
Abstract
Background
Obesity is a very heterogeneous disorder at both the clinical and molecular levels and with high heritability. Several monogenic forms and genes with strong effects have been identified for non-syndromic severe obesity. Novel therapeutic interventions are in development for some genetic forms, emphasizing the importance of determining genetic contributions.
Objective
We aimed to define the contribution of rare single-nucleotide genetic variants (RSVs) in candidate genes to non-syndromic severe early-onset obesity (EOO; body mass index (BMI) >+3 standard deviation score, <3 years).
Methods
Using a pooled DNA-sequencing approach, we screened for RSVs in 15 obesity candidate genes in a series of 463 EOO patients and 480 controls. We also analysed exome data from 293 EOO patients from the “Viva la Familia” (VLF) study as a replication dataset.
Results
Likely or known pathogenic RSVs were identified in 23 patients (5.0%), with 7 of the 15 genes (BDNF, FTO, MC3R, MC4R, NEGR1, PPARG and SIM1) harbouring RSVs only in cases (3.67%) and none in controls. All were heterozygous changes, either de novo (one in BDNF) or inherited from obese parents (seven maternal, three paternal), and no individual carried more than one variant. Results were replicated in the VLF study, where 4.10% of probands carried RSVs in the overrepresented genes. RSVs in five genes were either absent (LEP) or more common in controls than in cases (ADRB3, LEPR, PCSK1 and PCSK2) in both obese datasets.
Conclusions
Heterozygous RSVs in several candidate genes of the melanocortin pathway are found in ~5.0% patients with EOO. These results support the clinical utility of genetic testing to identify patients who might benefit from targeted therapeutic intervention.
Similar content being viewed by others


Introduction
Obesity is the most prevalent chronic childhood disease in the occidental world and a risk factor for later obesity- and metabolic-related disorders. It is a heterogeneous multifactorial disease with high heritability (50–75%) that is undoubtedly higher in severe early-onset cases [1,2,3]. The genetic causes underlying obesity remain largely unknown with an important proportion of information missing regarding its heritability.
A small percentage of obesity occurs as part of a syndromic entity [4,5,6,7,8,9,10], but in the majority, obesity is not accompanied by other specific phenotypes. Highly penetrant rare genetic variants, mainly autosomal recessive, affecting at least eight genes [LEP (MIM 164160), LEPR (MIM 601007), MC4R (MIM 155541), PCSK1 (MIM 162150), POMC (MIM 176830), MC3R (MIM 155540), SIM1 (MIM 603128) or NTRK2 (MIM600456)] are reportedly found in 2–5% of non-syndromic obese patients [11,12,13,14,15,16,17]. However, multigenic or multifactorial inheritance is the best model to explain the aetiology of most cases. Common genetic variants involved in these forms of obesity have been identified by genome-wide and candidate-gene association studies. Genetic variants in genes such as ADRB3 (MIM 109691), BDNF (MIM 113505), FTO (MIM 610966), GHSR, NEGR1 (MIM 613173), PCSK2 (MIM 162151), PPARG (MIM 601487) and TMEM18 (MIM 613220) [18,19,20,21,22], among others, have been related to body mass index (BMI), but the strongest effect of a single common variant might explain about only 2% of the variance in BMI. Similar to other complex traits [23, 24], copy number variants (CNVs) also contribute to the genetic basis of obesity [25,26,27,28]. Identification of a potential genetic cause of severe obesity is important not only for individualized follow-up and genetic counselling but also because novel therapeutic approaches for genetically defined obesities are becoming available [29].
Next-generation sequencing (NGS) has facilitated identification of the molecular basis of Mendelian diseases, even with a reduced number of samples [30, 31]. However, large numbers of patients are needed to identify a significant mutation burden in a specific gene and prove its pathophysiological relevance in multifactorial or highly heterogeneous genetic disorders. Given the known population stratification, appropriate controls from the same geographic origin are also needed to discard population variants that might not be disease causative. Strategies using pooled DNA-sequencing have proven effective in minimizing the costs of tackling multifactorial or heterogeneous disorders using large numbers of samples [32].
In highly heterogeneous diseases such as obesity, severe cases with early onset are more likely explained by highly penetrant rare genetic variants. We have used a pooled strategy to sequence a panel of selected candidate genes in 463 patients with non-syndromic severe early-onset obesity (EOO) and 480 controls searching for rare sequence variants (RSVs) with high penetrance.
Subjects and methods
Subjects and samples
Severe early-onset obese patients were selected by using the criteria of a BMI Z-score above three according to age and sex for the Spanish population [33] at their first examination and that visited the clinic due to obesity that was reported to have an onset before 3 years of age.
Patients underwent a detailed clinical evaluation, including oriented clinical history, physical examination and complementary tests to rule out syndromic, endocrine or secondary forms of obesity. These evaluations included an interview with a dietitian to determine eating habits such as the frequency, speed and amount of food ingested. A molecular karyotype by microarray and a methylation-sensitive multiple-ligation probe amplification to discard specific entities that might clinically overlap with isolated obesity were also performed [5].
A sample of 463 subjects with severe EOO was selected. Blood was collected from patients and both parents when possible to study the segregation of the genetic variants with the phenotype in the family. DNA was isolated from total blood using the Gentra Puregene Blood kit (Qiagen) according to the manufacturer’s instructions.
A total of 480 adult subjects with normal weight (BMI <P75) from the same geographic origin, provided by Banco Nacional de ADN Carlos III (Universidad de Salamanca, Spain), were used as controls.
Candidate genes
To define the list of 15 candidate genes, a systematic literature review was done to identify: (1) genes with reported mutations in patients with obesity (LEP, LEPR, MC3R, MC4R, PCSK1, NTRK2 and SIM1) [11,12,13,14,15,16, 34], and (2) genes with single-nucleotide polymorphisms associated to obesity in genome-wide association studies (BDNF, FTO, NEGR1, GHSR, ADRB3, PPARG, PCSK2 and TMEM18A) [18,19,20,21,22].
Pooled DNA-sequencing
Targeted enrichment was used to capture coding regions from the 15 selected genes (SeqCap EZ Choice Enrichment Kits, Roche Sequencing). Instead of sequencing individual samples, a pooled DNA strategy was used. Each sample was included in two different pools and each pool had 20 different samples. A total of 48 pools from patients and 48 pools from controls were designed (Fig. 1). To identify which sample carried each variant, the results from each pool were overlapped to identify the two pools harbouring the same alterations. Using this pooling approach, one heterozygous variant in a sample is expected to appear in about 1/40 (2.5%) of the reads. To ensure that variants with a low percentage of reads were represented in the sequencing experiment, high coverage was mandatory. The massive sequencing was done with MiSeq (Illumina).

Summary of the strategy followed for single-nucleotide variant (SNV) analysis and rare sequence variant (RSV) identification
Variant calling
Reads were mapped to the human reference genome (UCSC hg19). We used the MUTEC software [35] to call RSVs represented in a low percentage of reads. Given the very high coverage, false-positive calls were expected in almost all positions mapped. Thus, several quality filters were used to discriminate real variants from false positives in an automatic manner (Fig. 1) and visual inspection of reads in the integrated genome viewer [36] allowed us to clearly discriminate in dubious cases. Six samples with seven previously detected variants were used for optimization of filtering parameters. Base quality index and strand bias were the best predictors to reduce false-positive detection rates. Since false positives tended to have much lower base quality values, only variants with individual Phred scores over 5 and an average base quality over 15 in the two different pools were selected. Variants with high strand bias were found to be false positives. Thus, variants with extreme strand bias (proportion of same strand reads <20% or >80%) were discarded as likely false positives.
Custom-made Perl scripts were used to automate sample identification crossing the pools where the same variant was detected. The proportion of variants in each pool was also considered in the pipeline as an estimation of allele frequency. Although obesity is a highly heterogeneous disorder, the same RSV might appear twice (present in two alleles from either the same or different samples), expected in 5% of the reads. Consequently, the first step was filtering the variants present in between 1 and 10% of the mapped reads. Variants with a lower percentage were probably artefacts and variants with a higher percentage (present in three or more samples in the same pool) were thought too common to be considered highly penetrant RSVs for this heterogeneous disease.
In positions where the coverage was under 1000×, it was particularly difficult to discriminate between real variants and false positives. Therefore, we individually assessed all variants in positions with coverage under 1000×.
Prioritization and analysis of variants
To establish the burden of RSVs identical approaches were used in each group (Fig. 1). Intronic variants were excluded due to the difficulty in interpreting their clinical significance and only exonic variants and those putatively affecting splicing (±6) were selected. We discarded variants with population frequencies in any public database (gnomAD, 1000 genomes, Kaviar, Spanish variant server) [37] higher than 1/1000 or described in a homozygous state in at least one individual, assuming that variants with a relatively high frequency in the general population are unlikely high penetrant variants related to the severe phenotype of EOO.
All nonsense or frameshift, canonical splicing and missense variants predicted as damaging (Condel >0.522 and CADD >20) [38, 39] were considered as highly likely to be pathogenic (from now on called likely pathogenic variants). All previously reported mutations with experimentally validated functional consequences were also included [40]. To define pathogenicity, we also considered the constraint metrics for variation tolerance of each gene. Variation-intolerant genes were considered those with pLi (probability of loss-of-function intolerance) >0.50 and missense Z-score >0 [37].
Variant validation and co-segregation
To validate RSVs detected by pooled DNA-sequencing, PCR amplicons from individual DNA samples were obtained and sequenced by Sanger technology. The same technique was used to analyse parental samples when available to define inheritance pattern and possible co-segregation of the RSV with the phenotype in each family.
Replication cohort
Exomes from 293 unrelated patients with EOO from the Viva la Familia (VLF) study [41] were used to replicate the results found in our initial cohort. VLF comprises obese probands with BMI >95th percentile, between 4 and 19 years old, from Hispanic families in Houston. Sequence read archive (SRA) files were downloaded from publicly available servers (dbGAP: phs000616.v2.p2). Variant calling in the 15 candidate genes was done with GATK [42] and filtering was performed using the same criteria as above.
Ethics statement
The project was approved by the Medical Ethical Committee of Hospital Infantil Universitario Niño Jesús in Madrid, Spain and is in accordance with the “Ethical Principles for Medical Research Involving Human Subjects” adopted in the Declaration of Helsinki by the World Medical Association (64th WMA General Assembly, Fortaleza, Brazil, October 2013) and Spanish data protection act (Ley Orgánica 15/1999 de Protección de Datos). Written informed consent was obtained from all patients or their legal guardian after a complete description of the study.
Results
RSV detection sensitivity and specificity
The mean sequence coverage of patient pools was 2396× (SD: 574) and 3428× (SD: 660) in controls, with >98% of targeted sites showing coverage above 400×. The sensitivity and specificity of this approach were assessed. Six of the seven known variants in samples included in the study were blindly identified with the filters mentioned above, yielding a sensitivity of 86%. The only variant not identified had low-quality scores. As proof of concept for high specificity, 23 variants detected with the pooled DNA approach were selected and all (23/23, 100%) validated by Sanger sequencing. Although the pooled DNA approach may not detect a small percentage of real variants, it does not capture false positives. These results demonstrate that the strategy used is efficient to identify RSVs in large cohorts of samples.
Burden of RSVs in EOO
A total of 73 different RSVs were selectively identified in only one group (Table 1): 43 variants in 48 subjects of the patient group and 30 variants in 31 controls. This difference was statistically significant (odds ratio (OR) = 1.61; p = 0.0342; Table 1 and Supplemental Tables 1 and 2). We used very stringent criteria of pathogenicity to better define the putative clinically relevant variants: all loss-of-function (nonsense, frameshift and canonical splice-site variants) and missense variants predicted as pathogenic by two stringent algorithms (Condel and CADD) were considered as likely pathogenic. Likely or known pathogenic (previously validated) RSVs were found in 23 patients and 8 controls (OR = 2.98; p = 0.0055). Most RSVs were present in a single sample, but a few cases with two samples harbouring the same RSV were found and three patients shared a variant in FTO.
A subset of sequenced genes accumulated the differential mutational load found in patients compared to controls: BDNF, FTO, MC3R, MC4R, NEGR1, PPARG and SIM1. Analysing this group of genes separately, a total of 30 patients with RSVs were identified in patients versus four controls (OR = 7.78; p < 0.0001; Table 1) and four of these seven overrepresented genes (BDNF, NEGR1, PPARG and SIM1) are variation intolerant (Supplemental Table 3). Analysing the likely pathogenic variants in the subset of genes, RSVs were identified in 17 patients (3.67%) and no controls (p < 0.0001; Table 1). Two of the variants identified were present in two unrelated patients (Table 2). All 17 likely pathogenic variants identified in the overrepresented subset of genes heterozygous and no individual carried more than one likely pathogenic variant (Tables 1 and 2).
In the remaining subset of genes, 18 RSVs were identified in patients and 27 in controls (not statistically significant), including 6 likely pathogenic variants in patients and 8 in controls (not statistically significant; Supplemental Tables 1 and 2).
Heterozygous RSVs in patients with EOO
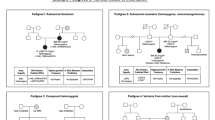
Among the genes with higher mutational load in patients, MC4R was the most frequent with seven RSVs in patients (1.51%). Of these, five have been reported as pathogenic by demonstration of partial (H76R) or complete (S127L, T150I, A259V and P272L) loss of function [13, 40]. The two RSVs not previously described include a nonsense change interrupting the protein in the 22nd codon (R22X) and a missense change (V52A) predicted as likely benign. In the five cases with parental samples available, the RSV was inherited from obese mothers. Two missense variants in MC4R were detected in the control group, both predicted as benign and not previously described in patients. Three patients were also found to have RSVs in a related gene, MC3R, with the likely pathogenic RSV (R302W) identified in two unrelated patients (Table 2).
SIM1 also showed a high mutational load in patients (n = 6) compared to controls (n = 1). Two of the six variants identified in patients are strongly predicted to be pathogenic. One affects a canonical splice site in intron 3 (c.352 + 1G>A) and the other is a missense variant on a highly conserved residue of the first Perl–Arnt–Sim of the protein (Q206K) found in two siblings concordant with the phenotype. These RSVs were inherited from an obese father (splicing variant) and obese mother (missense variant) (Table 2). The variant detected in a control subject and four of the RSVs in patients are not predicted to be pathogenic by our criteria.
In the BDNF gene, three RSVs were found in four patients and none in controls. Two of the variants are predicted to be pathogenic. One of these RSVs was not found in parental samples, indicating that it occurred de novo in the patient. Parenthood was proven using microsatellite markers. The other RSV (I231V) was found in two unrelated cases, paternally inherited in the case with parental samples available (Table 2).
We detected three RSVs (two likely pathogenic) in PPARG in patients, as well as one single likely pathogenic missense RSV in FTO and NEGR1, and none in controls.
Clinical features of RSV carriers
The main phenotypic features of patients harbouring likely pathogenic RSVs in the selected genes are detailed in Table 2. The six patients harbouring pathogenic RSVs in MC4R were males with a BMI above + 5.5 standard deviation score (SDS), orexigenic impulsivity, hyperinsulinism, some overgrowth (height >+1 SDS over target height) and advanced bone age (14 months on average), a phenotype previously defined in patients with MC4R mutations [43, 44]. The patient with the A259V variant had macroorchydism (8 cc volume testes despite Tanner stage I), which is not a common feature of MC4R deficiency, given the lack of MC4R expression in the testis [45]. The patient with the novel nonsense mutation (R22X) had an extremely high BMI (+18.8 SDS), uncontrollable orexigenic impulsivity, overgrowth (+3 SDS), advanced bone age (51 months over chronological age) and hyperinsulinism. His mother, also a carrier of the R22X mutation, had a BMI of 51.3 kg/m2. The patient with a predicted benign RSV at MC4R (V52A) was a girl with milder obesity (+4.2 BMI-SDS), no overgrowth (height at −1 SDS), average bone age, and hyperinsulinism.
The two patients with likely pathogenic RSVs in MC3R had milder obesity (BMI around +4 SDS) and no overgrowth. One had insulin resistance.
Patients harbouring SIM1 RSVs had BMIs of 4.3 and 5.6 SDS, increased longitudinal growth (+0.7 and +1.3 SDS over standardized target height), mild hyperphagia, insulin resistance, and impaired glucose tolerance.
Patients with RSVs in the BDNF gene had BMIs ranging from +3.3 to +6.1 SDS. Patients with PPARG RSVs including one sibling (BMI +4.1 to +5.5 SDS) showed insulin resistance and three out of four had dyslipidaemia.
Replication cohort
The results of the VLF study are shown in Table 1 and Supplemental Table 4. The frequency of RSVs in VLF replicated the results for BDNF, FTO, MC3R, MC4R and SIM1. No RSVs with the established criteria were detected in NEGR1 or PPARG in the VLF cohort, but likely pathogenic RSVs were identified in GHSR and TMEM18, absent in controls. The increased rate of RSVs between VLF subjects (32/293, 10.92%) and controls (31/480, 6.46%) in all genes was similar to that of the Spanish EOO dataset (OR = 1.69; p = 0.0306). The separate analysis of the overrepresented subset of genes (BDNF, FTO, MC3R, MC4R, NEGR1, PPARG and SIM1) yielded a significantly higher proportion of VLF patients with RSVs (16/293) than controls (4/480; OR = 6.55; p = 0.0002). This difference was also statistically significant when considering only likely pathogenic variants (6/293 VLF patients versus 0/480 controls; p = 0.0029; Supplemental Table 5).
Discussion
Analysis of an elevated number of individuals is necessary to identify genetic factors with strong effects in heterogeneous diseases with important environmental influence, such as obesity. Pooled DNA-sequencing reduces the number of sequencing reactions, thus reducing costs without decreasing the number of samples. This approach must be optimized to identify all relevant variants and discern between real variants and artefacts. Sequencing coverage above 400 reads in most positions would yield >10 reads of any single RSV diluted in a pool of 20 different samples. Since sequence artefacts are detected in most mapped positions, quality filters were required to discriminate between real variants and false positives. The percentage of reads with the variant, strand bias and Phred score had the greatest discriminatory ability. Full validation rate by Sanger sequencing (23 of 23 variants) proved the high specificity and suitability of the pooling strategy.
We failed to detect one of seven7 previously defined RSVs (sensitivity 86%). This incomplete detection rate might invalidate using pooling strategies for diagnostic purposes, but it should not affect the results of this study. The average coverage of the patient pools was lower than that of the control pools (2396× vs. 3428×). If this difference were to generate bias when detecting RSVs, it would favour RSV detection in controls. A more similar coverage between groups might even increase the difference in burden between patients and controls.
We focused on RSVs in 15 candidate genes that could have a strong effect in a small subgroup of patients according to information available at the onset of this study. This strategy might have missed some variants with higher frequencies that also contribute to obesity. RSVs were identified in 10.36% of patients with half of them (5.0%) most likely being pathogenic, a burden significantly higher in patients than in controls. This burden was attributable to a subset of seven genes (BDNF, FTO, MC3R, MC4R, NEGR1, PPARG and SIM1). The replication study corroborated these results indicating that highly penetrant RSVs in these genes contribute to part of the missing heritability of EOO.
When patients are grouped according to the gene mutated, most groups are too small to establish conclusive clinical molecular correlations, but some common features can be discerned. Among the overrepresented genes, MC4R was the most frequent (1.51% of patients), similar to reports revealing heterozygous and homozygous mutations in MC4R accounting for 1–6% of severe obesity in humans [40, 43, 44]. Patients with RSVs in MC4R had hyperphagia with compulsive eating, very severe obesity (mean BMI + 8.9 SDS), overgrowth, accelerated skeletal maturation and metabolic disturbances including insulin resistance. Behavioural disturbances occurred mainly associated with food seeking. It is of note that all patients described here with RSVs at MC4R were male. The literature does not support the possibility that in obese subjects this gene is exclusively affected in males. However, there may be a global bias in the parental transmission of RSVs. To determine this possibility, a larger sample size is required, as well as additional investigation. It is also of note that there was a lack of significant differences in gene burden between the two relatively close ethnic groups compared, European Spaniards and Hispanics in the USA.
Like MC4R, MC3R has a critical role in regulating energy balance [46]. It was recently reported to regulate the upper and lower limits of an individual’s homeostatic set-points and the response to external metabolic challenges [47]. MC3R-knockout mice were shown to have dysregulation of energy expenditure, feeding responses and neuroendocrine responses to metabolic challenges such as fasting, high fat diet intake, pregnancy and the loss of estrogens. For example, these mice lost more weight during fasting than wild-type mice, but gained more weight when put on a high fat diet [47]. The three patients (0.65%) with missense mutations in MC3R had a similar degree of obesity, which was milder than those harbouring RSVs in MC4R, and no overgrowth or advanced skeletal maturation. These patients did not exhibit measurable hyperphagia, similar to what was observed in MC3R-deficient mice [47]. However, as with mice, an inadequate diet could contribute to the excess weight gain. Only one of our patients had hyperinsulinism and hyperuricaemia; this patient also harboured a variant in the other allele that did not fulfil the defined criteria as its frequency is 1/945 in the European population. However, if this second variant is functionally relevant, inheritance would be compatible with a recessive or codominant model as reported for MC4R. In the codominant mode of inheritance, both monoallelic and biallelic mutations can cause the disorder and patients with biallelic mutations are more affected than their heterozygous relatives [48].
BDNF is a pro-survival factor in the brain that also participates in appetite regulation. Heterozygous BDNF-knockout mice are obese, hyperphagic and hyperactive [49] and disruption and deletion of BDNF have been described in patients with obesity. WAGR syndrome is a contiguous gene deletion syndrome on chromosome 11p13 characterized by Wilms’ tumour, Aniridia, Genitourinary anomalies and Mental retardation, with obesity present in patients with larger deletions encompassing BDNF along with the other critical genes (WAGRO syndrome) [10]. Disruption of BDNF expression in a patient with a paracentric inversion and deletion of the entire BDNF locus in a mother and child were associated with hyperphagia, severe obesity and mildly impaired cognition with or without attention deficits and hyperactivity [50,51,52]. However, no point mutations in BDNF have been reported to date in obese patients. In our series, three patients (0.65%) harboured two different missense RSVs in this gene predicted as pathogenic, with none detected in controls. Two unrelated cases carried the same variant (I231V) and another carried a de novo variant (C141G9). Obese patients with BDNF variants presented mild to severe hyperphagia and insulin resistance with dyslipidaemia in two cases. No behavioural or learning issues detected.
Six RSVs in SIM1 were found in our cohort and two were predicted as likely pathogenic. A third case was the affected brother of case 1782, concordant for the Q206K RSV. The phenotype of all three cases included a BMI-SDS of +4 to +5, no intellectual or behavioural abnormalities, and insulin resistance with impaired glucose tolerance. Sim1 haploinsufficiency in mice induces hyperphagia, obesity and central nervous system developmental abnormalities [53]. Deletions [54, 55] and translocations [56] affecting SIM1 cause severe obesity in association with intense orexigenic impulsivity and other features resembling Prader–Willi syndrome. Loss-of-function point mutations in SIM1 with variable expressivity and incomplete penetrance also produce a “Prader–Willi-like” phenotype [57], but also non-syndromic obesity [58]. Thus the described RSVs could contribute to the postulated increased intra-family risk for non-syndromic obesity [58].
Significant differences in RSV load between patients and controls were detected in NEGR1 (3 patients, 0 controls), FTO (4 patients, 1 control) and PPARG (3 patients, 0 controls), with five of the RSVs predicted as likely pathogenic. Although association studies have documented their role in obesity, no highly penetrant RSVs have been described to date in these genes. Our data indicate that some RSVs at NEGR1, FTO and PPARG may behave as highly penetrant alleles causative of EOO, although the number of patients is too small to define distinctive phenotypes. The patient with a likely pathogenic RSV at NEGR1 had speech and behavioural problems and hyperuricaemia, while the three patients harbouring RSVs in PPARG (including one sibling) had insulin resistance and associated dyslipidaemia, in agreement with the role of PPARG variants in predisposition to metabolic syndrome [59].
Weaker evidence for definite implication was obtained for GHSR, NTRK2 and TMEM18. Likely pathogenic RSVs at NTRK2 were identified in one of our patients and two from the VLF study, but also in one control. Single patients with RSVs at GHSR and TMEM18, absent in controls, were found in the VLF cohort. Whether these monoallelic RSVs contribute to the obesity phenotype awaits studies in larger series and functional studies. De novo NTRK2 mutations, a highly intolerant gene to both loss-of-function (pLI = 1) and missense variants (miss-z = 4.35), have been previously associated with severe obesity and developmental delay [17]. Thus, some heterozygous partially functional variants could cause a phenotype of non-syndromic obesity without significant developmental delay.
RSVs in five genes were either absent (LEP) or more common in controls than in cases (ADRB3, LEPR, PCSK1 and PCSK2) in both datasets. Biallelic mutations in LEP, LEPR and PCSK1 with a recessive model of inheritance have been described in severely obese patients. In a study of 300 obese subjects, biallelic LEP mutations were identified with a frequency of 3% [48], but 3/4th of the patients with biallelic mutations were homozygous, with mutations in identical-by-descent regions in consanguineous families [48]. Thus, the prevalence of biallelic mutations in recessive genes in populations that are not inbred, such as the two cohorts studied here, might be quite low [60]. POMC was not included in this study based on the assumed adrenal insufficiency associated to pathogenic variants in this gene. However, POMC is now being analysed in this cohort in another study.
It is clear that functional analyses at the cellular, tissue and organism levels are required to define the physiopathogenic mechanisms of all variants reported here. However, we show strong epidemiological and statistical evidence by defining the genes harbouring heterozygous deleterious changes in our large cohort of EOO patients and then validating the results in a replication dataset. Moreover, there is experimental evidence for the abnormal function of some of the reported RSVs [13, 40, 61]. Many genes involved in appetite and metabolic control appear to be sensible to dosage [50, 62, 63], with rescue of haploinsufficiency of Mc4r and Sim1 recently shown to revert the obese phenotype in mice [64]. Thus, although the molecular mechanisms underlying how these newly described RSVs affect body weight remain to be established, there is precedence for heterozygous affectation of these genes being functionally important. Moreover, the overall prevalence of monoallelic RSVs found in our cohort is similar to that of a recent publication [65] where a diagnosis rate of genetic obesity was reported in 3.9% of patients (48/1230) and 7.3% of the paediatric subgroup (12/164). It should also be emphasized that the interaction of these genetic variants with the environment is of utmost importance with morbid obesity being more likely to occur in these cases when dietary habits are inadequate.
In summary, we documented a higher burden of likely pathogenic heterozygous RSVs in several candidate genes in patients with severe EOO compared to controls. The yield for RSV detection is relatively high, around 5% of patients with EOO. Identification of a potential genetic cause of the phenotype in each case is very important for individualized follow-up and genetic counselling to the family. In addition, definition of the specific defect at the molecular level may be instrumental, as this group of individuals may be relatively refractory to weight loss through diet and exercise and novel therapeutic approaches for genetically defined obesities are becoming available. For instance, highly potent second-generation MC4R agonists lead to weight loss in individuals with MC4R deficiency [40]. Thus, several patients in this study could possibly benefit from this therapy. Our results reinforce the role of the leptin–melanocortin pathway in obesity and bring to light other genes that may carry highly penetrant obesogenic single allele variants, such as FTO, NEGR1 and PPARG. The availability of an evidence-based algorithm for diagnostic analysis of early-onset obesity would be of great clinical interest. However, the obese phenotype of the children reported here, as well as in other studies, is non-specific, and an algorithm that could accurately suggest which genes should be studied in each case is difficult even if phenotype–genotype relationships lead to a better definition of the different clinical conditions. Instead, given the currently available and affordable tools for diagnostic genomic analysis, a genotype-first approach using expanded panel of genes with effective copy number analyses should be indicated in these children with early-onset obesity.
References
Friedman JM. Obesity in the new millennium. Nature. 2000;40:632–4.
Walley AJ, Asher JE, Froguel P. The genetic contribution to non-syndromic human obesity. Nat Rev Genet. 2009;10:431–42.
Llewellyn CH, Trzaskowski M, Plomin R, Wardle J. Finding the missing heritability in pediatric obesity: the contribution of genome-wide complex trait analysis. Int J Obes. 2013;37:1506–9.
Hatada I, Ohashi H, Fukushima Y, Kaneko Y, Inoue M, Komoto Y, et al. An imprinted gene p57KIP2 is mutated in Beckwith-Wiedemann syndrome. Nat Genet. 1996;14:171–3.
Martos-Moreno GÁ, Serra-Juhé C, Pérez-Jurado LA, Argente J. Underdiagnosed Beckwith–Wiedemann syndrome among early onset obese children. Arch Dis Child. 2014;99:965–7.
Nicholls RD, Knoll JHM, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in non-deletion Prader–Willi syndrome. Lett Nat. 1989;342:281–5.
Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, El-Maarri O, Horsthemke B. Epimutations in Prader–Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet. 2003;72:571–7.
Sanz-Fernández M, Muñoz-Calvo MT, Pozo-Román J, Martos-Moreno GÁ, Argente J. Clinical and radiological findings in a case of pseudohypoparathyroidism type 1a: Albright hereditary osteodystrophy. An Pediatr. 2015;82:439–41.
Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, Hoskins BE, et al. Triallelic inheritance in Bardet–Biedl Syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–9.
Han JC, Liu QR, Jones M, Levinn RL, Menzie CM, Jefferson-George KS, et al. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N Engl J Med. 2008;359:918–27.
Ranadive S, Vaisse C. Lessons from extreme human obesity: monogenic disorders. Endocrinol Metab Clin North Am. 2008;37:733–51.
Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–103.
Granell S, Serra-Juhé C, Martos-Moreno GÁ, Díaz F, Pérez-Jurado LA, Baldini G, et al. A novel melanocortin-4 receptor mutation MC4R-P272L associated with severe obesity has increased propensity to be ubiquitinated in the ER in the face of correct folding. PLoS ONE. 2012;7:e50894.
Montague CT, Farooqi IS, Wareham NJ, Soos MA, Rau H, Wareham NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–8.
Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20:113–4.
Yeo GSH, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111–2.
Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7:1187–9.
Willer CJ, Speliotes EK, Loos RJF, Li S, Lindgren CM, Heid IM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34.
Herbert A, Gerry NP, McQueen MB, Heid IM, Pfeufer A, Illig T, et al. A common genetic variant is associated with adult and childhood obesity. Science. 2006;312:279–83.
Turcot V, Lu Y, Highland HM, Schurmann C, Justice AE, Fine RS, et al. Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat Genet. 2018;50:26–41.
Meyre D, Delplanque J, Chèvre JC, Lecoeur C, Lobbens S, Gallina S, et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet. 2009;41:157–9.
Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41:18–24.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–73.
Stefansson H, Rujescu D, Cichon S, Pietiläinen OP, Ingason A, Steinberg S, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–6.
Jarick I, Vogel CIG, Scherag S, Schäfer H, Hebebrand J, Hinney A, et al. Novel common copy number variation for early onset extreme obesity on chromosome 11q11 identified by a genome-wide analysis. Hum Mol Genet. 2011;20:840–52.
Serra-Juhé C, Martos-Moreno GÁ, Bou de Pieri F, Flores R, González JR, Rodríguez-Santiago B, et al. Novel genes involved in severe early-onset obesity revealed by rare copy number and sequence variants. PLoS Genet. 2017;13:1–19.
Wheeler E, Huang N, Bochukova E, Keogh JM, Lindsay S, Garg S, et al. Genome-wide SNP and CNV analysis identifies common and low-frequency variants associated with severe early-onset obesity. Nat Genet. 2013;45:513–7.
Bochukova EG, Huang N, Keogh J, Henning E, Purmann C. Large, rare chromosomal deletions associated with severe early- onset obesity. Nature. 2010;463:666–70.
Grandone A, Di Sessa A, Umano GR, Toraldo R, Miraglia Del Giudice E. New treatment modalities for obesity. Best Pract Res Clin Endocrinol Metab. 2018;32:535–49.
Mardis ER. A decade’s perspective on DNA sequencing technology. Nature. 2011;470:198–203.
Zhang J, Chiodini R, Badr A, Zhang G. The impact of next-generation sequencing on genomics. J Genet Genom. 2011;38:95–109.
Zuzarte PC, Denroche RE, Fehringer G, Katzov-Eckert H, Hung RJ, McPherson JD. A two-dimensional pooling strategy for rare variant detection on next-generation sequencing platforms. PLoS ONE. 2014;9:e93455.
Hernández M, Castellet J, Narvaiza JL, Rincón JM, Ruiz I, Sánchez E, et al. Curvas y tablas de crecimiento. 2nd edn. Madrid: Instituto de Investigación sobre Crecimiento y Desarrollo, Fundación Faustino Orbegozo, Editorial Garsi; 1988.
Ristow M, Müller-Wieland D, Pfeiffer A, Krone W, Kahn R. Obesity associated with a mutation in a genetic regulator of adipocyte differentiation. N Engl J Med. 2015;339:953–9.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9.
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative Genomics Viewer. Nat Biotechnol. 2011;29:24–26.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–9.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Collet TH, Dubern B, Mokrosinski J, Connors H, Keogh JM, Mendes de Oliveira E, et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol Metab. 2017;6:1321–9.
Butte NF, Cai G, Cole SA, Comuzzie AG. Viva la Familia Study: genetic and environmental contributions to childhood obesity and its comorbidities in the Hispanic population. Am J Clin Nutr. 2006;84:646–54.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2009;20:254–60.
Govaerts C, Srinivasan S, Shapiro A, Zhang S, Picard F, Clement K, et al. Obesity-associated mutations in the melanocortin 4 receptor provide novel insights into its function. Peptides. 2005;26:1909–19.
Martinelli CE, Keogh JM, Greenfield JR, Henning E, van der Klaauw AA, Blackwood A, et al. Obesity due to melanocortin 4 receptor (MC4R) deficiency is associated with increased linear growth and final height, fasting hyperinsulinemia, and incompletely suppressed growth hormone secretion. J Clin Endocrinol Metab. 2011;96:181–8.
Chhajlani V. Distribution of cDNA for melanocortin receptor subtypes in human tissues. Biochem Mol Biol Int. 1996;38:73–80.
Zegers D, Beckers S, Hendrickx R, Van Camp JK, Van Hoorenbeeck K, Desager KN, et al. Prevalence of rare MC3R variants in obese cases and lean controls. Endocrine. 2013;44:386–90.
Ghamari-Langroudi M, Cakir I, Lippert RN, Sweeney P, Litt MJ, Ellacott KLJ, et al. Regulation of energy rheostasis by the melanocortin-3 receptor. Sci Adv. 2018;4. https://doi.org/10.1126/sciadv.aat0866.
Farooqi IS, Wangensteen T, Collins S, Kimber W, Matarese G, Keogh JM, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. 2007;356:237–47.
Fox EA, Biddinger JE, Jones KR, McAdams J, Worman A. Mechanism of hyperphagia contributing to obesity in brain-derived neurotrophic factor knockout mice. Neuroscience. 2013;229:176–99.
Gray J, Yeo GSH, Cox JJ, Morton J, Adlam AL, Keogh JM, et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55:3366–71.
Jouan L, Gauthier J, Dion PA, Rouleau GA. Rare variants in complex traits: novel identification strategies and the role of de novo mutations. Hum Hered. 2013;74:215–25.
Harcourt BE, Bullen DVR, Kao KT, Tassoni D, Alexander EJ, Burgess T, et al. Maternal inheritance of BDNF deletion, with phenotype of obesity and developmental delay in mother and child. Am J Med Genet A. 2018;176:194–200.
Michaud JL, Boucher F, Melnyk A, Gauthier F, Goshu E, Lévy E, et al. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet. 2001;10:1465–73.
Villa A, Urioste M, Bofarull J, Martínez-Frías M. De novo interstitial deletionq16.2q21 on chromosome 6. Am J Med Genet. 1995;55:379–83.
Faivre L, Cormier-Daire V, Lapierre JM, Colleaux L, Jacquemont S, Geneviéve D, et al. Deletion of the SIM1 gene (6q16.2) in a patient with a Prader–Willi-like phenotype. J Med Genet. 2002;39:594–6.
Holder JL, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9:101–8.
Bonnefond A, Raimondo A, Stutzmann F, Ghoussaini M, Ramachandrappa S, Bersten DC, et al. Loss-of-function mutations in SIM1 contribute to obesity and Prader-Willi – like features. J Clin Invest. 2013;123:3037–41.
Ramachandrappa S, Raimondo A, Cali AMG, Keogh JM, Henning E, Saeed S, et al. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J Clin Invest. 2013;123:1–9.
Rocha RM, Barra GB, ÉCCC Rosa, Garcia ÉC, Amato AA, Azevedo MF. Prevalence of the rs1801282 single nucleotide polymorphism of the PPARG gene in patients with metabolic syndrome. Arch Endocrinol Metab. 2015;59:297–302.
Paolini B, Maltese PE, Del Ciondolo I, Tavian D, Missaglia S, Ciuoli C, et al. Prevalence of mutations in LEP, LEPR, and MC4R genes in individuals with severe obesity. Genet Mol Res. 2016;19:15. https://doi.org/10.4238/gmr.15038718.
Moore BS, Mirshahi T. Genetic variants help define the role of the MC4R C-terminus in signaling and cell surface stability. Sci Rep. 2018;8:10397.
Lee YS, Poh LK, Kek BL, Loke KY. The role of melanocortin 3 receptor gene in childhood obesity. Diabetes. 2007;56:2622–30.
Drabkin M, Birk OS, Birk R. Heterozygous versus homozygous phenotype caused by the same MC4R mutation: novel mutation affecting a large consanguineous kindred. BMC Med Genet. 2018;19:135.
Matharu N, Rattanasopha S, Tamura S, Maliskova L, Wang Y, Bernard A, et al. CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency. Science. 2018. pii: eaau0629.
Kleinendorst L, Massink MPG, Cooiman MI, Savas M, van der Baan-Slootweg OH, Roelants RJ, et al. Genetic obesity: next-generation sequencing results of 1230 patients with obesity. J Med Genet. 2018;55:578–86.
Acknowledgements
JA was funded by the Spanish Ministry of Health (FIS-PI13/02195 and PI16/00485, co-funded by FEDER), the Fundación de Endocrinología y Nutrición and the “Centro de Investigación Biomédica en Red” for obesity and nutrition (CIBEROBN) of the Instituto de Salud Carlos III, Spain. LAP-J was funded by the Spanish Ministry of Health (FIS-PI1302481, co-funded by FEDER), the Generalitat de Catalunya (2014SRG1468), the Institució Catalana de Recerca i Estudis Avançats (ICREA Academia programme), the Spanish Ministry of Economy and Competiveness “Programa de Excelencia María de Maeztu” (MDM-2014-0370) and the Centro de Investigación Biomédica en Red for rare diseases (CIBERER) of the Instituto de Salud Carlos III, Spain. JAC was funded by grants from the Spanish Ministry of Science and Innovation (BFU2017-82565-C21-R2). We would like to thank Francisca Díaz and Sandra Canelles for their excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
LAPJ is a partner and scientific advisor of qGenomics Laboratory. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Serra-Juhé, C., Martos-Moreno, G.Á., Bou de Pieri, F. et al. Heterozygous rare genetic variants in non-syndromic early-onset obesity. Int J Obes 44, 830–841 (2020). https://doi.org/10.1038/s41366-019-0357-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-019-0357-5
- Springer Nature Limited
This article is cited by
-
Genetics and epigenetics in the obesity phenotyping scenario
Reviews in Endocrine and Metabolic Disorders (2023)
-
Glucagon-like peptide-1 analog therapy in rare genetic diseases: monogenic obesity, monogenic diabetes, and spinal muscular atrophy
Acta Diabetologica (2023)
-
Testing for rare genetic causes of obesity: findings and experiences from a pediatric weight management program
International Journal of Obesity (2022)
-
Two novel variants in DYRK1B causative of AOMS3: expanding the clinical spectrum
Orphanet Journal of Rare Diseases (2021)
-
Genetic Determinants of Childhood Obesity
Molecular Diagnosis & Therapy (2020)