
Abstract
The human pathogen Helicobacter pylori infects more than half of the world’s population and is a paradigm for persistent yet asymptomatic infection but increases the risk for chronic gastritis and gastric adenocarcinoma. For successful colonization, H. pylori needs to subvert the host cell death response, which serves to confine pathogen infection by killing infected cells and preventing malignant transformation. Infection of gastric epithelial cells by H. pylori provokes direct and fast activation of the proinflammatory and survival factor NF-κB, which regulates target genes, such as CXCL8, BIRC3 and TNFAIP3. However, it is not known how H. pylori exploits NF-κB activation and suppresses the inflammatory response and host apoptotic cell death, in order to avert the innate immune response and avoid cell loss, and thereby enhance colonization to establish long-term infection. Here we assign for the first time that H. pylori and also Campylobacter jejuni-induced ubiquitin-editing enzyme A20 bifunctionally terminates NF-κB activity and negatively regulates apoptotic cell death. Mechanistically, we show that the deubiquitinylase activity of A20 counteracts cullin3-mediated K63-linked ubiquitinylation of procaspase-8, therefore restricting the activity of caspase-8. Interestingly, another inducible NF-κB target gene, the scaffold protein p62, ameliorates the interaction of A20 with procaspase-8. In conclusion, pathogen-induced de novo synthesis of A20 regulates the shut-off of the survival factor NF-κB but, on the other hand, also impedes caspase-8-dependent apoptotic cell death so as to promote the persistence of pathogens.
Similar content being viewed by others


Main
Helicobacter pylori is a Gram-negative bacterium that colonizes the human gastric mucosa.1 A balance between moderate H. pylori colonization and mild host responses leads in most of the cases to an unrecognized long-term infection within a healthy individual.2 On the other hand, H. pylori prevalence varies remarkably between and within populations based on the role of socioeconomic-related environmental factors. Thus, in a number of cases, persistent infection by H. pylori contributes to a plethora of gastrointestinal diseases, for instance, chronic active gastritis and gastric ulcers, and represents a major risk factor for the development of gastric cancer.3 In addition, bacterial virulence factors such as the cytotoxin-associated gene pathogenicity island-encoded protein CagA, which is translocated into the host cell via the bacterial type IV secretion system (T4SS), and the vacuolating cytotoxin VacA have been implicated in the severity of gastric malignancies.4
The ubiquitous transcription factor NF-κB affects multiple physiological processes of which the regulation of immune and inflammatory responses is the most conserved among different species. The mammalian NF-κB family consists of five members, RelA, RelB, c-Rel, NFKB1 (p105/p50) and NFKB2 (p100/p52), which form various homodimeric and heterodimeric NF-κB complexes. These remain sequestered, hence inactive, in the cytoplasm by the inhibitors of NF-κB (IκBs). Upon H. pylori infection, the IKK complex becomes activated by phosphorylation involving the kinases TAK1 and MEKK3.5 Thereafter, the activated IKK complex phosphorylates IκBα leading to its proteasomal degradation and the subsequent nuclear translocation of the NF-κB complex. NF-κB is due to its gene-regulatory capacity, a cell survival factor that could contribute to cancer development.6
Regulated cell death is the execution of an intracellular programme to maintain cellular homeodynamics. In recent years, the view on regulated cell death was broadened to include many different modes, such as autophagy, apoptosis, necroptosis, mitotic catastrophe and pyroptosis.7 The best-studied cell death programme is apoptosis. An apoptotic signal can either be induced extrinsically by death receptors such as CD95 or TRAIL-R1/R2 or intrinsically via the mitochondria. Stimulation by the respective death ligand results in the formation of a death-inducing signalling complex (DISC), where procaspase-8 is activated leading to induction of the caspase cascade followed by the demolition of the cell. At the DISC, procaspase-8 is activated by dimerization in the death effector domain chains followed by internal cleavage. Procaspase-8a/b (p55/p53) processing at the DISC results in generation of the N-terminal cleavage products p43/p41, the prodomains p26/p24, as well as the C-terminal cleavage products p30, p18 and p10. The active caspase-8 heterotetramer p10/p18 generated at the DISC initiates apoptosis.8
In response to H. pylori infection, gastric epithelial cells have been reported to show moderate apoptosis. Both VacA as well as a membrane-associated protein γ-glutamyl transpeptidase have been implicated in triggering the intrinsic apoptotic pathway.9, 10 Interestingly, CagA has been described to counteract the apoptosis-inducing ability of VacA.11 On the other hand, induction of the extrinsic apoptotic pathway by H. pylori has also been demonstrated that was attributed to the upregulation of CD95L, TRAIL and their respective receptors.12, 13 These studies collectively indicate that the host cell, when infected with H. pylori, can execute both extrinsic and intrinsic pathways to effect apoptotic cell death.
A20 (TNFAIP3) is an immediate early target gene of NF-κB that is involved in the termination of NF-κB activation as part of a negative feedback loop.14 The deubiquitinylase (DUB) activity in the ovarian tumour domain as well as the ubiquitin E3 ligase activity in the fourth zinc finger of A20 are important for this role. However, A20 is also able to inhibit the activation of NF-κB in a non-catalytic manner by interacting directly with NEMO via polyubiquitin chains and thus preventing the activating phosphorylation of IKKβ by its upstream kinase TAK1.15 Furthermore, A20 is also implicated in the regulation of apoptosis.16, 17, 18 Lately, a role of A20 in other modes of regulated cell death, such as necroptosis and autophagy, has been proposed.19, 20
p62 (sequestosome-1) is a ubiquitin-binding protein serving as a scaffold/adaptor molecule that is transcriptionally regulated by NF-κB.21, 22 Studies have demonstrated the involvement of p62 in NF-κB activation by IL-1β and mTORC1 activation as well as autophagy.23, 24, 25 More recently, the contribution of p62 to the anti-inflammatory capabilities of NF-κB by limiting the NLRP3-inflammasome activation has been reported.26
The activation of NF-κB in H. pylori infection is a well-studied process, but to date, the molecular crosstalk between NF-κB signalling and the host apoptotic cell death is unclear. The present study was undertaken to address the role of the NF-κB target gene A20 in H. pylori infection in gastric epithelial cells with emphasis on the host apoptotic cell death. We provide evidence, using CRISPR/Cas9 A20 knockout cells, that A20 mitigates not only the proinflammatory NF-κB but also the apoptotic response of the host cell to bacterial pathogens. We also show that the inhibition of apoptotic cell death involves the DUB activity of A20 and the scaffold protein p62 to facilitate effective ubiquitin editing of procaspase-8.
Results
The ubiquitin-editing enzyme A20 suppresses H. pylori T4SS-dependent and CagA-independent NF-κB activation
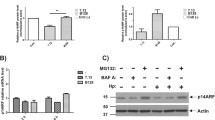
Our previous work showed that H. pylori induces NF-κB in a T4SS-dependent and CagA-independent manner.27, 28 Immunoblot (IB) analysis of the expression of the ubiquitin-editing enzyme A20 revealed an increase in A20 expression within 4 h followed by a gradual decrease up to 24 h in cells infected with H. pylori (Figure 1a). This was preceded by the fast activation of NF-κB as indicated by the phosphorylation and degradation of IκBα and subsequent phosphorylation of RelA (Figure 1a). It is known that the A20 gene promoter can also be regulated by other transcription factors, including C/EBPβ and USF1.29, 30 In order to demonstrate the dependency of A20 gene regulation on NF-κB activation, we examined A20 expression in cells treated with an IKK inhibitor prior to infection. Herein, inhibition of NF-κB led to a suppression of A20 mRNA and protein expression (Figures 1b and c). In addition, A20 expression was induced by H. pylori wt and CagA-deficient but not T4SS-deficient (virB7) isogenic strains (Figure 1d). The inhibitory role of A20 in TNF-stimulated NF-κB activation has been investigated previously.14, 15 To study the impact of A20 on NF-κB regulation in H. pylori-infected cells, we depleted A20 using siRNA as well as generated A20-knockout cells using the CRISPR/Cas9 system. Unless stated otherwise, parent AGS cells and A20-knockout AGS cells will be referred to as wt and A20KO cells, respectively. We observed enhanced NF-κB activation in siRNA-transfected cells (Figure 1e and Supplementary Figure S1a) and A20KO cells (Figure 1f and Supplementary Figure S1b). The same effect was also observed for another human pathogen, namely Campylobacter jejuni (Figure 1g), and TNF stimulation (Figure 1h). In addition, NF-κB-dependent transactivation of luciferase activity was increased in the A20KO cells in response to H. pylori infection as well as TNF stimulation (Figure 1i). Accordingly, H. pylori-infected A20KO cells also showed higher transcript levels of NF-κB target genes CXCL8 and BIRC3 (Figure 1j). In A20KO cells reconstituted with A20, attenuated NF-κB activation was observed in comparison to control A20KO cells (empty vector only), thereby confirming the inhibitory effect of A20 on H. pylori-induced NF-κB activation (Figure 1k).

A20 upregulation after H. pylori-induced NF-κB activation in gastric epithelial cells. (a) AGS cells were infected with H. pylori for the indicated times. Cell lysates were subjected to IB for analysis of proteins involved in NF-κB activation. (b) Total RNA was isolated after H. pylori infection with and without IKK inhibitor. Changes in A20 transcript (TNFAIP3) expression were investigated by quantitative PCR. Data shown depict the average of triplicate determinations. (c) Cell lysates were harvested after H. pylori infection in the absence or presence of IKK inhibitor for the indicated times and analysed in IB. (d) Cells were infected with different isogenic H. pylori strains wt, virB7 or cagA followed by IB analysis. GAPDH served as a loading control. (e-i) The effect of A20 depletion by siRNA transfection (e) or CRISPR/Cas9-mediated knockout (f-i) on the activity of NF-κB upon H. pylori infection (e, f), C. jejuni infection (g) or TNF stimulation (h) was analysed by IB (e-h) or transactivation assay (i). Representative IBs from at least two independent experiments with similar outcomes are shown. The transactivation assay was performed at least five times. Error bars, S.D., *P<0.02, #P<0.05. (j) Wt and A20KO_1 cells were infected with H. pylori for 3 h. Total RNA was isolated and changes in the level of NF-κB target genes CXCL8 and BIRC3 were determined by quantitative PCR. Data shown depict the average of triplicate determinations. (k) A20KO_1 cells were transfected with either pCMV (as empty vector control) or full-length active A20 (pA20) plasmids 24 h prior to H. pylori infection for the times shown. Cell lysates were harvested and analysed by IB
A20 impedes apoptotic cell death in pathogen-infected cells
Compelling evidence exists for the involvement of H. pylori and the survival factor NF-κB in the development of gastric cancer.31, 32, 33 Further, H. pylori infection is accompanied by moderate apoptosis in different cell lines.34, 35 Thus we studied the impact of NF-κB activity on apoptotic cell death in H. pylori-infected cells. Infection by H. pylori and treatment with CD95L showed apoptotic cell death, contrary to IL-1β and TNF stimulation (Figure 2a), as indicated by the detection of active caspase-8 and -3. This finding was supported by imaging flow cytometric analysis showing an increase in cells positively stained for cleaved caspase-3 after infection (Figure 2b). We also observed the onset of apoptosis in infected MKN-45 cells, thus excluding cell line-specific effects (Figure 2c). Furthermore, we observed enhanced processing of procaspase-8 and -3 in infected A20KO cells (Figure 2d and Supplementary Figure S2a). When A20 was reintroduced into the A20KO cells by overexpression, infection by H. pylori led to less cleaved caspase-8 and -3 (Figure 2e and Supplementary Figure S2b). Our results therefore strongly implicate that, in part, apoptotic cell death in H. pylori infection involves a regulation by A20. The processing of procaspase-8 and -3 was blocked by the addition of procaspase-8-specific inhibitor Z-IETD-FMK in a concentration-dependent manner (Figure 2f), indicating a caspase-dependent mechanism for the observed cell death. This was substantiated by the Annexin V/PI staining, which showed a decrease in the percentage of apoptotic cells in wt (15–9% cell death) and A20KO cells (22–10% cell death) upon inhibition of procaspase-8 (Figure 2g). Herein, 20 μM of Z-IETD-FMK had a stronger impact on the apoptotic cell death induced in A20KO cells as compared with the wild-type cells (wt 38% reduction versus A20KO 52% reduction, Figure 2g), suggesting that A20 affects procaspase-8-dependent apoptotic cell death. Our findings imply that a strong and sustained activity of the survival factor NF-κB in the absence of A20 is not capable to suppress the apoptotic cell death. To investigate whether this observation is specific for H. pylori, we studied infection with C. jejuni, which also showed increased activation of NF-κB in A20KO cells. C. jejuni infection elicited enhanced processing of procaspase-8 to its active p18 fragment in A20KO cells (Figure 2h). Our results demonstrate that A20 suppresses apoptotic cell death in cells infected with different pathogens, which suggests a specific role of A20 in attenuating host cell survival apart from the termination of NF-κB activity.

A20 impedes apoptotic cell death in pathogen-infected cells. (a) AGS cells were infected with H. pylori or treated with different stimuli for the times indicated followed by IB analysis of procaspase-8 and -3 cleavage. (b) Procaspase-3 activation was determined by imaging flow cytometry of immunostained cleaved caspase-3 (n=3 for H. pylori infection). Treatment with staurosporine (ST, final concentration 1 μM) served as a positive control. (c) MKN-45 cells were infected with H. pylori and analysed by Annexin V/PI staining using imaging flow cytometry. Error bar, S.D.; *P<0.005; **P<0.0005. (d) Wt and A20KO_1 cells were infected with H. pylori for the indicated time points before analysis by IB. (e) A20KO_1 cells were transfected with either pCMV (as empty vector control) or full-length active A20 (pA20) plasmids 24 h prior to H. pylori infection for a further 24 h. Cell lysates were harvested and analysed by IB. Wt and A20KO_1 cells were pretreated with Z-IETD-FMK for 15 min followed by H. pylori infection for 24 h before analysis by IB (f) or Annexin V/PI staining using imaging flow cytometry (g). Representative images and dot plots for the cellular staining are shown. Percentage of apoptotic cells represents the sum of annexin V-positive and double positive cells. Error bar, S.D., *P<0.005; **P<0.0005. The concentrations of Z-IETD-FMK used were 10 μM and 20 μM for panel (f) and 20 μM for panel (g). (h) Wt and A20KO_1 cells were infected with C. jejuni followed by IB analysis of procaspase-8 and -3 cleavage. Representative IBs from two independent experiments are shown
DUB activity of A20 counteracts efficient procaspase-8 activation by removal of K63-linked polyubiquitin in H. pylori infection
Thus far, our findings implicated a function of A20 in the regulation of procaspase-8 activation in infected cells. Next we investigated the interaction between endogenous A20 and procaspase-8. In co-immunoprecipitation (co-IP) studies, we detected A20 interaction with procaspase-8, which was inducible upon H. pylori infection (Figure 3a). In vitro translation using FLAG-tagged A20 and HA-tagged procaspase-8 revealed a direct interaction between A20 and procaspase-8 (Figure 3b).

The DUB activity of A20 limits procaspase-8 activation by removing K63-linked polyubiquitin upon H. pylori infection. (a and b) AGS cells were infected with H. pylori and a caspase-8 co-IP was performed (a) or in vitro translated FLAG-A20 and HA-caspase-8 were co-incubated followed by IPs with the respective anti-FLAG or anti-HA antibody (b) and analysed by IB. In IgG, an isotype-matched antibody was used. (c–e) Wt and A20KO_1 cells (c) or wt cells treated with 10 μM Z-IETD-FMK (d) or A20KO_1 cells complemented with pCMV (empty vector control), pC103A (A20 DUB mutant), pC624/627A (A20 E3 ligase mutant) or pA20 (full-length active A20) by transfection (e) were infected with H. pylori and caspase-8 IP under denaturing conditions was performed and analysed in IB for endogenous K63-linked polyubiquitin (c–e). Representative IBs from two independent experiments are shown
During apoptotic cell death, the K63-linked polyubiquitinylation of procaspase-8 supported its full activation and processing.16 In the presence of H. pylori, we observed an increase in the endogenous K63-linked polyubiquitinylation of procaspase-8 when we performed caspase-8 IP under denaturing conditions, an effect that was even more pronounced in A20KO cells (Figure 3c and Supplementary Figure S3a). Notably, we also observed an apparent decrease in the endogenous procaspase-8 polyubiquitinylation from 9 to 24 h after infection, which corresponded to an increase in the signal of cleaved caspase-8 (Figure 3c). We hypothesized that the decrease in K63-linked polyubiquitinylation is due to the processing of polyubiquitinylated procaspase-8 to its active fragment, thus reducing the pool of polyubiquitinylated procaspase-8. This hypothesis was supported when we observed an increase in the K63-linked polyubiquitinylation of procaspase-8 in infected cells co-incubated with Z-IETD-FMK (Figure 3d). Collectively, our data imply that the DUB activity of A20 could be responsible for removing the K63-linked polyubiquitin from procaspase-8, leading to a decrease in caspase-8 activity. We then performed complementation experiments by overexpressing full-length active A20 (pA20), A20 DUB mutant (pC103A) or A20 E3 ligase mutant (pC624/627A) in A20KO cells followed by caspase-8 IP under denaturing conditions. Infection with H. pylori led to an increase in the K63-linked polyubiquitinylation of procaspase-8 in empty vector control A20KO cells as well as A20KO cells, which overexpressed the A20 DUB mutant, and only a marginal K63-linked polyubiquitin signal was observed in A20KO cells reconstituted with the A20 E3 ligase mutant or active A20 (Figure 3e and Supplementary Figure S3b), confirming our hypothesis that the DUB activity of A20 is important for the ubiquitin editing of procaspase-8. In accordance with a previous report, we investigated whether the cullin3 (Cul3) RING-based E3 ligase is responsible for the polyubiquitinylation of procaspase-8.16 Cul3 co-IP revealed an interaction with A20 and procaspase-8 (Figure 4a). Moreover, the K63-linked polyubiquitinylation of procaspase-8 was abolished in Cul3-suppressed cells after infection, suggesting that Cul3 is responsible for ligating K63-linked polyubiquitin to procaspase-8 upon H. pylori infection (Figure 4b and Supplementary Figure S4a), as has been shown before in TRAIL-treated cells by Jin et al.16 Further analysis using Annexin V/PI staining showed that Cul3-knockdown cells were less apoptotic in comparison to mock- or scrambled siRNA-transfected cells after infection (Figure 4c and Supplementary Figure S4b), hence associating the K63-linked polyubiquitinylation of procaspase-8 directly to the efficiency of the induction of apoptotic cell death by H. pylori.

Depletion of Cul3 affects the apoptotic cell death of cells infected with H. pylori. (a) AGS cells were infected with H. pylori and subjected to a Cul3 co-IP followed by IB analysis of interacting proteins. (b) Cul3-depleted cells by siRNA transfection were infected with H. pylori. Caspase-8 IP under denaturing conditions was performed and the K63-linked ubiquitinylation status of procaspase-8 was analysed in IB. Representative IBs from two experiments are shown. (c) The impact of Cul3 depletion on the percentage of apoptotic cells was evaluated using Annexin V/PI staining followed by imaging flow cytometry. Representative images and dot plots for the cellular staining are shown. Percentage of apoptotic cells represents the sum of annexin V-positive and double positive cells. Error bar, S.D., *P<0.01; **P<0.005
Scaffold protein p62 enables A20 to more efficiently remove polyubiquitin from procaspase-8 in H. pylori-infected cells
Having shown that the DUB activity of A20 removes polyubiquitin from procaspase-8, we were interested whether other proteins apart from A20 contribute to procaspase-8 activation. Herein, an NF-κB target gene, p62 has been described to be a polyubiquitin-binding protein serving as a scaffold or molecular bridge,22 and based on this function, it mediates the aggregation of ubiquitinylated procaspase-8.16 Similar to A20, p62 gene regulation was induced by H. pylori (Figure 5a). Co-IPs using A20 antibody detected an interaction with procaspase-8 and p62 (Figure 5b). Furthermore, infection of cells followed by caspase-8 co-IP and IB analysis revealed not only an interaction with both A20 and p62 but also that the suppression of p62 by siRNA led to lesser interaction of A20 with procaspase-8 (Figure 5c and Supplementary Figure S5a). Interestingly, p62 could be co-immunoprecipitated with A20 in the absence of procaspase-8, suggesting that this interaction is not mediated by ubiquitin (Figure 5d and Supplementary Figure S5b). Hence, our data imply that the ability of A20 to remove K63-linked polyubiquitin from procaspase-8 is dependent on the presence of p62. Indeed, our further investigation showed an increase in the signal of processed procaspase-8 (Figure 5e and Supplementary Figure S5c) and increased signal for K63-linked polyubiquitinylated procaspase-8 (Figure 5f and Supplementary Figure S5d) in p62-depleted cells after infection. Taken together, our observations suggest that p62 promotes efficient interaction of A20 with procaspase-8 to remove K63-linked polyubiquitin chains from procaspase-8 in pathogen infection.

p62 enhances interaction between A20 and procaspase-8 for effective editing of K63-linked polyubiquitinylation of procaspase-8. AGS cells were infected with H. pylori followed by IB analysis of p62 expression (a) or co-IP with anti-A20 antibody was performed and analysed by IB for interacting proteins (b). (c) p62-depleted cells by siRNA transfection were infected with H. pylori. Procaspase-8 co-IP was performed and analysed by IB for interacting proteins. (d) Caspase-8-depleted cells by siRNA transfection were infected with H. pylori. A20 co-IP was performed and analysed by IB for interacting proteins. (e and f) The effect of p62 depletion by siRNA transfection on procaspase-8 processing (e) and K63-linked ubiquitinylation of procaspase-8 after caspase-8 IP under denaturing conditions (f) was analysed by IB. Representative IBs from two independent experiments are shown. (g) Schematic representation of the findings in this study. Infection with H. pylori leads to the activation of NF-κB (1), which in turn upregulates A20 (2). A20 fulfills two opposing functions, one that inhibits further activation of NF-κB (3), thus providing a negative feedback loop and the other, supported by p62, removes K63-linked polyubiquitin from procaspase-8 (4) to negatively regulate the apoptotic response of the host cell
Discussion
In the present study, we provide direct evidence that the NF-κB-regulated and de novo synthesized ubiquitin-editing enzyme A20 suppresses caspase-8 activity and apoptotic cell death in H. pylori-infected cells. We observed an interaction between procaspase-8 and the E3 ligase Cul3, which catalyses the conjugation of K63-linked ubiquitin to procaspase-8. The A20’s DUB activity reverses the K63-linked ubiquitinylation of procaspase-8, a process that is supported by the novel functional complex of procaspase-8/p62/A20.
Our results indicate that as one of the consequences, infection by H. pylori causes host apoptotic cell death via the extrinsic apoptotic pathway (Figures 2f and g), which is attributed to the upregulation of CD95L, TRAIL and their respective receptors.12, 13 Although apoptosis of host cells is beneficial for the bacterium as it provides a source of nutrients, H. pylori also needs to maintain the integrity of the gastric epithelium for colonization.36 To this end, infection activates NF-κB signalling, leading in most cases to a placid inflammatory response. Our data (Figures 1e, f and 2d) imply that NF-κB-induced A20 inhibits NF-κB activation and subsequently attenuates the host cell apoptotic response. These two seemingly disparage effects of A20 highlight a novel mechanism how H. pylori benefits from the host cellular response. We have made similar observations in C. jejuni-infected cells (Figure 2h), suggesting that other pathogens could also exploit the host cellular response to their own advantage in a similar manner. This is in our opinion an important feature of pathogen infection as it contributes to the ability of the pathogen to persist in a hostile environment. Interestingly, an earlier study has reported that cIAP2, another NF-κB target gene, also prevents apoptosis in H. pylori infection.37 In addition, previous studies have shown that pathogens could capitalize on the ability of A20 to inhibit NF-κB signalling in order to subvert the host immune response. Leishmania donovani, Mycobacterium fortuitum and the measles virus, for instance, suppress the TLR-mediated response by upregulating A20 and therefore inhibiting the activity of NF-κB.38, 39, 40
We demonstrated here that the Cul3-dependent K63-linked polyubiquitinylation of procaspase-8 (Figure 4b) was removed by the DUB activity of A20 (Figure 3e), affirming that A20 has a protective function against apoptotic cell death in response to H. pylori infection. NF-κB activation in infected cells also resulted in the upregulation of p62 (Figure 5a), which also takes part in the autophagy of microbial pathogens.41 Jin et al.16 have elucidated the role of p62 as a scaffold protein to support effective processing of procaspase-8 after Cul3-dependent ubiquitinylation in TRAIL-stimulated cells. We report here that, in the context of pathogen-induced apoptotic cell death, the presence of p62 favours interaction between A20 and procaspase-8 (Figure 5c), therefore enabling A20 to remove K63-linked polyubiquitin from procaspase-8 (Figure 5f). Additionally, we also presented data showing that the interaction of p62 and A20 is independent of ubiquitinylated procaspase-8 (Figure 5d), suggesting that p62 could serve as an adaptor between procaspase-8 and A20. This result is supported by the well-known feature of A20 to recruit adaptor molecules in order to fulfill its function.42, 43 In fact, it could be that p62 exerts different functions with respect to the apoptotic cell death in pathogen infection as compared with TRAIL, that is, that different triggers could affect the balance between ubiquitinylation and deubiquitinylation of procaspase-8. Whether other A20 adaptor molecules such as ABINs are also involved in pathogen-infected cells remains to be elucidated.44
In conclusion, we showed that H. pylori infection induces the expression of NF-κB-regulated target genes A20 and p62. A20 negatively impacts NF-κB activity and subsequently promotes cell survival due to the suppression of caspase-8 activity by forming a complex with p62 and procaspase-8 (Figure 5g). Our study highlights the complexity of the interplay between NF-κB signalling and apoptotic cell death due to the involvement of A20, which has distinct functions in the regulation of each of the two processes. The intriguing question remains how the host cellular response is influenced by other factors such that it favours infection by the pathogen in question. This warrants further investigation as other molecules, such as RIP1 and TRADD, exist that are also involved in both the NF-κB and cell death pathways and deeper insight into the complex regulation of host responses towards persistent pathogen infections will provide means of intervention to contain the infection.
Materials and methods
Cell culture
AGS (ATCC CRL-1739) and MKN-45 cells (DSMZ ACC 409) were routinely cultivated in RPMI-1640 medium supplemented with glutamine and 10 or 20% FCS, respectively, and incubated at 37 °C in a 5% CO2 humidified incubator. Cells were seeded at a density of 3.5 × 105 per 60 mm culture dish or 1.4 × 106 per 100 mm culture dish for H. pylori or C. jejuni infection (MOI 100), TNF (10 ng/ml) (PeproTech, Hamburg, Germany), CD95L (250 nM) (PeproTech) or staurosporine (1 μM) (Enzo Life Sciences GmbH, Lörach, Germany) treatment. Cell culture medium was changed to fresh RPMI-1640 containing 10% FCS 24 h prior to treatment of the cells. H. pylori wt (P1 strain), isogenic strains cagA (CagA-deficient) and virB7 (T4SS-deficient) were cultivated under microaerophilic conditions at 37 °C on agar plates containing 10% horse serum and vancomycin for wt and vancomycin/streptomycin for cagA and virB7 for 72 h. Bacteria was replated and cultivated for further 48 h before use for experiments. C. jejuni was cultivated on Campylobacter blood-free selective Agar Base (Oxoid, Germany, Munich) containing Campylobacter growth supplement (Oxoid) 48 h prior to infection. The procaspase-8-specific inhibitor Z-IETD-FMK and the IKK inhibitor VII used in some experiments were purchased from VWR (Darmstadt, Germany) and Merck (Germany, Darmstadt), respectively. Cells were preincubated with Z-IETD-FMK (10 μM or 20 μM) or IKK inhibitor VII (10 μM) for 15 min prior to H. pylori infection.
Transfection of siRNAs and plasmids
Twenty-four hours prior to siRNA transfections, AGS cells were seeded at a density of 1.5 × 105 per 60 mm culture dish. Transfection was performed using the jetPRIME Kit (VWR, Germany) according to the manufacturer’s protocol. Cell culture medium was changed to fresh RPMI-1640 containing 10% FCS prior to transfection. The siRNAs were used at a final concentration of 40 nM for A20, 40 nM or 20 nM for procaspase-8, 20 nM for Cul3 and 10 nM for p62. For plasmid transfection, cells were seeded at a density of 1.4 × 106 per 100 mm dish 24 h prior to transfection. The amount of plasmid used per 100 mm dish was 2 μg. Plasmid transfection was performed using Effectene transfection reagent (Qiagen, Hilden, Germany) according to the suggested protocol from the manufacturer for the 100 mm dish format. The siRNAs and plasmids were transfected into the cells for 24 and 6 h, respectively, before the cell culture medium was changed to fresh RPMI-1640 medium containing 10% FCS followed by further incubation for 18–24 h. Mock control are cells treated with only the transfection reagent. For the siRNA-negative control, scrambled siRNA (scr) (AllStars, Qiagen) was used. The following siRNAs were used: A20si5 (J-009919-05-0005) was from Dharmacon (Lafayette, CO, USA). A20si9 (SI05018601) and Casp8si11 (SI02661946) were from Qiagen. p62siB 5′-GGACCCAUCUGUCUUCAAAdTdT-3′, p62siC 5′-GCAUUGAAGUUGAUAUCGAdTdT-3′, Cul3siA 5′-GAGAAGAUGUACUAAAUUCdTdT-3′, Cul3siD 5′-GAGAUCAAGUUGUACGUUAdTdT-3′ and Casp8si1 5′-GGAGCUGCUCUUCCGAAUU-3′ were from Eurofins Genomics (Ebersberg, Germany).
SDS-PAGE and Western blotting
Cells were lysed for 15 min on ice in lysis buffer (50 mM Tris/HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, 10 mM K2HPO4, 10% glycerol, 1% Triton X-100 and 0.5% NP-40) containing phosphatase inhibitors (1 mM sodium vanadate, 1 mM sodium molybdate, 20 mM sodium fluoride, 10 mM sodium pyrophosphate, 1 mM AEBSF and 20 mM 2-phosphoglycerate) and protease inhibitor mix (complete, EDTA-free, Roche, Mannheim, Germany). Cell lysates were obtained after centrifugation at 13 000 r.p.m. for 10 min at 4 °C. Protein concentration was estimated using the BCA Protein Assay Kit (Thermo Fisher Scientific, Darmstadt, Germany). SDS-PAGE was performed in Tris-Glycine buffer gels and transferred onto PVDF membranes (Merck, Darmstadt, Germany) and blocked for 1 h at room temperature using 5% skim milk in TBS containing 0.1% Tween (TBS-T). The primary antibodies were incubated overnight in either 5% BSA or 5% skim milk in TBS-T at 4 °C. The membranes were washed thrice in TBS-T and incubated with the appropriate HRP-conjugated secondary antibody for 1 h at room temperature at a dilution of 1:5000 in 5% skim milk in TBS-T, followed by three washes in TBS-T. The membranes were developed using a chemiluminescent substrate (Millipore). The band pattern was visualized using the ChemoCam Imager (Intas, Göttingen, Germany).
Antibodies used in this work were as follows: phospho-RelA (#3031), phospho-IκBα (#9246), IκBα (#4812), caspase-8 (#9746), caspase-3 (#9692), and cleaved caspase-3 (#9661) were from Cell Signaling Technology (Beverly, MA, USA). FLAG-tag (F3165) was from Sigma-Aldrich (Taufkirchen, Germany). A20 (sc-166692) for IB or IP, caspase-8 (sc-6136 or sc-393776) for IP and HA-probe (sc-805) were from Santa Cruz (Santa Cruz, TX, USA). A20 (IMG-161A) for IB use was from Imgenex (San Diego, CA, USA). GAPDH (MAB374) and K63-polyubiquitin (05-1308) were from Millipore. K63-polyubiquitin (clone HWA4C4) was from BioLegend (San Diego, CA, USA). Flagellin was from Acris Antibodies (Herford, Germany). CagA (HPM-5001-5) was from Austral Biologicals (San Ramon, CA, USA). p62 (MBL-M162-3) was from BIOZOL (Eching, Germany). Cullin-3 (611848) was from BD Transduction Laboratories (Heidelberg, Germany). The secondary anti-rabbit-HRP or anti-mouse-HRP antibodies were from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA, USA).
Immunoprecipitation
Cells were detached by trypsin, washed once with PBS and lysed for 15 min on ice in IP buffer (30 mM Tris/HCl pH 7.5, 150 mM NaCl, 10% glycerol and 1% Triton X-100) containing protease and phosphatase inhibitors. Cell lysates were obtained after centrifugation at 13 000 r.p.m. for 10 min at 4 °C. For co-IP, 2 mg cell lysate was incubated overnight at 4 °C with 1 μg IP antibody or isotype IgG. Precipitation of protein complexes was performed by adding protein A/G magnetic beads (Thermo Fisher Scientific, Darmstadt, Germany) for 1 h at 4 °C. The beads were washed three times in IP buffer and twice in PBS. Elution of beads was achieved by incubation with 2 × sample buffer for 20 min at room temperature. Eluate was transferred to a clean tube and heated for 5 min at 95 °C. For IP under denaturing conditions, cells were lysed in IP buffer containing 1% SDS and heated at 95 °C for 15 min followed by centrifugation at 13 000 r.p.m. for 15 min. Cell lysates were diluted 1:10 in IP buffer before IP.
Site-directed mutagenesis
The FLAG-tagged A20 expressing plasmid (RC221337-OR) was purchased from BioCat (Heidelberg, Germany). Silencing of the DUB or E3 ligase activity of A20 was achieved using the Site-Directed Mutagenesis Kit QuickChange II XL (Agilent, Frankfurt am Main, Germany). Primers were designed to replace C103 or C624/627 by alanine to mutate the DUB and E3 ligase activity, respectively. The primers are as follows: C103A 5′-GAACGGTGACGGCAATGCCCTCATGCATGCCAC-3′ and C624/627A 5′-GACTCCAGAAAACAAGGGCTTTGCCACACTGGCTTTCATCGAGTACAGAGAAAA-3′. Successful mutation was verified by sequencing.
Generation of A20 knockout cells using CRISPR/Cas9 technology
AGS cells were seeded in six-well plates and transfected with either 1 μg of the A20 double nickase plasmid (Santa Cruz) or the respective control double nickase plasmid (Santa Cruz). The transfection reagent jetPRIME was used according to the manufacturer’s protocol. After 24 h incubation, the medium was changed to selection medium containing 0.5 μg/ml Puromycin. After 10 days of selection, single colonies were selected by limiting dilution in 96-well plates and expanded.
Imaging flow cytometry
Analysis of AGS cells for cleaved caspase-3 or Annexin V/PI staining was performed using the FlowSight instrument and the IDEAS software version 6.0 (Amnis/Merck Millipore, Darmstadt, Germany). The cells were harvested using trypsin and either stained with an Annexin V-FITC/PI Kit (MabTag, Friesoythe, Germany) or fixed in 3% formaldehyde in PBS for 10 min followed by permeabilization in 90% ethanol. The fixed cells were washed in incubation buffer (0.5% BSA in PBS) twice and stained with anti-cleaved caspase-3 antibody conjugated to AlexaFluor647 (#9602, Cell Signaling Technology) for 1 h at room temperature. All samples were recorded in the respective channels and compensation was performed before analysis. For the analysis of cell death, the Annexin V-positive and double positive cells were summed up to give the percentage of apoptotic cells. The number of cleaved caspase-3-positive cells was given as the percentage of single cells.
In vitro translation assay
The plasmids for FLAG-A20 (RC221337-OR) and HA-caspase-8 (OHu25469C) were purchased from BioCat and GenScript (Hong Kong, China), respectively. The proteins were in vitro translated using the PURExpress Kit (New England Biolabs, Frankfurt am Main, Germany) for 2 h at 37 °C, then mixed for 1 h at 37 °C followed by IP using the respective anti-FLAG or anti-HA antibody. The IPs were separated by SDS-PAGE.
Transactivation assay
AGS cells were seeded in 24-well plates at a density of 6 × 104 cells per well in RPMI-1640 culture medium supplemented with 10% FCS. The firefly luciferase plasmid containing five copies of an NF-κB response element (Promega, Madison, WI, USA) was mixed with Renilla Luciferase plasmid at a ratio 50 : 1 and transfected using Attractene transfection reagent (Qiagen) for 48 h. After H. pylori infection or TNF stimulation for 3 h, luciferase activity was estimated in cell lysates using the Dual-Luciferase Reporter Assay System (Promega) with a Lumat LB 9507 luminometer (Berthold Technologies, Bad Wildbad, Germany). The inducible firefly luciferase activity was normalized relative to Renilla’s activity, and fold changes in stimulated samples were calculated compared with non-stimulated cells.
Quantitative PCR
Total RNA was isolated using the NucleoSpin RNAII Kit (Macherey-Nagel, Berlin, Germany). The total RNA was reverse transcribed into cDNA using the RT2 First Strand Kit (Qiagen). Quantitative PCR was performed using the Taqman gene expression assay for A20 (Hs00234713_m1) and GAPDH (Hs03929097_g1) for normalization or the qPCR GreenMaster with highROX (Jena Bioscience, Jena, Germany) with the following primer pairs. IL-8 (5′-AGATGTCAGTGCATAAAGACA-3′ forward; 5′-TATGAATTCTCAGCCCTCTTCAAAAA-3′ reverse), cIAP2 and RPL13A for normalization (from the primer library HPA-1, Biomol, Hamburg, Germany). The comparative threshold method (ΔΔCt) was used to quantify relative changes of the target mRNA. Data were expressed as fold change in mRNA.
Statistics
All quantitative data were presented as mean±S.D. Statistical analysis was performed using Student’s T-test (OriginPro 2015, OriginLab, Northampton, MA, USA). P<0.05 was regarded as significant.
References
Kusters JG, van Vliet AHM, Kuipers EJ . Pathogenesis of Helicobacter pylori Infection. Clin Microbiol Rev 2006; 19: 449–490.
Blaser MJ, Atherton JC . Helicobacter pylori persistence: biology and disease. J Clin Invest 2004; 113: 321–333.
Shanks AM, El-Omar EM . Helicobacter pylori infection, host genetics and gastric cancer. J Dig Dis 2009; 10: 157–164.
Backert S, Clyne M . Pathogenesis of Helicobacter pylori infection. Helicobacter 2011; 16 (Suppl 1): 19–25.
Sokolova O, Maubach G, Naumann M . MEKK3 and TAK1 synergize to activate IKK complex in Helicobacter pylori infection. Biochim Biophys Acta 2014; 1843: 715–724.
Perkins ND . The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer 2012; 12: 121–132.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19: 107–120.
Schleich K, Warnken U, Fricker N, Ozturk S, Richter P, Kammerer K et al. Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol Cell 2012; 47: 306–319.
Cover TL, Krishna US, Israel DA, Peek RM . Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res 2003; 63: 951–957.
Kim K-M, Lee S-G, Park M-G, Song J-Y, Kang H-L, Lee W-K et al. gamma-Glutamyltranspeptidase of Helicobacter pylori induces mitochondria-mediated apoptosis in AGS cells. Biochem Biophys Res Commun 2007; 355: 562–567.
Oldani A, Cormont M, Hofman V, Chiozzi V, Oregioni O, Canonici A et al. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog 2009; 5: e1000603.
Martin JH, Potthoff A, Ledig S, Cornberg M, Jandl O, Manns MP et al. Effect of H. pylori on the expression of TRAIL, FasL and their receptor subtypes in human gastric epithelial cells and their role in apoptosis. Helicobacter 2004; 9: 371–386.
Jones NL, Day AS, Jennings HA, Sherman PM . Helicobacter pylori induces gastric epithelial cell apoptosis in association with increased Fas receptor expression. Infect Immun 1999; 67: 4237–4242.
Shembade N, Ma A, Harhaj EW . Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 2010; 327: 1135–1139.
Skaug B, Chen J, Du F, He J, Ma A, Chen Zhijian J . Direct, noncatalytic mechanism of IKK inhibition by A20. Mol Cell 2011; 44: 559–571.
Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009; 137: 721–735.
Bellail AC, Olson JJ, Yang X, Chen ZJ, Hao C . A20 ubiquitin ligase-mediated polyubiquitination of RIP1 inhibits caspase-8 cleavage and TRAIL-induced apoptosis in glioblastoma. Cancer Discov 2012; 2: 140–155.
Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity 2010; 33: 181–191.
Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol 2015; 16: 618–627.
Shi CS, Kehrl JH . TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal 2010; 3: ra42.
Vadlamudi RK, Shin J . Genomic structure and promoter analysis of the p62 gene encoding a non-proteasomal multiubiquitin chain binding protein. FEBS Lett 1998; 435: 138–142.
Geetha T, Wooten MW . Structure and functional properties of the ubiquitin binding protein p62. FEBS Lett 2002; 512: 19–24.
Duran A, Amanchy R, Linares Juan F, Joshi J, Abu-Baker S, Porollo A et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 2011; 44: 134–146.
Zotti T, Scudiero I, Settembre P, Ferravante A, Mazzone P, D’Andrea L et al. TRAF6-mediated ubiquitination of NEMO requires p62/sequestosome-1. Mol Immunol 2014; 58: 27–31.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282: 24131–24145.
Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016; 164: 896–910.
Schweitzer K, Sokolova O, Bozko PM, Naumann M . Helicobacter pylori induces NF-[kappa]B independent of CagA. EMBO Rep 2010; 11: 10–11.
Sokolova O, Borgmann M, Rieke C, Schweitzer K, Rothkotter HJ, Naumann M . Helicobacter pylori induces type 4 secretion system-dependent, but CagA-independent activation of IkappaBs and NF-kappaB/RelA at early time points. Int J Med Microbiol 2013; 303: 548–552.
Tiruppathi C, Soni D, Wang DM, Xue J, Singh V, Thippegowda PB et al. The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat Immunol 2014; 15: 239–247.
Lai T-Y, Wu S-D, Tsai M-H, Chuang EY, Chuang L-L, Hsu L-C et al. Transcription of Tnfaip3 is regulated by NF-κB and p38 via C/EBPβ in activated macrophages. PLoS ONE 2013; 8: e73153.
Soutto M, Belkhiri A, Piazuelo MB, Schneider BG, Peng D, Jiang A et al. Loss of TFF1 is associated with activation of NF-kappaB-mediated inflammation and gastric neoplasia in mice and humans. J Clin Invest 2011; 121: 1753–1767.
Hatakeyama M . Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 2004; 4: 688–694.
Fox JG, Rogers AB, Ihrig M, Taylor NS, Whary MT, Dockray G et al. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Res 2003; 63: 942–950.
Shibayama K, Doi Y, Shibata N, Yagi T, Nada T, Iinuma Y et al. Apoptotic signaling pathway activated by Helicobacter pylori infection and increase of apoptosis-inducing activity under serum-starved conditions. Infect Immun 2001; 69: 3181–3189.
Zhang H, Fang D-C, Lan C-H, Luo Y-H . Helicobacter pylori infection induces apoptosis in gastric cancer cells through the mitochondrial pathway. J Gastroenterol Hepatol 2007; 22: 1051–1056.
Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T et al. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 2007; 2: 250–263.
Yanai A, Hirata Y, Mitsuno Y, Maeda S, Shibata W, Akanuma M et al. Helicobacter pylori induces antiapoptosis through nuclear factor-κB activation. J Infect Dis 2003; 188: 1741–1751.
Srivastav S, Kar S, Chande AG, Mukhopadhyaya R, Das PK . Leishmania donovani exploits host deubiquitinating enzyme A20, a negative regulator of TLR signaling, to subvert host immune response. J Immunol 2012; 189: 924–934.
Lee GJ, Lee H-M, Kim TS, Kim JK, Sohn KM, Jo E-K . Mycobacterium fortuitum induces A20 expression that impairs macrophage inflammatory responses. Pathog Dis 2016; 74: ftw015.
Yokota S-i, Okabayashi T, Yokosawa N, Fujii N . Measles virus P protein suppresses Toll-like receptor signal through up-regulation of ubiquitin-modifying enzyme A20. FASEB J 2008; 22: 74–83.
Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH . The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol 2009; 183: 5909–5916.
Parvatiyar K, Barber GN, Harhaj EW . TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem 2010; 285: 14999–15009.
Shembade N, Parvatiyar K, Harhaj NS, Harhaj EW . The ubiquitin-editing enzyme A20 requires RNF11 to downregulate NF-kappaB signalling. EMBO J 2009; 28: 513–522.
Gao L, Coope H, Grant S, Ma A, Ley SC, Harhaj EW . ABIN1 protein cooperates with TAX1BP1 and A20 proteins to inhibit antiviral signaling. J Biol Chem 2011; 286: 36592–36602.
Acknowledgements
The work was supported by grants of the German Research Foundation to M.N. (CRC 854 A04), D.S. (CRC 854 A05) and S.B. (CRC 1181 A04) and by stipends from the Medical Faculty of the Otto von Guericke University to M.C.C.L. and M.H.F.
Author contributions
M.N. conceived the study, supervised the analysis, wrote the manuscript and secured funding. M.C.C.L. and G.M. established the methodologies, performed experiments and wrote the manuscript. M.H.F., R.D., J.B. and O.S. performed experiments. S.B. and I.N.L. provided reagents. S.B., D.S. and I.N.L. provided expertise and feedback.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Lim, M., Maubach, G., Sokolova, O. et al. Pathogen-induced ubiquitin-editing enzyme A20 bifunctionally shuts off NF-κB and caspase-8-dependent apoptotic cell death. Cell Death Differ 24, 1621–1631 (2017). https://doi.org/10.1038/cdd.2017.89
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.89
- Springer Nature Limited
This article is cited by
-
Cytoplasmic TP53INP2 acts as an apoptosis partner in TRAIL treatment: the synergistic effect of TRAIL with venetoclax in TP53INP2-positive acute myeloid leukemia
Journal of Experimental & Clinical Cancer Research (2024)
-
The Complexity of Being A20: From Biological Functions to Genetic Associations
Journal of Clinical Immunology (2024)
-
NF-κB/RelA controlled A20 limits TRAIL-induced apoptosis in pancreatic cancer
Cell Death & Disease (2023)
-
USP48 and A20 synergistically promote cell survival in Helicobacter pylori infection
Cellular and Molecular Life Sciences (2022)
-
Helicobacter pylori-induced reactive oxygen species direct turnover of CSN-associated STAMBPL1 and augment apoptotic cell death
Cellular and Molecular Life Sciences (2022)