BS, N.oxBS) OxBS .df <- data.frame(OxBS .EN) OxBS .df <- OxBS .df[complete.cases(OxBS .df),] OxBS .df <- cbind(rownames(OxBS .df), data.frame(OxBS .df), row.names = NULL) colnames(OxBS .df) <- c("probeID","NoTx.5mC","DAC.5mC","DAC.aa.5mC","NoTx.5hmC ","DAC.5hmC ","DAC.aa.5hmC ")
-
10.
Calculate β-values for each drug treatment relative to each other for both 5mC and 5hmC.
OxBS .df <- mutate(OxBS .df, DAC.5hmC .db = DAC.5hmC - NoTx.5hmC ) OxBS .df <- mutate(OxBS .df, DACaa.5hmC .db = DAC.aa.5hmC - NoTx.5hmC ) OxBS .df <- mutate(OxBS .df, DAC.5mC.db = DAC.5mC - NoTx.5mC) OxBS .df <- mutate(OxBS .df, DACaa.5mC.db = DAC.aa.5mC - NoTx.5mC) OxBS .df <- mutate(OxBS .df, DACaa.5mC.DAC.db = DAC.aa.5mC - DAC.5mC) OxBS .df <- mutate(OxBS .df, DACaa.5hmC .DAC.db = DAC.aa.5hmC - DAC.5hmC )
3.3.3 Comparison of TAB and OxBS Array Results
To directly compare results derived from TAB array and OxBS array among samples, we merged the calculated 5mC and 5hmCβ-values from each analysis for probes that maintained high QC standards between both arrays (n = 448,954 CpGs). Multi-dimensional scaling (MDS) analysis among all probes revealed that samples clustered based on drug treatments and cytosine modification rather than by platform (TAB versus OxBS), indicating that our results between the approaches were consistent (Fig. 5a). Next, we directly compared β-values of 5mC and 5hmC for samples that were the same between the two arrays: NoTx1, DAC1, and DAC1 + VitC (Fig. 5b). Overall, 5mC β-values were consistent between TAB and OxBS array with Pearson correlation coefficients above 0.9 (Fig. 5b, top). While 5hmCβ-values were not as consistent as 5mC, we noted that DAC1 + VitC, the sample with known higher amounts of 5hmC compared to NoTx1 and DAC1, yielded the highest Pearson correlation coefficient (R = 0.365) (Fig. 5b, bottom), suggesting that when 5hmC is abundant, both platforms may more consistently capture this distribution. We believe that the lack of strong correlation between TAB and OxBS5hmCβ-values is due to the low abundance of this cytosine modification and the difference in how the β-values are determined in TAB (directly) versus OxBS array (indirectly).

Comparison of TAB array and OxBS array results for determining drug-induced passive vs. active DNA demethylation. (a) Multidimensional scaling (MDS) of β-values among all the shared CpG probes (n = 448,954 CpGs) from TAB array (square) and OxBS array (circle) analysis. 5mC and 5hmC labeling denotes the cytosine modification that is clustered. (b) β-Value density scatterplots for 5mC (top) and 5hmC (bottom) as derived from OxBS array (x-axis) and TAB array (y-axis) for the same biological sample. R = Pearson correlation coefficient between OxBS and TAB array β-values. (c) Number of significantly differentially modified cytosines (both increase and decrease in modification) for TAB array (left) and OxBS array (right). Differentially modified CpGs for TAB array were determined by limma significance testing among biological duplicates (adjusted p-value ≤ 0.01, log2 fold-change ≥ 1). Differentially modified CpGs for OxBS array were determined by applying β-value cut-offs among a single biological replicate (5mC: |β-value| ≥ 0.2; 5hmC: |β-value| ≥ 0.1). (d) Percentage of CpG probes from TAB array (left) and OxBS array (right) that meet criteria for specific classification of cytosine modification behavior (5hmC and 5mC). +/− = significant increase/decrease in modification as described in c; 0 = no significant change
3.3.3.1 General Pipeline
-
1.
Merge the β-value results from both TAB array and OxBS array at individual CpG loci using the following command and merge by “probeID” (common identifier between both datasets):
TAB.Ox <- merge(x = TABbetas_df, y = OxBS .df, by = "probeID")
-
2.
Perform multidimensional scaling (MDS) analysis on all sample β-values using the following command from minfi to determine variance and relative separation among samples:
TAB.Ox.mds <- TAB.Ox[,c(2:25)] TAB.Ox.mds <- data.matrix(TAB.Ox.mds) plotMDS(TAB.Ox.mds, top = 500000, gene.selection = "common")
-
3.
Perform Pearson correlation between all sample β-values and visualize the correlation using the following commands:
correlation.table <- TAB.Ox[,c(2:25)] res2 <- cor(correlation.table, method = c("pearson")) head(res2) ## NoTx1_TAB DAC1_TAB DAC2_BS DAC2_aa_BS DAC1_aa_TAB NoTx2_TAB ## NoTx1_TAB 1.0000000 0.5643396 0.3490599 0.3351804 0.4845934 0.7459988 ## DAC1_TAB 0.5643396 1.0000000 0.3358964 0.3162146 0.4842038 0.5913501 ## DAC2_BS 0.3490599 0.3358964 1.0000000 0.9899646 0.4206427 0.3245357 ## DAC2_aa_BS 0.3351804 0.3162146 0.9899646 1.0000000 0.3919641 0.3077355 ## DAC1_aa_TAB 0.4845934 0.4842038 0.4206427 0.3919641 1.0000000 0.5281145 ## NoTx2_TAB 0.7459988 0.5913501 0.3245357 0.3077355 0.5281145 1.0000000 ## DAC2_TAB DAC2_aa_TAB NoTx1_BS DAC1_BS DAC1_aa_BS NoTx2_BS ## NoTx1_TAB 0.6304929 0.5435206 0.3126113 0.3471773 0.3327407 0.3062050 ## DAC1_TAB 0.5972861 0.4992690 0.3139309 0.3342330 0.3134234 0.3098587 ## DAC2_BS 0.3495153 0.4635381 0.9814008 0.9959075 0.9897792 0.9803097 ## DAC2_aa_BS 0.3286710 0.4296738 0.9705574 0.9901041 0.9956422 0.9696005 ## DAC1_aa_TAB 0.5281480 0.6878447 0.4090525 0.4181847 0.3881634 0.4049876 ## NoTx2_TAB 0.6858835 0.6020530 0.2822104 0.3220271 0.3049796 0.2749049 ## NoTx1_5mC DAC1_5mC DAC1_aa_5mC NoTx2_5mC DAC2_5mC DAC2_aa_5mC ## NoTx1_TAB 0.1979439 0.2625503 0.2047185 0.2322715 0.2511905 0.1988773 ## DAC1_TAB 0.2535767 0.1724687 0.1847842 0.2525685 0.2437360 0.1920930 ## DAC2_BS 0.9692103 0.9813832 0.9343618 0.9689641 0.9853086 0.9389595 ## DAC2_aa_BS 0.9597779 0.9788413 0.9499092 0.9600163 0.9786455 0.9603676 ## DAC1_aa_TAB 0.3616385 0.3511735 0.1005694 0.3569280 0.3449820 0.2161602 ## NoTx2_TAB 0.1980618 0.2311349 0.1603694 0.1727235 0.2151134 0.1506239 ## NoTx.5mC DAC.5mC DAC.aa.5mC NoTx.5hmC DAC.5hmC DAC.aa.5hmC ## NoTx1_TAB 0.2951723 0.3191521 0.2837059 0.1247655159 0.1550317 0.1778982 ## DAC1_TAB 0.3062280 0.3267851 0.2832649 0.0783602799 0.1163134 0.1519621 ## DAC2_BS 0.9741743 0.9853362 0.9331337 0.0003636201 0.1795046 0.3208518 ## DAC2_aa_BS 0.9642960 0.9841005 0.9522282 -0.0162461095 0.1416026 0.2771144 ## DAC1_aa_TAB 0.4017782 0.3994346 0.2941971 0.0639621609 0.1772982 0.3645255 ## NoTx2_TAB 0.2606820 0.2832240 0.2437400 0.1614722777 0.1898653 0.1997464 smoothScatter(TAB.Ox$DAC1_aa_TAB~TAB.Ox$DAC.aa.5hmC , nbin = 2000, bandwidth = 0.00001, colramp = colorRampPalette(c(blue2red(12))), nrpoints = 100, xlim = c(-0.02,1), ylim = c(0,1), xlab = "", ylab = "", axes = TRUE, frame.plot = FALSE)
3.3.4 Determining Active Versus Passive DNA Demethylation Using TAB and OxBS Arrays
For each individual platform, we next determined which cytosines were significantly differentially modified for 5mC and 5hmC relative to the other drug treatment samples. For TAB array analysis, we assayed both biological duplicates of each drug treatment, which allowed us to conduct significance testing for each cytosine modification using limma [54, 55]. We considered a CpG as differentially modified if the adjusted p-value ≤0.01 and the log2 fold-change ≥1.0. As would be expected from global analysis, DAC + VitC exhibited gains in 5hmC relative to both NoTx and DAC (Fig. 5c, left). Both DAC and DAC + VitC drug treatments also demonstrated significant loss of 5mC relative to NoTx, and DAC + VitC additionally had a number of CpGs that significantly lost 5mC relative to DAC (Fig. 5c). We performed OxBS arrays on a single set of drug treatments, so rather than conduct significance testing across biological duplicates, we calculated β-values among the samples and set the following thresholds for determining differential modifications: 5hmC |β-value| ≥0.1; 5mC |β-value| ≥0.2. Consistent with our comparison of TAB array and OxBS array β-values, the pattern of differentially modified cytosines, as queried by OxBS array, was almost identical to that of TAB array (Fig. 5c). Notably, OxBS array differential analysis did call more probes significant using our set criteria; however, this was most likely due to our inability to call statistical significance, as we only submitted one of the drug treatment sets for OxBS array analysis, indicating the importance of querying biological replicates when possible.
Our ultimate goal for TAB array and OxBS array analysis was to distinguish the degree of active versus passive DNA demethylation in drug treatments. To do this, we classified the collective behavior of cytosine nucleotides, both 5mC and 5hmC, for each individual platform. Using the criteria for determining differential modifications as discussed for TAB array and OxBS array analysis (Fig. 5c), we classified a CpG’s collective behavior by asking how 5hmC changed in one drug treatment group relative to another, and then asking how 5mC changed as well. If the criteria was not met for determining differential modifications, we classified this as “no change” in the modification. For example, if 5hmC at an individual CpG increased (“+”) in DAC + VitC relative to NoTx, and 5mC at the same CpG decreased (“−”), we considered this a CpG that was susceptible to active DNA demethylation (Fig. 5d, middle circles, red). However, if 5hmC did not have a significant change (“0”), but 5mC decreased (“−”), then we would consider this passive loss of DNA methylation (Fig. 5d, blue). No change in 5mC or 5hmC at a CpG locus is represented by dark green. All classifications that had a measurable number of probes that behaved in the given manner are shown in the legend for Fig. 5d. Overall, for both TAB array and OxBS array analysis, DAC treatment compared to NoTx demonstrated predominately passive DNA demethylation (Fig. 5d, top, blue). Addition of VitC to DAC treatments (DAC + VitC) successfully induced active DNA demethylation in addition to passive DNA demethylation relative to NoTx as queried by both platforms (Fig. 5d, middle, red/blue). Finally, by comparing DAC + VitC to DAC, we observed that while passive DNA demethylation is largely conserved with DAC treatments (highlighted by the increase of “no change” in dark green), the primary difference between DAC + VitC and DAC is the induction of 5mC conversion to 5hmC and an increase in active DNA demethylation with the addition of VitC (Fig. 5d, bottom see Note 19).
3.3.4.1 TAB Array Pipeline
-
1.
For statistical testing, we use the standard workflow within limma to compare sample groups [54,55,56,57]. First, transform β-values to M-values, and transform the data frame into a data matrix.
myMs <- logit2(TABbetas_df) myMs <- data.matrix(myMs)
-
2.
Next, set up a design matrix that places each sample into its corresponding treatment group and modification group. For this analysis, treat 5mC and 5hmC values as separate groups.
design <- model.matrix(~0+factor(c(1–9))) colnames(design) <-c("NoTx_TAB","DAC_TAB","DAC_aa_TAB","NoTx_BS","DAC_BS","DAC_aa_BS","NoTx_5mC","DAC_5mC","DAC_aa_5mC")
-
3.
Construct a contrast matrix for the samples to be compared, and then proceed with the standard limma workflow to calculate the statistical significance. For simplicity of comparisons and to get individual statistics for each comparison, make each contrast matrix individually and combine all statistical data at the end.
fit <- lmFit(myMs, design) #Bisulfite comparisons #DAC vs NoTx contrast.matrix <- makeContrasts(DAC_BS-NoTx_BS, levels = design) fit3 <- contrasts.fit(fit, contrast.matrix) fit3 <- eBayes(fit3) DAC.NoTx.BS <- topTable(fit3, num = Inf) DAC.NoTx.BS <- cbind(rownames(DAC.NoTx.BS), data.frame(DAC.NoTx.BS), row.names = NULL) colnames(DAC.NoTx.BS) <- c("probeID","logFC.DACvNoTx.BS","AveExpr.DACvNoTx.BS","t.DACvNoTx.BS","P.Val.DACvNoTx.BS","adj.P.Val.DACvNoTx.BS","B.DACvNoTx.BS") DAC.NoTx.BS <- DAC.NoTx.BS[,c(1, 2, 6)] #DAC_aa vs NoTx contrast.matrix <- makeContrasts(DAC_aa_BS-NoTx_BS, levels = design) fit4 <- contrasts.fit(fit, contrast.matrix) fit4 <- eBayes(fit4) DACaa.NoTx.BS <- topTable(fit4, num = Inf) DACaa.NoTx.BS <- cbind(rownames(DACaa.NoTx.BS), data.frame(DACaa.NoTx.BS), row.names = NULL) colnames(DACaa.NoTx.BS) <- c("probeID","logFC.DACaavNoTx.BS","AveExpr.DACaavNoTx.BS","t.DACaavNoTx.BS","P.Val.DACaavNoTx.BS","adj.P.Val.DACaavNoTx.BS","B.DACaavNoTx.BS") DACaa.NoTx.BS <- DACaa.NoTx.BS[,c(1, 2, 6)] #DAC_aa vs DAC contrast.matrix <- makeContrasts(DAC_aa_BS-DAC_BS, levels = design) fit5 <- contrasts.fit(fit, contrast.matrix) fit5 <- eBayes(fit5) DACaa.DAC.BS <- topTable(fit5, num = Inf) DACaa.DAC.BS <- cbind(rownames(DACaa.DAC.BS), data.frame(DACaa.DAC.BS), row.names = NULL) colnames(DACaa.DAC.BS) <- c("probeID","logFC.DACaa.DAC.BS","AveExpr.DACaa.DAC.BS","t.DACaa.DAC.BS","P.Val.DACaa.DAC.BS","adj.P.Val.DACaa.DAC.BS","B.DACaa.DAC.BS") DACaa.DAC.BS <- DACaa.DAC.BS[,c(1, 2, 6)] #TAB comparisons #DAC vs NoTx contrast.matrix <- makeContrasts(DAC_TAB-NoTx_TAB, levels = design) fit6 <- contrasts.fit(fit, contrast.matrix) fit6 <- eBayes(fit6) DAC.NoTx.TAB <- topTable(fit6, num = Inf) DAC.NoTx.TAB <- cbind(rownames(DAC.NoTx.TAB), data.frame(DAC.NoTx.TAB), row.names = NULL) colnames(DAC.NoTx.TAB) <- c("probeID","logFC.DACvNoTx.TAB","AveExpr.DACvNoTx.TAB","t.DACvNoTx.TAB","P.Val.DACvNoTx.TAB","adj.P.Val.DACvNoTx.TAB","B.DACvNoTx.TAB") DAC.NoTx.TAB <- DAC.NoTx.TAB[,c(1, 2, 6)] #DAC_aa vs NoTx contrast.matrix <- makeContrasts(DAC_aa_TAB-NoTx_TAB, levels = design) fit7 <- contrasts.fit(fit, contrast.matrix) fit7 <- eBayes(fit7) DACaa.NoTx.TAB <- topTable(fit7, num = Inf) DACaa.NoTx.TAB <- cbind(rownames(DACaa.NoTx.TAB), data.frame(DACaa.NoTx.TAB), row.names = NULL) colnames(DACaa.NoTx.TAB) <- c("probeID","logFC.DACaavNoTx.TAB","AveExpr.DACaavNoTx.TAB","t.DACaavNoTx.TAB","P.Val.DACaavNoTx.TAB","adj.P.Val.DACaavNoTx.TAB","B.DACaavNoTx.TAB") DACaa.NoTx.TAB <- DACaa.NoTx.TAB[,c(1, 2, 6)] #DAC_aa vs DAC contrast.matrix <- makeContrasts(DAC_aa_TAB-DAC_TAB, levels = design) fit8 <- contrasts.fit(fit, contrast.matrix) fit8 <- eBayes(fit8) DACaa.DAC.TAB <- topTable(fit8, num = Inf) DACaa.DAC.TAB <- cbind(rownames(DACaa.DAC.TAB), data.frame(DACaa.DAC.TAB), row.names = NULL) colnames(DACaa.DAC.TAB) <- c("probeID","logFC.DACaa.DAC.TAB","AveExpr.DACaa.DAC.TAB","t.DACaa.DAC.TAB","P.Val.DACaa.DAC.TAB","adj.P.Val.DACaa.DAC.TAB","B.DACaa.DAC.TAB") DACaa.DAC.TAB <- DACaa.DAC.TAB[,c(1, 2, 6)] #True 5mC comparisons #DAC vs NoTx contrast.matrix <- makeContrasts(DAC_5mC-NoTx_5mC, levels = design) fit9 <- contrasts.fit(fit, contrast.matrix) fit9 <- eBayes(fit9) DAC.NoTx.5mC <- topTable(fit9, num = Inf) DAC.NoTx.5mC <- cbind(rownames(DAC.NoTx.5mC), data.frame(DAC.NoTx.5mC), row.names = NULL) colnames(DAC.NoTx.5mC) <- c("probeID","logFC.DACvNoTx.5mC","AveExpr.DACvNoTx.5mC","t.DACvNoTx.5mC","P.Val.DACvNoTx.5mC","adj.P.Val.DACvNoTx.5mC","B.DACvNoTx.5mC") DAC.NoTx.5mC <- DAC.NoTx.5mC[,c(1, 2, 6)] #DAC_aa vs NoTx contrast.matrix <- makeContrasts(DAC_aa_5mC-NoTx_5mC, levels = design) fit10 <- contrasts.fit(fit, contrast.matrix) fit10 <- eBayes(fit10) DACaa.NoTx.5mC <- topTable(fit10, num = Inf) DACaa.NoTx.5mC <- cbind(rownames(DACaa.NoTx.5mC), data.frame(DACaa.NoTx.5mC), row.names = NULL) colnames(DACaa.NoTx.5mC) <- c("probeID","logFC.DACaavNoTx.5mC","AveExpr.DACaavNoTx.5mC","t.DACaavNoTx.5mC","P.Val.DACaavNoTx.5mC","adj.P.Val.DACaavNoTx.5mC","B.DACaavNoTx.5mC") DACaa.NoTx.5mC <- DACaa.NoTx.5mC[,c(1, 2, 6)] #DAC_aa vs DAC contrast.matrix <- makeContrasts(DAC_aa_5mC-DAC_5mC, levels = design) fit11 <- contrasts.fit(fit, contrast.matrix) fit11 <- eBayes(fit11) DACaa.DAC.5mC <- topTable(fit11, num = Inf) DACaa.DAC.5mC <- cbind(rownames(DACaa.DAC.5mC), data.frame(DACaa.DAC.5mC), row.names = NULL) colnames(DACaa.DAC.5mC) <- c("probeID","logFC.DACaa.DAC.5mC","AveExpr.DACaa.DAC.5mC","t.DACaa.DAC.5mC","P.Val.DACaa.DAC.5mC","adj.P.Val.DACaa.DAC.5mC","B.DACaa.DAC.5mC") DACaa.DAC.5mC <- DACaa.DAC.5mC[,c(1, 2, 6)] NCCIT.stats <- merge(x = DAC.NoTx.BS, y = DACaa.NoTx.BS, by = "probeID") NCCIT.stats <- merge(x = NCCIT.stats, y = DACaa.DAC.BS, by = "probeID") NCCIT.stats <- merge(x = NCCIT.stats, y = DAC.NoTx.TAB, by = "probeID") NCCIT.stats <- merge(x = NCCIT.stats, y = DACaa.NoTx.TAB, by = "probeID") NCCIT.stats <- merge(x = NCCIT.stats, y = DACaa.DAC.TAB, by = "probeID") NCCIT.stats <- merge(x = NCCIT.stats, y = DAC.NoTx.5mC, by = "probeID") NCCIT.stats <- merge(x = NCCIT.stats, y = DACaa.NoTx.5mC, by = "probeID") NCCIT.stats <- merge(x = NCCIT.stats, y = DACaa.DAC.5mC, by = "probeID") head(NCCIT.stats, 2) ## probeID logFC.DACvNoTx.BS adj.P.Val.DACvNoTx.BS logFC.DACaavNoTx.BS ## 1 1 -2.3111577 9.042276e-06 -2.3341208 ## 2 10 0.9194189 6.677093e-01 0.4857809 ## adj.P.Val.DACaavNoTx.BS logFC.DACaa.DAC.BS adj.P.Val.DACaa.DAC.BS ## 1 5.231595e-06 -0.02296312 0.9999978 ## 2 8.419191e-01 -0.43363805 0.9999978 ## logFC.DACvNoTx.TAB adj.P.Val.DACvNoTx.TAB logFC.DACaavNoTx.TAB ## 1 0.02229206 0.999997 0.31428085 ## 2 -0.49531260 0.999997 0.02696331 ## adj.P.Val.DACaavNoTx.TAB logFC.DACaa.DAC.TAB adj.P.Val.DACaa.DAC.TAB ## 1 0.4024360 0.2919888 0.4181687 ## 2 0.9999993 0.5222759 0.9046001 ## logFC.DACvNoTx.5mC adj.P.Val.DACvNoTx.5mC logFC.DACaavNoTx.5mC ## 1 -1.708069 8.033274e-05 -1.82091491 ## 2 3.187525 5.854615e-02 0.05450381 ## adj.P.Val.DACaavNoTx.5mC logFC.DACaa.DAC.5mC adj.P.Val.DACaa.DAC.5mC ## 1 2.495906e-05 -0.1128459 0.7522229 ## 2 9.772170e-01 -3.1330210 0.1663701
-
4.
Combine the statistics for differential methylation with the calculated β-values.
TABbetas_df <- cbind(rownames(TABbetas_df), data.frame(TABbetas_df), row.names = NULL) colnames(TABbetas_df) <- c("probeID","NoTx1_TAB","DAC1_TAB","DAC2_BS","DAC2_aa_BS","DAC1_aa_TAB","NoTx2_TAB","DAC2_TAB","DAC2_aa_TAB", "NoTx1_BS","DAC1_BS","DAC1_aa_BS","NoTx2_BS","NoTx1_5mC","DAC1_5mC","DAC1_aa_5mC","NoTx2_5mC","DAC2_5mC","DAC2_aa_5mC") NCCIT.final <- merge(x = TABbetas_df, y = NCCIT.stats, by = "probeID")
-
5.
Using adjusted p-values ≤0.01 and LogFC ≥1, define the direction of the change for each modification or note if the change is not significant.
NCCIT.final <- mutate(NCCIT.final, DACvsTx_5mC_direction = ifelse(adj.P.Val.DACvNoTx.5mC <= 0.01 & logFC.DACvNoTx.5mC >= 1, "Up", ifelse(adj.P.Val.DACvNoTx.5mC <= 0.01 & logFC.DACvNoTx.5mC <= -1, "Down", "NotSig"))) NCCIT.final <- mutate(NCCIT.final, DACaavsTx_5mC_direction = ifelse(adj.P.Val.DACaavNoTx.5mC <= 0.01 & logFC.DACaavNoTx.5mC >= 1, "Up", ifelse(adj.P.Val.DACaavNoTx.5mC <= 0.01 & logFC.DACaavNoTx.5mC <=- 1, "Down", "NotSig"))) NCCIT.final <- mutate(NCCIT.final, DACvsTx_5hmC _direction = ifelse(adj.P.Val.DACvNoTx.TAB <= 0.01 & logFC.DACvNoTx.TAB >= 1, "Up", ifelse(adj.P.Val.DACvNoTx.TAB <= 0.01 & logFC.DACvNoTx.TAB <= -1, "Down", "NotSig"))) NCCIT.final <- mutate(NCCIT.final, DACaavsTx_5hmC _direction = ifelse(adj.P.Val.DACaavNoTx.TAB <= 0.01 & logFC.DACaavNoTx.TAB >= 1, "Up", ifelse(adj.P.Val.DACaavNoTx.TAB <= 0.01 & logFC.DACaavNoTx.TAB <=- 1, "Down", "NotSig"))) NCCIT.final <- mutate(NCCIT.final, DACaavsDAC_5mC_direction = ifelse(adj.P.Val.DACaa.DAC.5mC <= 0.01 & logFC.DACaa.DAC.5mC >= 1, "Up", ifelse(adj.P.Val.DACaa.DAC.5mC <= 0.01 & logFC.DACaa.DAC.5mC <= -1, "Down", "NotSig"))) NCCIT.final <- mutate(NCCIT.final, DACaavsDAC_5hmC _direction = ifelse(adj.P.Val.DACaa.DAC.TAB <= 0.01 & logFC.DACaa.DAC.TAB >= 1, "Up", ifelse(adj.P.Val.DACaa.DAC.TAB <= 0.01 & logFC.DACaa.DAC.TAB <=- 1, "Down", "NotSig"))) table(NCCIT.final$DACaavsTx_5hmC _direction) ## ## Down NotSig Up ## 848 410813 45727
-
6.
Using if-else statements, define the collective behavior of 5hmC and 5mC for each individual probe using the significance criteria defined above. Only DAC + VitC relative to NoTx is shown as an example.
NCCIT.final <- mutate(NCCIT.final, DACaavsTx.states = ifelse(DACaavsTx_5hmC _direction == "Up" & DACaavsTx_5mC_direction == "NotSig", "State1", ifelse(DACaavsTx_5hmC _direction == "NotSig" & DACaavsTx_5mC_direction == "Up", "State2", ifelse(DACaavsTx_5hmC _direction == "Up" & DACaavsTx_5mC_direction == "Up","State3", ifelse(DACaavsTx_5hmC _direction == "Up" & DACaavsTx_5mC_direction == "Down","State4", ifelse(DACaavsTx_5hmC _direction == "Down" & DACaavsTx_5mC_direction == "Up", "State5", ifelse(DACaavsTx_5hmC _direction == "Down" & DACaavsTx_5mC_direction == "Down","State6", ifelse(DACaavsTx_5hmC _direction == "NotSig" & DACaavsTx_5mC_direction == "Down","State7", ifelse(DACaavsTx_5hmC _direction == "Down" & DACaavsTx_5mC_direction == "NotSig", "State8", ifelse(DACaavsTx_5hmC _direction == "NotSig" & DACaavsTx_5mC_direction == "NotSig", "State9", "else")))))))))) table(NCCIT.final$DACaavsTx.states) ## ## State1 State2 State4 State5 State6 State7 State8 State9 ## 622 22869 45105 7 431 217602 410 170342
3.3.4.2 OxBS Array Pipeline
-
1.
Using the calculated β-values, define the direction of change for each modification or note if the change is not significant using the following criteria:
5hmC
|β-value| ≥0.1
5mC
|β-value| ≥0.2
OxBS .df <- mutate(OxBS .df, DACvsTx_5mC_direction = ifelse(DAC.5mC.db >= 0.2, "Up", ifelse(DAC.5mC.db <= -0.2, "Down", "NotSig"))) OxBS .df <- mutate(OxBS .df, DACvsTx_5hmC _direction = ifelse(DAC.5hmC .db >= 0.1, "Up", ifelse(DAC.5hmC .db <= -0.1, "Down", "NotSig"))) OxBS .df <- mutate(OxBS .df, DACaavsTx_5mC_direction = ifelse(DACaa.5mC.db >= 0.2, "Up", ifelse(DACaa.5mC.db <= -0.2, "Down", "NotSig"))) OxBS .df <- mutate(OxBS .df, DACaavsTx_5hmC _direction = ifelse(DACaa.5hmC .db >= 0.1, "Up", ifelse(DACaa.5hmC .db <= -0.1, "Down", "NotSig"))) OxBS .df <- mutate(OxBS .df, DACaavsDAC_5mC_direction = ifelse(DACaa.5mC.DAC.db >= 0.2, "Up", ifelse(DACaa.5mC.DAC.db <= -0.2, "Down", "NotSig"))) OxBS .df <- mutate(OxBS .df, DACaavsDAC_5hmC _direction = ifelse(DACaa.5hmC .DAC.db >= 0.1, "Up", ifelse(DACaa.5hmC .DAC.db <= -0.1, "Down", "NotSig"))) table(OxBS .df$DACaavsTx_5hmC _direction) ## ## Down NotSig Up ## 1614 453418 108176
-
2.
Using if-else statements, define the collective behavior of 5hmC and 5mC for each individual probe using the significance criteria defined above. Only DAC + VitC relative to NoTx is shown as an example.
OxBS .df <- mutate(OxBS .df, DACaavsTx.states = ifelse(DACaavsTx_5hmC _direction == "Up" & DACaavsTx_5mC_direction == "NotSig", "State1", ifelse(DACaavsTx_5hmC _direction == "NotSig" & DACaavsTx_5mC_direction == "Up", "State2", ifelse(DACaavsTx_5hmC _direction == "Up" & DACaavsTx_5mC_direction == "Up","State3", ifelse(DACaavsTx_5hmC _direction == "Up" & DACaavsTx_5mC_direction == "Down","State4", ifelse(DACaavsTx_5hmC _direction == "Down" & DACaavsTx_5mC_direction == "Up", "State5", ifelse(DACaavsTx_5hmC _direction == "Down" & DACaavsTx_5mC_direction == "Down","State6", ifelse(DACaavsTx_5hmC _direction == "NotSig" & DACaavsTx_5mC_direction == "Down","State7", ifelse(DACaavsTx_5hmC _direction == "Down" & DACaavsTx_5mC_direction == "NotSig", "State8", ifelse(DACaavsTx_5hmC _direction == "NotSig" & DACaavsTx_5mC_direction == "NotSig", "State9", "else")))))))))) table(OxBS .df$DACaavsTx.states) ## ## State1 State4 State6 State7 State8 State9 ## 3359 104817 100 221062 1514 232356
4 Notes
-
1.
500 ng will allow for amplification of 4–5 genomic loci in technical duplicate.
-
2.
Add an additional centrifugation step following the last ethanol wash to remove any excess ethanol left in the column. Excess ethanol will adversely affect the results of this analysis.
-
3.
Bisulfite-converted gDNA should be used for HRM analysis immediately after completion of bisulfite conversion, as DNA will begin to degrade and results will be adversely affected.
-
4.
gDNA isolated from HCT116 DKO1 cells (genetic hypomorph of DNMT1 and genetic knockout of DNMT3B alleles) serves as a good positive control for lack of cytosine modifications at all genomic loci.
-
5.
Make sure to mix all gDNA samples extremely well before applying to the assay plate, as comparisons among samples are dependent on the amount of gDNA loaded.
-
6.
Measure remaining gDNA by NanoDrop to ensure accurate concentrations and amount loaded into the assay wells.
-
7.
We determined that the calculation using polynomial second order regression fit our standard curve best for our analysis of %5hmC (data not shown).
-
8.
Even loading of gDNA across samples is crucial. To achieve this, use the average of at least two Nanodrop readings and thoroughly mix samples and wells. In addition, it is helpful to move quickly through the denaturation and neutralization steps to ensure even processing across samples. If an alternative starting concentration of gDNA is desired, adjust accordingly, but account for a minimum of 20 μL dead volume in each well of the 96 well plate to improve pipetting accuracy.
-
9.
Always apply liquid to the membrane while the vacuum is off. If individual wells do not clear, pipetting a few times will allow them to flow through.
-
10.
Perform washes in liberal amounts of 1× PBST and rock with sufficient vigor to ensure thorough and even washing.
-
11.
We tried several different methods of purifying gDNA following β-GT and Tet oxidation reactions (including phenol:chloroform purification with ethanol precipitation and standard DNA purification kits) and determined that KAPA Pure Beads most reliably gave the best yield of gDNA. As the amount of gDNA to be submitted to core facilities for EPIC array processing is crucial, it is important to use a method of DNA recovery that will provide the best overall yield as measured by Invitrogen Qubit dsDNA HS assay.
-
12.
To avoid pulling beads with the withdrawal of the supernatant, leave a small amount of volume at the bottom of the tube. Addition of 80% ethanol will take the small proportion of beads at the bottom of the tube and efficiently capture in the magnetic field so that accidentally taking beads will not be an issue in the removal of the washes.
-
13.
Tet oxidation reagent #1 and #2 can be made ahead of time, aliquoted, and stored at −80 °C for future, one-time use.
-
14.
All Tet oxidation reagents should remain in the dark as these components are light-sensitive.
-
15.
For protein production of TET2-CD enzyme, please see Chapter 13 by Dr. Gerd Pfeifer’s group in this edition of Methods in Molecular Biology.
-
16.
Contact the core facility that will be processing samples on the EPIC array prior to submission. Most core facilities that process EPIC arrays require a certain amount and quality of gDNA, and TAB-treated gDNA typically does not meet these standards. Discuss with the core facility the upstream modification that will be done with the gDNA, and how much gDNA is expected to be submitted. Our laboratory typically recovers 280–330 ng of TAB-treated gDNA from the initial 500 ng of gDNA that was put into the reaction. If TET2-CD enzyme is in ample supply, we recommend doubling the Tet oxidation reaction for each sample and pooling the reactions together prior to KAPA Pure Beads DNA purification.
-
17.
For the initial spike-in of the standard to the T4-βGT reaction, add only 5mC to one reaction and only 5hmC to a different reaction. Both the 5mC and 5hmC standard from ZYMO have the same sequence, so determining efficiency of the TAB oxidation reaction for both standards needs to be separated in space. The standards are also modified at every cytosine (CpG and CpH) in the sequence. We designed bisulfite primers using MethPrimer that would still allow for amplification following bisulfite conversion of this DNA molecule:
-
Forward Primer: TTTAAAGATGTAGGGGTAAAAGTTA
-
Reverse Primer: ACCAAATTTAATTCCTTTCAAC
-
-
18.
For validation of TAB reactions on the 5mC standard, we submit at least 30 colonies to ensure that all 5mC was successfully oxidized.
-
19.
Of particular note, we would like to comment on deciding between TAB array and OxBS array for quantifying 5hmC in a sample population. As previously mentioned, 5hmC abundance is very low in comparison to 5mC (almost 14-fold on average), with the exception of ESCs and brain tissue [10, 11, 58]. In our analysis, the OxBS array was inefficient at detecting 5hmC in the NoTx and DAC samples due to the low level of 5hmC; however, it was much better at capturing this modification in the DAC + VitC sample in which VitC successfully induced an increase in the mark (Fig. 4c), suggesting that a threshold of 5hmC abundance is needed in a sample population for OxBS array to reliably quantify this mark. Indeed, by coupling this rationale with our ELISA-based global quantification results (Fig. 2c), we hypothesize that a threshold of at least 0.1% total 5hmC is needed in a gDNA sample to be detected by OxBS array, as our DAC + VitC samples exceeded this threshold while our DAC and NoTx samples did not. To further investigate this notion, we downloaded OxBS array data from a patient cohort of brain tissue samples and performed the subtraction calculation on these data (GSE138597) [59]. While in our analysis of NoTx and DAC treated samples we observed a large percentage of probes with negative 5hmCβ-values following subtraction (Fig. 4c), the brain samples with known high levels of 5hmC only demonstrated 9% of negative 5hmCβ-values on average (data not shown). We believe that samples with low 5hmC abundance processed by OxBS array analysis are more susceptible to this issue than if they are processed by TAB array, as OxBS array provides an indirect measurement of 5hmC while TAB array provides a direct measurement. Further work needs to be conducted to determine what exact global 5hmC threshold needs to be met to allow accurate quantification of 5hmC by the OxBS array. We recommend that the decision for use of TAB array or OxBS array should be made following global quantification of 5hmC levels in a sample. If the global amount of 5hmC is very low, we recommend the TAB array approach, as it can directly measure low levels of 5hmC present in a sample population. If the global amount of 5hmC is relatively high, then we recommend the OxBS approach, as reagent cost and sample processing time is limited in comparison to TAB array.
Change history
25 July 2021
In the original version of this book, chapters 3, 7, and 17 were published non-open access. It has now been changed to open access under a CC BY 4.0 license and the copyright holder has been updated to “The Author(s).” This book has also been updated with these changes.
References
Greenberg MVC, Bourc’his D (2019) The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol 20:590–607
Bostick M, Kim JK, Estève P-O et al (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317:1760–1764
Robert M-F, Morin S, Beaulieu N et al (2003) DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat Genet 33:61–65
Sharif J, Muto M, Takebayashi S et al (2007) The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450:908–912
Spada F, Haemmer A, Kuch D et al (2007) DNMT1 but not its interaction with the replication machinery is required for maintenance of DNA methylation in human cells. J Cell Biol 176:565–571
Laurent L, Wong E, Li G et al (2010) Dynamic changes in the human methylome during differentiation. Genome Res 20:320–331
Ziller MJ, Gu H, Müller F et al (2013) Charting a dynamic DNA methylation landscape of the human genome. Nature 500:477–481
Kriaucionis S, Heintz N (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324:929–930
Ooi SKT, Bestor TH (2008) The colorful history of active DNA demethylation. Cell 133:1145–1148
Tahiliani M, Koh KP, Shen Y et al (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935
Ito S, D’Alessio AC, Taranova OV et al (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466:1129–1133
Ito S, Shen L, Dai Q et al (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333:1300–1303
He Y-F, Li B-Z, Li Z et al (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333:1303–1307
Shen L, Wu H, Diep D et al (2013) Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 153:692–706
Weber AR, Krawczyk C, Robertson AB et al (2016) Biochemical reconstitution of TET1-TDG-BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat Commun 7:10806
Iurlaro M, Ficz G, Oxley D et al (2013) A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol 14:R119
Spruijt CG, Gnerlich F, Smits AH et al (2013) Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152:1146–1159
Wu X, Zhang Y (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18:517–534
Gu T-P, Guo F, Yang H et al (2011) The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477:606–610
Guo F, Li X, Liang D et al (2014) Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell 15:447–459
Mayer W, Niveleau A, Walter J et al (2000) Demethylation of the zygotic paternal genome. Nature 403:501–502
Oswald J, Engemann S, Lane N et al (2000) Active demethylation of the paternal genome in the mouse zygote. Curr Biol 10:475–478
Shen L, Inoue A, He J et al (2014) Tet3 and DNA replication mediate demethylation of both the maternal and paternal genomes in mouse zygotes. Cell Stem Cell 15:459–471
Guo F, Yan L, Guo H et al (2015) The transcriptome and DNA methylome landscapes of human primordial germ cells. Cell 161:1437–1452
Seisenberger S, Andrews S, Krueger F et al (2012) The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell 48:849–862
Koh KP, Yabuuchi A, Rao S et al (2011) Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 8:200–213
Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome — biological and translational implications. Nat Rev Cancer 11:726–734
Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol 14:R115
Robertson KD (2005) DNA methylation and human disease. Nat Rev Genet 6:597–610
Zhou W, Dinh HQ, Ramjan Z et al (2018) DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat Genet 50:591–602
Gerecke C, Schumacher F, Edlich A et al (2018) Vitamin C promotes decitabine or azacytidine induced DNA hydroxymethylation and subsequent reactivation of the epigenetically silenced tumour suppressor CDKN1A in colon cancer cells. Oncotarget 9:32822–32840
Gillberg L, Ørskov AD, Nasif A et al (2019) Oral vitamin C supplementation to patients with myeloid cancer on azacitidine treatment: Normalization of plasma vitamin C induces epigenetic changes. Clin Epigenetics 11:143
Liu M, Ohtani H, Zhou W et al (2016) Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc Natl Acad Sci U S A 113:10238–10244
Blaschke K, Ebata KT, Karimi MM et al (2013) Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500:222–226
Putiri EL, Tiedemann RL, Thompson JJ et al (2014) Distinct and overlapping control of 5-methylcytosine and 5-hydroxymethylcytosine by the TET proteins in human cancer cells. Genome Biol 15:R81
Bibikova M, Le J, Barnes B et al (2009) Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics 1:177–200
Bibikova M, Barnes B, Tsan C et al (2011) High density DNA methylation array with single CpG site resolution. Genomics 98:288–295
Nazor KL, Boland MJ, Bibikova M et al (2014) Application of a low cost array-based technique — TAB-Array — for quantifying and mapping both 5mC and 5hmC at single base resolution in human pluripotent stem cells. Genomics 104:358–367
Pidsley R, Zotenko E, Peters TJ et al (2016) Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol 17:208
Stewart SK, Morris TJ, Guilhamon P et al (2015) oxBS-450K: a method for analysing hydroxymethylation using 450K BeadChips. Methods 72:9–15
Jin S-G, Kadam S, Pfeifer GP (2010) Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res 38:e125–e125
Yu M, Han D, Hon GC et al (2018) Tet-assisted Bisulfite sequencing (TAB-seq). Methods Mol Biol 1708:645–663
Booth MJ, Branco MR, Ficz G et al (2012) Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 336:934–937
Kumaki Y, Oda M, Okano M (2008) QUMA: quantification tool for methylation analysis. Nucleic Acids Res 36:W170–W175
Montgomery J, Wittwer CT, Palais R et al (2007) Simultaneous mutation scanning and genotyping by high-resolution DNA melting analysis. Nat Protoc 2:59–66
Malentacchi F, Forni G, Vinci S et al (2009) Quantitative evaluation of DNA methylation by optimization of a differential-high resolution melt analysis protocol. Nucleic Acids Res 37:e86–e86
Wojdacz TK, Dobrovic A, Hansen LL (2008) Methylation-sensitive high-resolution melting. Nat Protoc 3:1903–1908
Li L-C, Dahiya R (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18:1427–1431
Sperger JM, Chen X, Draper JS et al (2003) Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc Natl Acad Sci U S A 100:13350–13355
Tiedemann RL, Putiri EL, Lee J-H et al (2014) Acute depletion redefines the division of labor among DNA methyltransferases in methylating the human genome. Cell Rep 9:1554–1566
Zhou W, Triche TJ, Laird PW et al (2018) SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res 46:e123–e123
Skvortsova K, Zotenko E, Luu P-L et al (2017) Comprehensive evaluation of genome-wide 5-hydroxymethylcytosine profiling approaches in human DNA. Epigenetics Chromatin 10:16
Xu Z, Taylor JA, Leung Y-K et al (2016) oxBS-MLE: an efficient method to estimate 5-methylcytosine and 5-hydroxymethylcytosine in paired bisulfite and oxidative bisulfite treated DNA. Bioinformatics 32:3667–3669
Maksimovic J, Phipson B, Oshlack A (2016) A cross-package Bioconductor workflow for analysing methylation array data. F1000Res 5:1281
Ritchie ME, Phipson B, Wu D et al (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47
Gentleman RC, Carey VJ, Bates DM et al (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5:R80
Huber W, Carey VJ, Gentleman R et al (2015) Orchestrating high-throughput genomic analysis with bioconductor. Nat Methods 12:115–121
Globisch D, Münzel M, Müller M et al (2010) Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One 5:e15367
Kochmanski J, Savonen C, Bernstein AI (2019) A novel application of mixed effects models for reconciling base-pair resolution 5-methylcytosine and 5-hydroxymethylcytosine data in neuroepigenetics. Front Genet 10:801
Acknowledgements
This work was supported in part by grants from the American Cancer Society—Michigan Cancer Research Fund to R.L.T. (PF-16-245-01-DMC) and the National Institutes of Health to S.B.R. (R35GM124736). We also acknowledge Julie Koeman and Marie Adams from the Van Andel Institute Genomics Core for their expertise with OxBS treatments and processing of the EPIC arrays. Finally, we would like to thank Dr. Alison Bernstein at Michigan State University for her guidance on processing OxBS array data.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this protocol

Cite this protocol
Tiedemann, R.L., Eden, H.E., Huang, Z., Robertson, K.D., Rothbart, S.B. (2021). Distinguishing Active Versus Passive DNA Demethylation Using Illumina MethylationEPIC BeadChip Microarrays. In: Bogdanovic, O., Vermeulen, M. (eds) TET Proteins and DNA Demethylation. Methods in Molecular Biology, vol 2272. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-1294-1_7
Download citation
DOI: https://doi.org/10.1007/978-1-0716-1294-1_7
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-1293-4
Online ISBN: 978-1-0716-1294-1
eBook Packages: Springer Protocols