
Abstract
Anticoagulant therapy is a mainstay in the management of patients with cardiovascular disease and related conditions characterized by a heightened risk for thrombosis. Acute coronary syndrome, chronic coronary syndrome, ischemic stroke, and atrial fibrillation are the most common. In addition to their proclivity for thrombosis, each of these four conditions is also characterized by local and systemic inflammation, endothelial/endocardial injury and dysfunction, oxidative stress, impaired tissue-level reparative capabilities, and immune dysregulation that plays a critical role in linking molecular events, environmental triggers, and phenotypic expressions. Knowing that cardiovascular disease and thrombosis are complex and dynamic, can the scientific community identify a common pathway or specific point of interface susceptible to pharmacological inhibition or alteration that is likely to be safe and effective? The contact factors of coagulation may represent the proverbial “sweet spot” and are worthy of investigation. The following review provides a summary of the fundamental biochemistry of factor XI, its biological activity in thrombosis, inflammation, and angiogenesis, new targeting drugs, and a pragmatic approach to managing hemostatic requirements in clinical trials and possibly day-to-day patient care in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
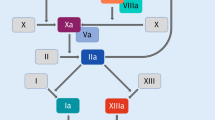
Contact activation pathway of coagulation
The contact activation pathway’s relevance for maintaining hemostatic capacity in vivo has been debated as patients deficient in factor (F) XII, PK (pre-kallikrein), or HK (high molecular weight kininogen) do not typically present with a bleeding phenotype, and Factor XI (FXI) deficiency (hemophilia C), is a mild- to- moderate, tissue-specific bleeding disorder. In addition, both FXII and FXI can be “bypassed” by other constituents of the coagulation pathway that can activate “downstream” proteins and produce sufficient thrombin to attain a fibrin clot [1].
In the past several decades, in vitro and animal model studies have suggested that some proteins of the contact pathway, specifically FXI and FXII, contribute to stable clot formation on aggregated platelets [2]. The procoagulant environment contributing to thrombus formation is not solely influenced by activation of the coagulation cascade and platelets but is also affected by the activity of other systems, including inflammatory and fibrinolytic systems that are influenced by FXI and FXII activity. Therefore, a comprehensive understanding of contact pathway proteins is required to guide future drug development and use in thrombosis-associated conditions.
Factor XI structure and function
Human Factor XI, a 160-kDa serine protease glycoprotein, circulates as a disulfide-linked dimer of two identical 607 amino acid subunits. Each polypeptide consists of a 35-kDa C-terminal light chain containing the trypsin-like catalytic domain, and an N-terminal 45-kDa heavy chain with four ~ 90 amino acid tandem repeats termed apple domains [3]. These apple domains confer binding to other proteins: A1 contains binding sites to HK and thrombin, A2 and A3 to FIX, A3 to the platelet receptor GP1bα and heparin, and A4 to FXII and the other subunit of FXI [4]. FXIa subsequently activates FIX to FIX a by the cleavage of two activation sites [5].
Evolutionary biology
It is widely thought that a series of gene duplications throughout evolution led to the complete clotting cascade, as seen in humans. Accordingly, clotting factors contain many of the same conserved domains and are closely related [6]. A simple version of the clotting cascade appeared in the earliest vertebrates, and as vertebrates, themselves have become more complex with more intricate and contained cardiovascular systems, the clotting cascade has evolved similarly. While thrombin was the first coagulation factor to emerge before the appearance of the first vertebrates, the contact pathway was the last to evolve, as genes for HK, FXII, and a single paralog of PK/XI are found later in vertebrate development [6].
Factor XI synthesis, storage, and expression
FXI is expressed mainly in the liver and is regulated by the transcription factor HNF4α, but FXI mRNA has also been detected in platelets, blood mononuclear cells, granulocytes, pancreas, and kidneys as well [7]. Hemophilia C, or FXI deficiency, is a rare bleeding disorder that affects approximately one in one million people. With over 190 reported mutations in the F11 gene [8], most are Cross-Reactive Material – (CRM −) (Type I) with mutations causing low FXI plasma levels. Fewer mutations are CRM + (Type II) with normal FXI levels but reduced coagulant activity mostly due to functional mutations. Historically, it has been difficult to predict bleeding tendency in FXI deficient patients as there has been a poor correlation between bleeding phenotype and plasma FXI levels. This important topic is discussed in detail in the following section.
Factor XII-mediated factor XI activation
FXII mediated activation of FXI is a pivotal step in the intrinsic pathway of coagulation, contributing to thrombus formation and hemostasis. Upon exposure to a negatively charged surface, such as activated platelets or subendothelial collagen, FXII undergoes a conformational change leading to its autoactivation to FXIIa (contact activation). FXIIa subsequently cleaves FXI to its activated form, FXIa, initiating a cascade of proteolytic events resulting in fibrin formation (Fig. 1) [9]. Mice lacking FXII or FXI are resistant to experimentally-induced thrombosis, suggesting that FXIIa activation of FXI contributes to thrombus formation in vivo [2]. Interestingly, FXII deficiency does not impair hemostasis, indicating that the FXIIa-mediated FXI activation contributes to thrombus formation while being dispensable for hemostatic processes. This mechanism underscores the significance of FXII-FXI interaction in maintaining hemostatic balance.

The intrinsic pathway of coagulation also referred to as the contact pathway is initiated following exposure of blood to negatively charged surfaces. The are a series of biochemical/enzymatic steps that follow, including activation of factors XII, XI, IX, and VIII that in complex with factor X (prothrombinase complex), and phospholipid convert prothrombin to thrombin (factor IIa). Thrombin then catalytically converts fibrinogen to fibrin- the predominant structural protein in a thrombus
Thrombin-mediated factor XI activation
The contribution of thrombin-mediated factor XI activation is primarily through the amplification of tissue factor-FVIIa (TF-FVIIa) which participates in both hemostasis and thrombosis (Fig. 2) [10]. The role of factor XI is particularly relevant in low TF organs and tissues, such as the placenta and uterus. The tissue specificity requirement for factor XI aligns biologically with observations made among persons with hemophilia C and other factor XI deficiency states will experience bleeding upon provocation of the same tissues that also possess high fibrinolytic activity including the nose, mouth, and genitourinary tract. These observations also highlight the importance of thrombin activatable fibrinolytic inhibitor (TAFI) [11].

Role of factor XI in thrombosis. Factor XI is particularly important for thrombin generation following activation of the intrinsic pathway of coagulation (see text). “Created with BioRender.com”
Factor XI and platelets
Because platelets provide a surface or template for factor XI mediated thrombin activation [12], and platelet-monocyte aggregates as well as micro-vesicles are a source of TF, thrombocytopenia and impaired extracellular vesicle formation contribute to bleeding phenotypes in patients with factor XI deficiency [13]. Thrombin activates FXI at a 5–10,000-fold enhancement when bound to the platelet surface [14]. While there are ~ 1500 binding sites for FXI on the surface of the platelet, FXIa binds to only ~ 250 sites [15], suggesting that FXI and FXIa bind in a different mechanism.
The relationship between factor XI, TF, and platelets is complex, and flow dynamics and shear stress must be taken into consideration to understand the relationship fully [16]. A relatively high concentration of platelets at the site of vascular injury, coupled with subendothelial TF exposure can physically inhibit TF-VIIa activity; this is particularly evident at high shear stress where platelet deposition in a physical boundary to TF complexing with factor FVII occurs more regularly. Proteins present at sites of vascular injury such as C1 inhibitor and platelet-released protease nexin 2 also inhibit fluid phase active factor XI [17].
Cellular and organ-specific tissue factor activity
TF is selectively expressed in cells and tissues [18], including nonvascular cells [19]. There is a nonuniform distribution, with high levels of TF in the brain, lung, and placenta; intermediate levels in the heart, gastrointestinal tract, kidney, uterus, and testes; and low levels in skeletal muscle, spleen, thymus, and the liver [19].
Factor XI, molecular biology, and thrombus architecture
Nakazawa and colleagues first identified cell-free (CF) nucleic acids, specifically cfRNA, that initiated coagulation by serving as a cofactor [20]. Kannemeier and colleagues performed a series of experiments to determine the functional significance of intracellular material exposed to blood following tissue injury [21]. Extracellular RNA-activated proteases of the contact system of coagulation, including factor XI itself exhibited strong RNA binding. Extracellular RNA also augmented (auto-) activation of proteases of the contact phase pathway of blood coagulation such as factors XII and XI, both exhibiting strong RNA binding.
Secondary structures of RNA, particularly hairpin-forming oligomers are highly procoagulant. There is an RNA length-contact activation-dependent relationship, however, even relatively short polyphosphates released from activated platelets accelerate factor V activation, inhibit the anticoagulant activity of tissue factor pathway inhibitor (TFPI), promote factor XI activation by thrombin, and are associated with the synthesis of thicker fibrin strands that are resistant in fibrinolysis [22]. Extracellular polyphosphates augment factor XI activation and often co-localize with extracellular DNA and RNA following cellular injury and in inflammatory environments [23].
Is hemophilia C a disease model for the safety of factor XI inhibitors?
Patients with hemophilia C have a deficiency of factor XI. Because bleeding tends to be mild, most often follows a hemostatic challenge, and in most cases does not correlate with factor XI antigen level or activity, complementary roles for other coagulation factors or hemostasis regulating pathways or systems must be considered (Fig. 3). Factor XI can activate coagulation factors X, V, and VIII. It also inhibits tissue factor pathway inhibitor (TFPI) [11].

Role of factor XI in hemostasis (see text). Factor XI plays an important role in thrombus consolidation and feedback activation following thrombin activation through the extrinsic or tissue factor pathway of coagulation. “Created with BioRender.com”
The activated partial thromboplastin time (APTT) does not correlate with the risk of bleeding in patients with hemophilia C. By contrast, if an APTT is performed with the contact pathway inhibited and using tissue factor (TF) rather than thrombin as an activator, clotting time correlates with a bleeding phenotype [24].
Hemophilia C and patient-specific factors
The management of patients with hemophilia C provides insights for the optimal selection of patients being considered for treatment with factor XI inhibitors. Guidelines for hemophilia C to include treatment of acute coronary syndrome recommend against coadministration of fibrinolytic therapy [25] and emphasize limiting the duration of dual antiplatelet therapy. Coagulation factor replacement therapy is an option in patients with a bleeding phenotype or further undergoing surgical revascularization. This is discussed in a subsequent section. Whether anti-TFPI antibody should be used in patients with hemophilia C is debated largely because of the infrequent need for replacement products; however, their availability is important and may also play a role in the development of factor XI inhibitors for a variety of clinical indications.
Hemophilia C and bleeding phenotypes
Investigation of hemophilia C and bleeding phenotypes provides useful information as the area of oral and parenteral factor XI inhibitors begins to take shape. Patients with and those without bleeding tendencies has similar factor XI antigen levels, prothrombin times, fibrinogen, factor XII, von Willebrand factor, and factor XIII concentrations. As previously discussed, clot structure and overall architecture differ among persons with a bleeding phenotype with lower fibrin density and clot stability following decalcification and addition of TF and phospholipids. Moreover, clot stability is highly impaired in the presence of tissue plasminogen activator [26]. In vitro models that combine the measurement of APTT in the rate of clot formation that can be assessed, and fibrinolysis assays may be able to identify patients at particularly high risk for bleeding.
Tissue Factor pathway inhibitor
Human TFPI is a protein composed of 3 Kunitz-type domains flanked by peptide segments. The K1 domain inhibits FVIIa complexed with TF, while the K2 domain inhibits factor Xa [27]. The TFPI-FXa complex is a potent inhibitor of TF-VII. The K3 domain participates in factor Xa kinetics on the vascular endothelial cell surface. Vascular endothelial cells are the primary site of full-length TFPI synthesis and release both constitutively and in response to vascular injury when hemostasis is required. Platelets also secrete TFPI that resides on the surface of activated platelets [28]. While the kinetics of TFPI, more specifically its clearance, is predominantly regulated by the reticuloendothelial system, vascular endothelial cells are involved with internalization and recycling particularly when TFPI is in complex with factor Xa [29].
Following thrombin generation, endothelial cells synthesize and release TFPI, increasing local anticoagulant effects [30]. Exogenous inhibition of coagulation factors Xa, IXa, and VIIa can increase cell surface TFPI [31], potentially augmenting their anticoagulant effects and intrinsic resistance to thrombus formation [32].
TFPI and bleeding phenotypes
A bleeding phenotype in factor XI deficiency states is characterized by reduced clot formation, decreased fiber network density, and increased susceptibility to fibrinolysis (Fig. 3). In vitro models of factor XI deficiency have shown that anti-TFPI antibody administration can increase thrombin generation [33]. Because patients with factor XI deficiency are dependent on TF-mediated thrombin generation, TFPI plasma concentration is believed to be important for characterizing and distinguishing patients with either a low or high likelihood of bleeding [34].
Why might hemophilia C increase TFPI levels?
Elevated TFPI activity has been described in patients with unclassified bleeding disorders [35] and in both hemophilia A and B [36]. By contrast, TFPI is not a major determinant of thrombin generation in healthy individuals [37].
Do factor XI inhibitors influence TFPI kinetics?
As briefly mentioned in the preceding section, TFPI is an anticoagulant protein that attenuates the initiation phase of coagulation before thrombin is generated [38]. TFPIs action is uniquely positioned to TF-expressing surfaces and sites of vascular injury. In addition, TFPI-α and TFPI-β localize to specific sites and dampen both cellular trafficking and signaling pathways governed by TF-mediated coagulation. A full appreciation of TFPI activity has led to the development of specific inhibitors for the management of hemophilia-associated bleeding [39] as well as bleeding complications that occur in rare bleeding disorders [40].
Because factor XI proteolytically cleaves TFPI, factor XI targeted therapies may increase TFPI thereby increasing overall anticoagulant effects [41]. A pro-fibrinolytic state may also ensue because of reduced TAFI [41]. Further investigation will be required to better understand the impact of factor XI targeted therapies on endogenous fibrinolysis and anticipated effects with short and long-term administration.
Factor XI and inflammation
Activation of the contact system has been shown to induce inflammatory reactions through activation of the classical cascade of the complement system, leading to anaphylatoxin production causing smooth muscle cell contraction, enhanced vascular permeability, and recruitment of inflammatory cells to produce a local immune response [42]. In addition, contact pathway activation leads to the release of BK, a small peptide that is cleaved from HK by kallikrein, which induces vascular permeability and binds to its receptor on endothelial cells to promote nitric oxide and prostacyclin release, inducing smooth muscle cell relaxation and vasodilation [43]. Constituents of the contact system can also bind to and activate immune cells directly, as kallikrein can induce neutrophil homing, aggregation, and degranulation [43].
Humans with VTE display a proteomics signature that supports an association between factor XI and inflammation. Specifically, factor XI activity is increased and correlates with proteins linked with thrombo-inflammation, oxidative stress, extracellular matrix formation, and apoptosis. Several examples include cell adhesion protein (Spon1), cytokine binding proteins, NF kappa beta, B-cell development, and Th17 cell differentiation [44]. There are similar associations in sepsis that cause systemic inflammation, cytokine release, and complement activation. Factor XI deficiency attenuates the inflammatory response to sepsis in animal models and contact-activated disseminated intravascular coagulation (DIC) [45].
Factor XI and the cardiovascular system
The liver and heart are known to exhibit one-sided dimensional and bidirectional signaling for physiologic homeostasis [46]. For example, patients with advanced cirrhosis have reduced ventricular diastolic and systolic performance. A condition known as cirrhotic cardiomyopathy has been proposed. In murine models, factor XI activates the bone morphogenic protein (BMP)- SMAD 1/5 pathway in the heart and protects against heart failure with preserved ejection fraction [47]. Increased expression of factor XI also reduces cardiac inflammation and fibrosis. Specifically, mice overexpressing factor XI have reduced macrophages, T cells, monocytes, and granulocytes in models of heart failure with preserved ejection fraction.
Inflammation is prevalent in the early stages of venous thrombosis, contributing to vascular remodeling, valve performance, and thrombus propagation. In animal models of VTE, factor XI inhibition is associated with endothelial cell monocyte and macrophage accumulation [48]. This is an area of ongoing investigation.
Factor XI and atherogenesis
Epidemiology-based observations suggest that factor XI deficiency is associated with reduced incidence of VTE, ischemic stroke, and myocardial infarction [49, 50]. There are data that also support a protective effect on atherogenesis, the proximate substrate for arterial thrombotic events [51]. Ngo and colleagues employed an LDL-R knockout, high-fat mouse model to test defective factor XI inhibition on atherogenesis. The identified reduced atherosclerotic lesion areas in the proximal aorta and aortic sinus with factor XI inhibition achieved with an antisense oligonucleotide for factor XI when compared to untreated controls. In an established atherosclerosis model, they also showed that factor XI inhibition reduced atherosclerotic lesion area and endothelial cell lipoprotein permeability [51].
Ganor et al. reported that Apoprotein E/FXI double knockout mice had a 32% reduction in atherosclerotic lesion area at 24 weeks compared with Apolipoprotein knockout mice [52].
Factor XI and thrombogenesis
While mice deficient in FXI do not exhibit abnormal bleeding, these mice are protected against thrombus formation in several models of arterial and venous thrombosis, as well as stroke. In several models of vascular injury, intravital microscopy and blood flow monitoring suggest that, while thrombus initiation is not interrupted on the vessel wall in FXI deficient mice, thrombi that are formed are unstable and break up before occlusion of a blood vessel can occur [53]. Protection from thrombosis is not limited to arterial injury, as FXI deficiency also confers a thrombo-protective effect in a transient middle cerebral artery occlusion model (MCAO), with a lower volume of infarcted brain tissue and less fibrin deposition in distal micro-vessels without hemorrhage [54].
Factor XI inhibitors in development
Based on the differential contribution of FXI in hemostasis and thrombus formation, particularly its interactions in the contact pathway, inhibition of its action has emerged and attracted attention as promising option for anticoagulation therapy. These inhibitors offer a unique therapeutic approach for preventing thrombotic events while potentially reducing the risk of bleeding complications compared to traditional anticoagulants. By specifically targeting Factor XI, these novel agents aim to interrupt the clotting process without affecting other factors essential for hemostasis. Several drugs have been developed or under-developing with different mechanisms. A summary of FXI inhibitors classes and drugs is shown in (Table 1) [55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75]. The general pharmacological characteristics of each class of factor XI inhibitors are summarized in (Table 2). These drugs have been tested in several conditions like total knee arthroplasty (TKA), end-stage renal disease (ESRD), atrial fibrillation (AF), acute ischemic stroke and acute myocardial infarction with promising efficacy and safety outcomes [55, 57,58,59, 62, 63, 76,77,78,79].
Prevention and management of bleeding
The experience in persons with hemophilia C suggests that bleeding events will be uncommon with factor XI inhibitors, however having a plan of management in place is wise should the need arise [80]. This is particularly important following major trauma, surgical procedures involving the genitourinary tract, and when there is a concomitant use of one or more platelet antagonists or fibrinolytic agents (Table 3).
Pharmacokinetic considerations
The optimal management must take the drug pharmacokinetics into consideration. Specifically, the circulating half-life of the drug and if applicable, the pharmacodynamically active major metabolite, site of metabolism, target (factor XI or XIa), target-drug dissociation constant, and circulating protein binding capacity. For example, a drug with a long circulating half-life that is non-protein bound (i.e., high free plasma concentration) would require a strategy of either reversal (of the drug), removal from the circulation, or replenishing thrombin generation using serial administration of replacement products that are sufficient to restore hemostasis, while at the same time minimizing the risk of thrombosis. By contrast, a factor XI inhibitor with a short circulating half-life that is highly protein-bound may only require a one-time administration of factor XI containing blood replacement products. As always, the severity of bleeding will be the primary determinant of management which could include close observation and local measures to support hemostasis for mild bleeding.
General approach
The initial step in the management of bleeding in patients receiving anticoagulant therapy, including a factor XI inhibitor is a careful assessment of clinical status, the need for supportive measures, and the site(s) of bleeding. Volume resuscitation may be required to normalize the blood pressure. Local control of a superficial source, including compression at vascular puncture sites may be sufficient in many cases, while non-compressible sites of bleeding may require systemic therapy or surgical interventions.
Intervention strategies
A pragmatic approach to the management of bleeding in patients receiving factor XI inhibitors has four fundamental tenets: reversal, removal, replacement, and restoration.
Reversal
The reversal of a drug inherently suggests that a specific antidote or decoy binding agent is available- ideally, co-developed with an anticoagulant with the express intent to have a drug-antidote pair or reversal agent for urgent or emergent circumstances that immediately require normal hemostasis. It is important to be aware that rapid reversal in the circulation will prompt the release of drug present within tissue stores (considered as a component of the volume of distribution) that have pharmacodynamic activity. The half-life or administration strategy of the reversal agent, serial bolus, or continuous infusion must be considered and designed accordingly.
Drug-antidote pairs as a proactive development strategy for anticoagulants is encouraged in the academic and private sectors. We believe that even optimal targets for inhibition still may increase bleeding risk-particularly when hemostasis is challenged by trauma, surgery, and use in combination with other antithrombotic agents. Developing a reversal agent after an anticoagulant has been approved for clinical use is costly and time consuming. Determining that a reversal agent is needed to address bleeding or mitigate bleeding in high-risk settings can also introduce uncertainty among clinicians and patients.
Removal
The removal of a drug from the circulation is inherently attractive when severe or life-threatening bleeding occurs or a surgical procedure associated with high-to-very high bleeding risk must be undertaken (e.g., neurosurgery). The most widely available strategy, hemodialysis, will not be a successful option for plasma protein-bound drugs or large molecules. The same may be true for plasmapheresis for protein-bound drugs, but large molecules can typically be removed.
Replacement
Intravenous infusion of clotting factors is designed to replenish normal plasma concentrations. It may also saturate a drugs binding sites thereby permitting endogenous clotting factors to participate in hemostasis [81].
Restoration
Restoration in the context of bleeding applies to thrombin generation and clot stability. Providing blood products that are known to generate thrombin by one or more mechanisms may be sufficient to stem bleeding, particularly in areas that are not readily accessible to manual compression or other local measures. This approach is also of benefit and frequently used in combination with an intervention to stop bleeding (e.g., cautery for a gastrointestinal source of bleeding). There are many determinants of clot stability, however, in the context of anticoagulant drugs in general and FXI inhibitors particularly, as well as years of experience with mitigating bleeding risk and managing bleeding among persons with hemophilia C attenuating endogenous fibrinolysis and allowing fibrin generated by thrombin’s activation of fibrinogen are the primary goals (see sections on recombinant factor VII, concizumab, and antifibrinolytic agents below).
Specific approach
Monoclonal antibodies
There are specific approaches to bleeding that are determined by the factor XI inhibitor and its associated mechanism of action. Factor XI replacement in the form of factor XI concentrate or fresh frozen plasma (FFP) is a preferred approach with the goal of saturating the drug thereby allowing unbound factor XI to participate in hemostasis. During treatment, factor XI should be measured to avoid very high free plasma concentrations and the potential risk of thrombosis. Factor XI replacement would not be an effective strategy for the management of osocimab-associated bleeding, given its mechanism of action that reduces factor XII-mediated factor XI activation.
Extraction of large molecules such as monoclonal antibodies is a familiar technique to the pharmaceutical industry for manufacturing purposes. These techniques could, at least theoretically, be used to remove a factor XI targeting monoclonal antibody. Extraction using reverse micelles (RM) of an anionic surfactant and a combination of anionic (AOT) and nonionic surfactants is possible [82]. The process, specifically for use on a case-by-case basis in the management of patients with life-threatening hemorrhage after receiving a factor XI monoclonal antibody, requires further investigation.
Small molecule inhibitors
Active site inhibitors that target factor XIa and not impacted by replacement products because factor XI itself is not reduced during drug therapy. Optimal management would require either reversal or removal of the drug; however, the currently available small molecules are not dialyzable, and monoclonal antibody reversal agents have not been included in their development. Restoration of thrombin generation using activated prothrombin complex concentrate, recombinant factor VIIa, and antifibrinolytic agents should be considered and administered according to the severity of bleeding.
Anti-sense oligonucleotides
This class of drug targets mRNA and inhibits the synthesis of factor XI. It is highly bound to plasma proteins with limited renal clearance and a very long duration of effect. Accordingly, a strategy to attenuate its effects would require long-term replacement. In acute settings, factor XI concentrate would be the optimal approach likely with concomitant antifibrinolytic therapy with a goal to immediately gain control over bleeding.
Factor XI concentrates
Factor XI concentrates are plasma-derived but contain a more consistent amount of factor XI when compared with fresh frozen plasma [83]. Recovery of factor XI from plasma is excellent, and the half-life is 40 h or more. The half-life of endogenously, hepatic-synthesized factor XI is 52 h.
There are virally inactivated factor XI concentrates available for clinical use outside of the United States. Both products also contain several other factors to minimize the procoagulant effects of administration such as Antithrombin, C1 esterase inhibitor, and heparin. The anticipated response is a 2% increase in factor XI for every unit per kilogram of factor XI concentrate. Accordingly, 15 units/kg is expected to increase the factor XI level to 30% or more. In many instances, this will be sufficient to restore hemostasis. Repeat dosing to increase factor XI to 50% or more should be considered if bleeding persists or recurs [84, 85].
Fresh frozen plasma
Fresh frozen plasma (FFP) is the fluid portion of a unit of whole blood frozen in a designated time frame, usually within 8 h. FFP contains all coagulation factors except platelets. Fresh frozen plasma contains fibrinogen (400 to 900 mg/unit), albumin, protein C, protein S, antithrombin, and TFPI. It is free of erythrocytes and leukocytes. FFP corrects coagulopathy by replacing or supplying plasma proteins in patients who are deficient in or have defective plasma proteins. A standard dose of 10 to 20 mL/kg (4 to 6 units in adults) will raise factor levels by approximately 20%. A rise of approximately 10% in various factors is sufficient to achieve hemostasis. Also, FFP provides some volume resuscitation, as each unit contains approximately 250 ml [86, 87].
There is approximately 1 unit of factor XI per mL of plasma. Accordingly, FFP given at a dose of 10 to 20 mL/kilogram should increase the factor XI level to 10–20% above baseline. The overall volume of fluid administered during replacement with FFP should always be taken into consideration in patients with a history of heart failure.
Activated prothrombin complex concentrate
Prothrombin complex concentrate (PCC) comes from the process of ion-exchange chromatography from the cryoprecipitate supernatant of large plasma pools and after the removal of antithrombin and factor XI. Processing techniques involving ion exchangers allow to produce either three-factor (i.e., factors II, IX, and X) or four-factor (i.e., factors II, VII, IX, and X) PCC.
While the absence of FXI from APCC makes it a less attractive alternative for the management of factor XI inhibitor-associated bleed, thrombin generation is still achievable.
A dose of 50 units/kg, with an additional 25 units/kg for uncontrolled bleeding is used in a variety of clinical settings, most often for warfarin-associated bleeding with an elevated INR. Its use in hemophilia has progressively lessened given the presence of alternatives that carry a lower risk of thrombosis [88].
Recombinant factor VII
Administration of pharmacologic doses of exogenous rFVIIa enhances thrombin generation on the platelet surface at the site of injury, independently of the presence of FVIII or FIX. Pharmacologic doses of rFVIIa induce hemostasis not only in hemophilia patients, but also in patients with thrombocytopenia, functional platelet defects, and bleeding triggered by surgery or trauma [89].
When administered at pharmacologic doses, rFVIIa binds to the surface of activated platelets in a TF-independent manner and promotes factor X activation, and thrombin generation on the activated platelet surface [90]. The binding of factor VIIa to platelets involves the glycoprotein Ib/IX/V complex and anionic phospholipids expressed on activated platelets.
In non-hemophilic conditions, platelet-bound rFVIIa increases the activation of both factor IX and FX and increases thrombin generation above normal levels [91]. Increased thrombin generation then promotes increased activation and local accumulation of platelets improving hemostasis.
A dose of rVIIa of 15 to 30 mcg/kg may be sufficient to restore hemostasis in patients with mild to moderate bleeding. Repeat dosing every 4–6 h should be considered for uncontrolled bleeding [92]. Higher doses may be needed in the treatment of intracranial hemorrhage (70–90 mcg/kg). It is important to recognize that factor VII has a very short half-life (~ 6 h or less).
Other considerations
Concizumab
Concizumab is a prophylactic therapy for patients with hemophilia. After subcutaneous administration, it binds to the Kunitz-2 binding domain of TFPI and prevents its binding to factor X. In turn, sufficient FXa can be produced by the TF-VIIa complex and participate in hemostasis [93]. The FDA has not approved concizumab in the United States, requesting more information on dosing and monitoring from the manufacturer. There are limited data on the treatment of hemophilia C and no data for the treatment of FXI inhibitors.
Emicizumab
Emicizumab is a humanized, specific monoclonal antibody that binds to factors IX and X, mediating factor X activation thereby bypassing the normal mechanism of FVIII-mediated activation that is missing in patients with hemophilia A [94, 95]. The drug is known to affect the accuracy of several laboratory tests that are used in coagulation. These include the activated partial thromboplastin time, activated clotting time, and all APTT-based assays such as FVIII activity and activated protein C (APC) resistance test. Several laboratory tests are not affected by the drug. These include prothrombin time, thrombin time, immune-based assays, and chromogenic assays other than FVIII. Concomitant administration of emicizumab and activated prothrombin complex concentrate is associated with thromboembolic events and thrombotic microangiopathy if the cumulative amount of the latter blood replacement product is greater than 100 units/kg/ 24 h [96]. There are limited data on the treatment of hemophilia C and no data for the treatment of FXI inhibitors.
Antifibrinolytic agents
The fibrinolytic system is designed to limit the amount of fibrin that is formed following vascular injury by converting plasminogen to plasmin [97]. Poorly regulated thrombus formation would have devastating consequences on tissue perfusion. By contrast, the management of hemophilias to include hemophilia C employ antifibrinolytic agents to generate fibrin, maintain vascular integrity and augment hemostasis. Several agents are widely available at relatively low cost and can be administered by varied routes depending on the site(s) of bleeding or the anticipated risk of bleeding from an invasive procedure. Clinicians should be aware of these agents and their use if factor XI inhibitors become part of the therapeutic armamentarium of anticoagulants in daily practice.
Tranexamic acid
Tranexamic acid is a synthetic analogue of lysine. It binds to lysine receptor sites on plasminogen blocking its conversion to the active enzyme plasmin and preventing fibrin degradation. It is more potent than epsilon aminocaproic acid. The available formulations and doses are summarized below:
Oral: 650 mg tablets in the United States and 500 mg in other countries
-
1 g 4 times daily
-
Mouthwash 5% for dental procedures
-
Intravenous for life-threatening hemorrhage—10 mg/kg every 6–8 h.
Epsilon aminocaproic acid
Epsilon aminocaproic acid, like tranexamic acid, is a lysine analogue that binds competitively to plasminogen, preventing its binding to fibrin, generation of plasmin, and the degradation of fibrin. The available formulations and doses are summarized below:
-
Oral: 5 to 6 g daily
-
Oral rinse: 15 mL solution containing 1.25 g/5 mL over 2 min.
Conclusion
Comprehensive understanding of the systems impacted by the contact pathway is crucial, as it influences numerous cardiovascular disorders. While FXI is thought to have a limited role in hemostasis where it is primarily activated to FXIa by thrombin, its role in thrombosis can vary depending on the event. When the extrinsic pathway is inhibited, the contact pathway plays a significant role in thrombus formation; however, FXI may not be essential for thrombosis at a ruptured plaque where a large amount of tissue factor can trigger independent thrombin generation. Moreover, FXI has additional participation in inflammation, angiogenesis, and fibrinolysis. As a result, the contact pathway, involving factor XI (FXI), has attracted great attention as a potential target for safer anticoagulants developments. Different classes of FXI inhibitors have been developed including antisense oligonucleotides, monoclonal antibodies, small synthetic molecules, natural peptides, and aptamers with different mechanisms. The clinical data has shown a promising effect of FXI inhibitors in preventing thrombosis without a concomitant increase in the bleeding burden. Additional large-scale clinical trials are needed to confirm the safety of these drugs on the long-term and support their use in different medical scenarios.
Data Availability
All data supporting the findings of this review are available within the paper and cited references.
References
Gailani D, Broze GJ Jr (1991) Factor XI activation in a revised model of blood coagulation. Science 253(5022):909–912. https://doi.org/10.1126/science.1652157
Cheng Q, Tucker EI, Pine MS, Sisler I, Matafonov A, Sun MF, White-Adams TC, Smith SA, Hanson SR, McCarty OJ, Renné T, Gruber A, Gailani D (2010) A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood 116(19):3981–3989. https://doi.org/10.1182/blood-2010-02-270918
Bouma BN, Griffin JH (1977) Human blood coagulation factor XI. Purification, properties, and mechanism of activation by activated factor XII. J Biol Chem 252(18):6432–6437
McMullen BA, Fujikawa K, Davie EW (1991) Location of the disulfide bonds in human coagulation factor XI: the presence of tandem apple domains. Biochemistry 30(8):2056–2060. https://doi.org/10.1021/bi00222a008
Walsh PN, Sinha D, Koshy A, Seaman FS, Bradford H (1986) Functional characterization of platelet-bound factor XIa: retention of factor XIa activity on the platelet surface. Blood 68(1):225–230
Doolittle RF (2009) Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol 74:35–40. https://doi.org/10.1101/sqb.2009.74.001
Podmore A, Smith M, Savidge G, Alhaq A (2004) Real-time quantitative PCR analysis of factor XI mRNA variants in human platelets. J Thromb Haemost 2(10):1713–1719. https://doi.org/10.1111/j.1538-7836.2004.00924.x
Zhang X, Lewandowska M, Aldridge M, Iglay K, Wolford E, Shapiro A (2023) Global epidemiology of factor XI deficiency: a targeted review of the literature and foundation reports. Haemophilia 29(2):423–434. https://doi.org/10.1111/hae.14687
Maas C, Renné T (2018) Coagulation factor XII in thrombosis and inflammation. Blood 131(17):1903–1909. https://doi.org/10.1182/blood-2017-04-569111
Gruber A, Hanson SR (2003) Factor XI-dependence of surface- and tissue factor-initiated thrombus propagation in primates. Blood 102(3):953–955. https://doi.org/10.1182/blood-2003-01-0324
Puy C, Tucker EI, Matafonov A, Cheng Q, Zientek KD, Gailani D, Gruber A, McCarty OJ (2015) Activated factor XI increases the procoagulant activity of the extrinsic pathway by inactivating tissue factor pathway inhibitor. Blood 125(9):1488–1496. https://doi.org/10.1182/blood-2014-10-604587
Baglia FA, Badellino KO, Li CQ, López JA, Walsh PN (2007) Factor XI binding to the platelet glycoprotein Ib-IX-V complex promotes factor XI activation by thrombin. J Biol Chem 282(39):29067. https://doi.org/10.1016/s0021-9258(20)58639-0
Nieuwland R, Gardiner C, Dignat-George F, Mullier F, Mackman N, Woodhams B, Thaler J (2019) Toward standardization of assays measuring extracellular vesicle-associated tissue factor activity. J Thromb Haemost 17(8):1261–1264. https://doi.org/10.1111/jth.14481
Greengard JS, Heeb MJ, Ersdal E, Walsh PN, Griffin JH (1986) Binding of coagulation factor XI to washed human platelets. Biochemistry 25(13):3884–3890. https://doi.org/10.1021/bi00361a022
Miller TN, Sinha D, Baird TR, Walsh PN (2007) A catalytic domain exosite (Cys527-Cys542) in factor XIa mediates binding to a site on activated platelets. Biochemistry 46(50):14450–14460. https://doi.org/10.1021/bi701310x
Fogelson AL, Hussain YH, Leiderman K (2012) Blood clot formation under flow: the importance of factor XI depends strongly on platelet count. Biophys J 102(1):10–18. https://doi.org/10.1016/j.bpj.2011.10.048
Humphreys SJ, Whyte CS, Mutch NJ (2023) “Super” SERPINs-A stabilizing force against fibrinolysis in thromboinflammatory conditions. Front Cardiovasc Med 10:1146833. https://doi.org/10.3389/fcvm.2023.1146833
Drake TA, Morrissey JH, Edgington TS (1989) Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol 134(5):1087–1097
Mackman N (2004) Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol 24(6):1015–1022. https://doi.org/10.1161/01.Atv.0000130465.23430.74
Nakazawa F, Kannemeier C, Shibamiya A, Song Y, Tzima E, Schubert U, Koyama T, Niepmann M, Trusheim H, Engelmann B, Preissner KT (2005) Extracellular RNA is a natural cofactor for the (auto-)activation of Factor VII-activating protease (FSAP). Biochem J 385(Pt 3):831–838. https://doi.org/10.1042/bj20041021
Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, Song Y, Tzima E, Kennerknecht E, Niepmann M, von Bruehl ML, Sedding D, Massberg S, Günther A, Engelmann B, Preissner KT (2007) Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A 104(15):6388-6393.https://doi.org/10.1073/pnas.0608647104
Kravtsov DV, Matafonov A, Tucker EI, Sun MF, Walsh PN, Gruber A, Gailani D (2009) Factor XI contributes to thrombin generation in the absence of factor XII. Blood 114(2):452–458. https://doi.org/10.1182/blood-2009-02-203604
Choi SH, Smith SA, Morrissey JH (2011) Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood 118(26):6963–6970. https://doi.org/10.1182/blood-2011-07-368811
Pike GN, Cumming AM, Hay CR, Bolton-Maggs PH, Burthem J (2015) Sample conditions determine the ability of thrombin generation parameters to identify bleeding phenotype in FXI deficiency. Blood 126(3):397–405. https://doi.org/10.1182/blood-2014-12-616565
Staritz P, de Moerloose P, Schutgens R, Dolan G (2013) Applicability of the European society of cardiology guidelines on management of acute coronary syndromes to people with haemophilia - an assessment by the ADVANCE working group. Haemophilia 19(6):833–840. https://doi.org/10.1111/hae.12189
Zucker M, Seligsohn U, Salomon O, Wolberg AS (2014) Abnormal plasma clot structure and stability distinguish bleeding risk in patients with severe factor XI deficiency. J Thromb Haemost 12(7):1121–1130. https://doi.org/10.1111/jth.12600
Bajaj MS, Birktoft JJ, Steer SA, Bajaj SP (2001) Structure and biology of tissue factor pathway inhibitor. Thromb Haemost 86(4):959–972
Maroney SA, Mast AE (2008) Expression of tissue factor pathway inhibitor by endothelial cells and platelets. Transfus Apher Sci 38(1):9–14. https://doi.org/10.1016/j.transci.2007.12.001
Becker RC, Alexander JH, Li Y, Robertson T, Kunitada S, Spencer FA, Yang H, Harrington RA (2004) Vascular endothelial tissue factor pathway inhibitor kinetics in culture following exposure to DX-9065a–a selective and direct factor Xa inhibitor. J Thromb Thrombolysis 18(3):193–197. https://doi.org/10.1007/s11239-005-0345-6
Ameri A, Kuppuswamy MN, Basu S, Bajaj SP (1992) Expression of tissue factor pathway inhibitor by cultured endothelial cells in response to inflammatory mediators. Blood 79(12):3219–3226
Crawley JT, Lane DA (2008) The haemostatic role of tissue factor pathway inhibitor. Arterioscler Thromb Vasc Biol 28(2):233–242. https://doi.org/10.1161/atvbaha.107.141606
Becker RC, Mahaffey KW, Yang H, Marian AJ, Furman MI, Michael Lincoff A, Hazen SL, Petersen JL, Reist CJ, Kleiman NS (2011) Heparin-associated anti-Xa activity and platelet-derived prothrombotic and proinflammatory biomarkers in moderate to high-risk patients with acute coronary syndrome. J Thromb Thrombolysis 31(2):146–153. https://doi.org/10.1007/s11239-010-0532-y
Reitsma SE, Holle LA, Bouck EG, Monroe DM, Mast AE, Burthem J, Bolton-Maggs PHB, Gidley GN, Wolberg AS (2023) Tissue factor pathway inhibitor is a potential modifier of bleeding risk in factor XI deficiency. J Thromb Haemost 21(3):467–479. https://doi.org/10.1016/j.jtha.2022.10.005
Puy C, Rigg RA, McCarty OJ (2016) The hemostatic role of factor XI. Thromb Res 141 Suppl 2(Suppl 2):S8-s11. https://doi.org/10.1016/s0049-3848(16)30354-1
MacDonald S, White D, Langdown J, Downes K, Thomas W (2020) Investigation of patients with unclassified bleeding disorder and abnormal thrombin generation for physiological coagulation inhibitors reveals multiple abnormalities and a subset of patients with increased tissue factor pathway inhibitor activity. Int J Lab Hematol 42(3):246–255. https://doi.org/10.1111/ijlh.13155
Chelle P, Montmartin A, Damien P, Piot M, Cournil M, Lienhart A, Genre-Volot F, Chambost H, Morin C, Tardy-Poncet B (2019) Tissue factor pathway inhibitor is the main determinant of thrombin generation in haemophilic patients. Haemophilia 25(2):343–348. https://doi.org/10.1111/hae.13679
Dielis AW, Castoldi E, Spronk HM, van Oerle R, Hamulyák K, Ten Cate H, Rosing J (2008) Coagulation factors and the protein C system as determinants of thrombin generation in a normal population. J Thromb Haemost 6(1):125–131. https://doi.org/10.1111/j.1538-7836.2007.02824.x
Mast AE (2016) Tissue factor pathway inhibitor: multiple anticoagulant activities for a single protein. Arterioscler Thromb Vasc Biol 36(1):9–14. https://doi.org/10.1161/atvbaha.115.305996
Patel-Hett S, Martin EJ, Mohammed BM, Rakhe S, Sun P, Barrett JC, Nolte ME, Kuhn J, Pittman DD, Murphy JE, Brophy DF (2019) Marstacimab, a tissue factor pathway inhibitor neutralizing antibody, improves coagulation parameters of ex vivo dosed haemophilic blood and plasmas. Haemophilia 25(5):797–806. https://doi.org/10.1111/hae.13820
Peterson JA, Maroney SA, Mast AE (2016) Targeting TFPI for hemophilia treatment. Thromb Res 141 Suppl 2(Suppl 2):S28-30. https://doi.org/10.1016/s0049-3848(16)30359-0
Visser M, Heitmeier S, Ten Cate H, Spronk HMH (2020) Role of factor xia and plasma Kallikrein in arterial and venous thrombosis. Thromb Haemost 120(6):883–993. https://doi.org/10.1055/s-0040-1710013
Hugli TE, Müller-Eberhard HJ (1978) Anaphylatoxins: C3a and C5a. Adv Immunol 26:1–53. https://doi.org/10.1016/s0065-2776(08)60228-x
Sainz IM, Pixley RA, Colman RW (2007) Fifty years of research on the plasma kallikrein-kinin system: from protein structure and function to cell biology and in-vivo pathophysiology. Thromb Haemost 98(1):77–83
Pallares Robles A, Ten Cate V, Schulz A, Prochaska JH, Rapp S, Koeck T, Panova-Noeva M, Heitmeier S, Schwers S, Leineweber K, Seyfarth HJ, Opitz CF, Spronk H, Espinola-Klein C, Lackner KJ, Münzel T, Andrade-Navarro MA, Konstantinides SV, Ten Cate H, Wild PS (2022) Association of FXI activity with thrombo-inflammation, extracellular matrix, lipid metabolism and apoptosis in venous thrombosis. Sci Rep 12(1):9761. https://doi.org/10.1038/s41598-022-13174-5
Tucker EI, Verbout NG, Leung PY, Hurst S, McCarty OJ, Gailani D, Gruber A (2012) Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood 119(20):4762–4768. https://doi.org/10.1182/blood-2011-10-386185
Møller S, Bernardi M (2013) Interactions of the heart and the liver. Eur Heart J 34(36):2804–2811. https://doi.org/10.1093/eurheartj/eht246
Cao Y, Wang Y, Zhou Z, Pan C, Jiang L, Zhou Z, Meng Y, Charugundla S, Li T, Allayee H, Seldin MM, Lusis AJ (2022) Liver-heart cross-talk mediated by coagulation factor XI protects against heart failure. Science 377(6613):1399–1406. https://doi.org/10.1126/science.abn0910
Jordan KR, Wyatt CR, Fallon ME, Woltjer R, Neuwelt EA, Cheng Q, Gailani D, Lorentz C, Tucker EI, McCarty OJT, Hinds MT, Nguyen KP (2022) Pharmacological reduction of coagulation factor XI reduces macrophage accumulation and accelerates deep vein thrombosis resolution in a mouse model of venous thrombosis. J Thromb Haemost 20(9):2035–2045. https://doi.org/10.1111/jth.15777
Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U (2008) Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood 111(8):4113–4117. https://doi.org/10.1182/blood-2007-10-120139
Sharman Moser S, Chodick G, Ni YG, Chalothorn D, Wang MD, Shuldiner AR, Morton L, Salomon O, Jalbert JJ (2022) The association between factor XI deficiency and the risk of bleeding, cardiovascular, and venous thromboembolic events. Thromb Haemost 122(5):808–817. https://doi.org/10.1055/s-0041-1735971
Ngo ATP, Jordan KR, Mueller PA, Hagen MW, Reitsma SE, Puy C, Revenko AS, Lorentz CU, Tucker EI, Cheng Q, Hinds MT, Fazio S, Monia BP, Gailani D, Gruber A, Tavori H, McCarty OJT (2021) Pharmacological targeting of coagulation factor XI mitigates the development of experimental atherosclerosis in low-density lipoprotein receptor-deficient mice. J Thromb Haemost 19(4):1001–1017. https://doi.org/10.1111/jth.15236
Shnerb Ganor R, Harats D, Schiby G, Gailani D, Levkovitz H, Avivi C, Tamarin I, Shaish A, Salomon O (2016) Factor XI deficiency protects against atherogenesis in apolipoprotein e/factor XI double knockout mice. Arterioscler Thromb Vasc Biol 36(3):475–481. https://doi.org/10.1161/atvbaha.115.306954
Wang X, Smith PL, Hsu MY, Gailani D, Schumacher WA, Ogletree ML, Seiffert DA (2006) Effects of factor XI deficiency on ferric chloride-induced vena cava thrombosis in mice. J Thromb Haemost 4(9):1982–1988. https://doi.org/10.1111/j.1538-7836.2006.02093.x
Renné T, Oschatz C, Seifert S, Müller F, Antovic J, Karlman M, Benz PM (2009) Factor XI deficiency in animal models. J Thromb Haemost 7(Suppl 1):79–83. https://doi.org/10.1111/j.1538-7836.2009.03393.x
Walsh M, Bethune C, Smyth A, Tyrwhitt J, Jung SW, Yu RZ, Wang Y, Geary RS, Weitz J, Bhanot S (2022) Phase 2 study of the factor XI antisense inhibitor IONIS-FXI(Rx) in patients with ESRD. Kidney Int Rep 7(2):200–209. https://doi.org/10.1016/j.ekir.2021.11.011
Willmann S, Marostica E, Snelder N, Solms A, Jensen M, Lobmeyer M, Lensing AWA, Bethune C, Morgan E, Yu RZ, Wang Y, Jung SW, Geary R, Bhanot S (2021) PK/PD modeling of FXI antisense oligonucleotides to bridge the dose-FXI activity relation from healthy volunteers to end-stage renal disease patients. CPT Pharmacometrics Syst Pharmacol 10(8):890–901. https://doi.org/10.1002/psp4.12663
Weitz JI, Bauersachs R, Becker B, Berkowitz SD, Freitas MCS, Lassen MR, Metzig C, Raskob GE (2020) Effect of osocimab in preventing venous thromboembolism among patients undergoing knee arthroplasty: the FOXTROT randomized clinical trial. JAMA 323(2):130–139. https://doi.org/10.1001/jama.2019.20687
Verhamme P, Yi BA, Segers A, Salter J, Bloomfield D, Büller HR, Raskob GE, Weitz JI (2021) Abelacimab for prevention of venous thromboembolism. N Engl J Med 385(7):609–617. https://doi.org/10.1056/NEJMoa2105872
Lorentz CU, Tucker EI, Verbout NG, Shatzel JJ, Olson SR, Markway BD, Wallisch M, Ralle M, Hinds MT, McCarty OJT, Gailani D, Weitz JI, Gruber A (2021) The contact activation inhibitor AB023 in heparin-free hemodialysis: results of a randomized phase 2 clinical trial. Blood 138(22):2173–2184. https://doi.org/10.1182/blood.2021011725
Nowotny B, Thomas D, Schwers S, Wiegmann S, Prange W, Yassen A, Boxnick S (2022) First randomized evaluation of safety, pharmacodynamics, and pharmacokinetics of BAY 1831865, an antibody targeting coagulation factor XI and factor XIa, in healthy men. J Thromb Haemost 20(7):1684–1695. https://doi.org/10.1111/jth.15744
Tucker EI, Marzec UM, White TC, Hurst S, Rugonyi S, McCarty OJ, Gailani D, Gruber A, Hanson SR (2009) Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood 113(4):936–944. https://doi.org/10.1182/blood-2008-06-163675
Weitz JI, Strony J, Ageno W, Gailani D, Hylek EM, Lassen MR, Mahaffey KW, Notani RS, Roberts R, Segers A, Raskob GE (2021) Milvexian for the prevention of venous thromboembolism. N Engl J Med 385(23):2161–2172. https://doi.org/10.1056/NEJMoa2113194
Shoamanesh A, Mundl H, Smith EE, Masjuan J, Milanov I, Hirano T, Agafina A, Campbell B, Caso V, Mas JL, Dong Q, Turcani P, Christensen H, Ferro JM, Veltkamp R, Mikulik R, De Marchis GM, Robinson T, Lemmens R, ... Hart RG (2022) Factor XIa inhibition with asundexian after acute non-cardioembolic ischaemic stroke (PACIFIC-Stroke): an international, randomised, double-blind, placebo-controlled, phase 2b trial. Lancet 400(10357):997–1007. https://doi.org/10.1016/s0140-6736(22)01588-4
Beale D, Dennison J, Boyce M, Mazzo F, Honda N, Smith P, Bruce M (2021) ONO-7684 a novel oral FXIa inhibitor: safety, tolerability, pharmacokinetics and pharmacodynamics in a first-in-human study. Br J Clin Pharmacol 87(8):3177–3189. https://doi.org/10.1111/bcp.14732
Chen R, Guan X, Hu P, Dong Y, Zhu Y, Zhang T, Zou J, Zhang S (2022) First-in-human study to assess the safety, pharmacokinetics, and pharmacodynamics of SHR2285, a small-molecule factor XIa inhibitor in healthy subjects. Front Pharmacol 13:821363. https://doi.org/10.3389/fphar.2022.821363
Perera V, Luettgen JM, Wang Z, Frost CE, Yones C, Russo C, Lee J, Zhao Y, LaCreta FP, Ma X, Knabb RM, Seiffert D, DeSouza M, Mugnier P, Cirincione B, Ueno T, Frost RJA (2018) First-in-human study to assess the safety, pharmacokinetics and pharmacodynamics of BMS-962212, a direct, reversible, small molecule factor XIa inhibitor in non-Japanese and Japanese healthy subjects. Br J Clin Pharmacol 84(5):876–887. https://doi.org/10.1111/bcp.13520
Pollack CV Jr, Kurz MA, Hayward NJ (2020) EP-7041, a factor XIa inhibitor as a potential antithrombotic strategy in extracorporeal membrane oxygenation: a brief report. Crit Care Explor 2(9):e0196. https://doi.org/10.1097/cce.0000000000000196
Al-Horani RA, Ponnusamy P, Mehta AY, Gailani D, Desai UR (2013) Sulfated pentagalloylglucoside is a potent, allosteric, and selective inhibitor of factor XIa. J Med Chem 56(3):867–878. https://doi.org/10.1021/jm301338q
Decrem Y, Rath G, Blasioli V, Cauchie P, Robert S, Beaufays J, Frère JM, Feron O, Dogné JM, Dessy C, Vanhamme L, Godfroid E (2009) Ir-CPI, a coagulation contact phase inhibitor from the tick Ixodes ricinus, inhibits thrombus formation without impairing hemostasis. J Exp Med 206(11):2381–2395. https://doi.org/10.1084/jem.20091007
Chen W, Carvalho LP, Chan MY, Kini RM, Kang TS (2015) Fasxiator, a novel factor XIa inhibitor from snake venom, and its site-specific mutagenesis to improve potency and selectivity. J Thromb Haemost 13(2):248–261. https://doi.org/10.1111/jth.12797
Assumpção TC, Ma D, Mizurini DM, Kini RM, Ribeiro JM, Kotsyfakis M, Monteiro RQ, Francischetti IM (2016) In vitro mode of action and anti-thrombotic activity of boophilin, a multifunctional kunitz protease inhibitor from the midgut of a tick vector of babesiosis, rhipicephalus microplus. PLoS Negl Trop Dis 10(1):e0004298. https://doi.org/10.1371/journal.pntd.0004298
Ma D, Mizurini DM, Assumpção TC, Li Y, Qi Y, Kotsyfakis M, Ribeiro JM, Monteiro RQ, Francischetti IM (2013) Desmolaris, a novel factor XIa anticoagulant from the salivary gland of the vampire bat (Desmodus rotundus) inhibits inflammation and thrombosis in vivo. Blood 122(25):4094–4106. https://doi.org/10.1182/blood-2013-08-517474
Li D, He Q, Kang T, Yin H, Jin X, Li H, Gan W, Yang C, Hu J, Wu Y, Peng L (2010) Identification of an anticoagulant peptide that inhibits both fXIa and fVIIa/tissue factor from the blood-feeding nematode ancylostoma caninum. Biochem Biophys Res Commun 392(2):155–159. https://doi.org/10.1016/j.bbrc.2009.12.177
Donkor DA, Bhakta V, Eltringham-Smith LJ, Stafford AR, Weitz JI, Sheffield WP (2017) Selection and characterization of a DNA aptamer inhibiting coagulation factor XIa. Sci Rep 7(1):2102. https://doi.org/10.1038/s41598-017-02055-x
Woodruff RS, Ivanov I, Verhamme IM, Sun MF, Gailani D, Sullenger BA (2017) Generation and characterization of aptamers targeting factor XIa. Thromb Res 156:134–141. https://doi.org/10.1016/j.thromres.2017.06.015
Büller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE, Segers A, Verhamme P, Weitz JI (2015) Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med 372(3):232–240. https://doi.org/10.1056/NEJMoa1405760
Piccini JP, Caso V, Connolly SJ, Fox KAA, Oldgren J, Jones WS, Gorog DA, Durdil V, Viethen T, Neumann C, Mundl H, Patel MR (2022) Safety of the oral factor XIa inhibitor asundexian compared with apixaban in patients with atrial fibrillation (PACIFIC-AF): a multicentre, randomised, double-blind, double-dummy, dose-finding phase 2 study. Lancet 399(10333):1383–1390. https://doi.org/10.1016/s0140-6736(22)00456-1
Sharma M, Molina CA, Toyoda K, Bereczki D, Kasner SE, Lutsep HL, Tsivgoulis G, Ntaios G, Czlonkowska A, Shuaib A, Amarenco P, Endres M, Diener HC, Gailani D, Kahl A, Donovan M, Perera V, Li D, Hankey GJ (2022) Rationale and design of the AXIOMATIC-SSP phase II trial: antithrombotic treatment with factor XIa inhibition to optimize management of acute thromboembolic events for secondary stroke prevention. J Stroke Cerebrovasc Dis 31(10):106742. https://doi.org/10.1016/j.jstrokecerebrovasdis.2022.106742
Rao SV, Kirsch B, Bhatt DL, Budaj A, Coppolecchia R, Eikelboom J, James SK, Jones WS, Merkely B, Keller L, Hermanides RS, Campo G, Ferreiro JL, Shibasaki T, Mundl H, Alexander JH (2022) A multicenter, phase 2, randomized, placebo-controlled, double-blind, parallel-group, dose-finding trial of the oral factor XIa inhibitor asundexian to prevent adverse cardiovascular outcomes after acute myocardial infarction. Circulation 146(16):1196–1206. https://doi.org/10.1161/circulationaha.122.061612
Salomon O, Zivelin A, Livnat T, Seligsohn U (2006) Inhibitors to factor XI in patients with severe factor XI deficiency. Semin Hematol 43(1 Suppl 1):S10-12. https://doi.org/10.1053/j.seminhematol.2005.11.018
Collins PW, Goldman E, Lilley P, Pasi KJ, Lee CA (1995) Clinical experience of factor XI deficiency: the role of fresh frozen plasma and factor XI concentrate. Haemophilia 1(4):227–231. https://doi.org/10.1111/j.1365-2516.1995.tb00080.x
George DA, Stuckey DC (2010) Extraction of monoclonal antibodies (IgG1) using anionic and anionic/nonionic reverse micelles. Biotechnol Prog 26(5):1352–1360. https://doi.org/10.1002/btpr.453
Ling G, Kagdi H, Subel B, Chowdary P, Gomez K (2016) Safety and efficacy of factor XI (FXI) concentrate use in patients with FXI deficiency: a single-centre experience of 19 years. Haemophilia 22(3):411–418. https://doi.org/10.1111/hae.12868
Bolton-Maggs P, Goudemand J, Hermans C, Makris M, de Moerloose P (2014) FXI concentrate use and risk of thrombosis. Haemophilia 20(4):e349-351. https://doi.org/10.1111/hae.12457
Bauduer F, de Raucourt E, Boyer-Neumann C, Trossaert M, Beurrier P, Faradji A, Peynet J, Borg JY, Chamouni P, Chatelanaz C, Henriet C, Bridey F, Goudemand J (2015) Factor XI replacement for inherited factor XI deficiency in routine clinical practice: results of the HEMOLEVEN prospective 3-year postmarketing study. Haemophilia 21(4):481–489. https://doi.org/10.1111/hae.12655
Alsammak MS, Ashrani AA, Winters JL, Pruthi RK (2017) Therapeutic plasma exchange for perioperative management of patients with congenital factor XI deficiency. J Clin Apher 32(6):429–436. https://doi.org/10.1002/jca.21532
Liumbruno G, Bennardello F, Lattanzio A, Piccoli P, Rossetti G (2009) Recommendations for the transfusion of plasma and platelets. Blood Transfus 7(2):132–150. https://doi.org/10.2450/2009.0005-09
Sørensen B, Spahn DR, Innerhofer P, Spannagl M, Rossaint R (2011) Clinical review: prothrombin complex concentrates–evaluation of safety and thrombogenicity. Crit Care 15(1):201. https://doi.org/10.1186/cc9311
Hedner U (2006) Mechanism of action, development and clinical experience of recombinant FVIIa. J Biotechnol 124(4):747–757. https://doi.org/10.1016/j.jbiotec.2006.03.042
Hoffman M, Monroe DM 3rd, Roberts HR (1998) Activated factor VII activates factors IX and X on the surface of activated platelets: thoughts on the mechanism of action of high-dose activated factor VII. Blood Coagul Fibrinolysis 9(Suppl 1):S61-65
Monroe DM (2008) Further understanding of recombinant activated factor VII mode of action. Semin Hematol 45(2 Suppl 1):S7-s11. https://doi.org/10.1053/j.seminhematol.2008.03.013
Duga S, Salomon O (2013) Congenital factor XI deficiency: an update. Semin Thromb Hemost 39(6):621–631. https://doi.org/10.1055/s-0033-1353420
Shapiro AD (2021) Concizumab: a novel anti-TFPI therapeutic for hemophilia. Blood Adv 5(1):279. https://doi.org/10.1182/bloodadvances.2019001140
Knight T, Callaghan MU (2018) The role of emicizumab, a bispecific factor IXa- and factor X-directed antibody, for the prevention of bleeding episodes in patients with hemophilia A. Ther Adv Hematol 9(10):319–334. https://doi.org/10.1177/2040620718799997
Barg AA, Budnik I, Avishai E, Brutman-Barazani T, Bashari D, Misgav M, Lubetsky A, Kuperman AA, Livnat T, Kenet G (2021) Emicizumab prophylaxis: prospective longitudinal real-world follow-up and monitoring. Haemophilia 27(3):383–391. https://doi.org/10.1111/hae.14318
Gelbenegger G, Schoergenhofer C, Knoebl P, Jilma B (2020) Bridging the missing link with emicizumab: a bispecific antibody for treatment of hemophilia A. Thromb Haemost 120(10):1357–1370. https://doi.org/10.1055/s-0040-1714279
Becker RC (1991) Seminars in thrombosis, thrombolysis and vascular biology. 4. Fibrinolysis. Cardiology 79(3):188–210. https://doi.org/10.1159/000174879
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ali, A.E., Becker, R.C. The foundation for investigating factor XI as a target for inhibition in human cardiovascular disease. J Thromb Thrombolysis (2024). https://doi.org/10.1007/s11239-024-02985-0
Accepted:
Published:
DOI: https://doi.org/10.1007/s11239-024-02985-0