
Abstract
The Nilaparvata muiri (Hemiptera: Delphacidae) is a sibling species of a destructive rice insect pest, the brown planthopper (BPH), Nilaparvata lugens. Here, we generated a high-quality chromosome-level genome assembly of N. muiri using a combination of the PacBio HiFi sequencing, Illumina short-read sequencing and Hi-C scaffolding technologies. The genome assembly (524.9 Mb) is anchored to 15 pseudochromosomes, with a scaffold N50 of 43.3 Mb and 99.1% BUSCO completeness. It contains 188.1 Mb repeat sequences and 13204 protein-coding genes. As a closely related species within the same genus as the significant pest, N. lugens, the chromosome-level genome assembly of N. muiri will provide important support for the better analysis of pathogenicity mechanisms of N. lugens based on comparative genomics.
Similar content being viewed by others
Background & Summary
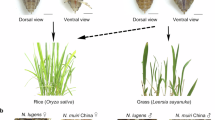
The Nilaparvata muiri (Hemiptera: Delphacidae) is a sibling species of the brown planthopper, a destructive rice insect pest, Nilaparvata lugens (Fig. 1), and is primarily found in China, Japan, South Korea, and Vietnam. N. muiri and N. lugens share a high degree of morphological similarity, have a broad overlapping distribution, and are active during similar periods (April to October)1. In the early 1980s, N. muiri was incorrectly identified as N. lugens, leading to an academic debate: Does the initial generation of N. lugens in China stem from local populations or from southeast Asia migration? Supporters for local populations cited evidence that N. lugens has been observed to overwinter in Leersia hexandra in China2. Subsequent research clarified that it was N. muiri, not N. lugens, that overwinters3. N. lugens cannot survive the cold conditions and does not overwinter in the colder parts of North Asia, such as Japan, Korea, and northern China4.

N. muiri and N. lugens. N. muiri and N. lugens are very similar morphologically. (a) N. muiri long-winged (macropterous) male adult. (b) N. lugens long-winged (macropterous) male adult63.
Despite high morphological similarity, they exhibit significantly different feeding behavior. N. muiri can only feed on gramineous weeds such as Leersia sayanuka and L. hexandra, not on rice, quite the opposite of N. lugens, which can only feed on rice5. In addition to conducting field surveys, black light trapping serves as another essential technique for monitoring N. lugens populations6. A recent report in Luhe, Nanjing shows that among the Nilaparvata specimens captured using black light traps, N. muiri accounting for up to 84.0%, 71.6%, 84.5% of the total in the years 2019, 2020, and 2021, respectively7. Traditional species-level identification of rice planthoppers, based on morphological characteristics, is expertise-dependent, labor-intensive, and time-consuming, occasionally leading to misidentifications. Large populations of N. muiri can profoundly affect the accurate monitoring and forecasting of the target insect pest, N. lugens8.
In this study, we assembled the first high quality chromosome-level genome of N. muiri using a combination of PacBio HiFi sequencing, Illumina short-read sequencing, and Hi-C sequencing technologies. The genome assembly (525 Mb) is anchored to 15 pseudochromosomes (Fig. 2), with a scaffold N50 of 43.32 Mb (Fig. 3). This genome assembly provides a valuable resource for N. muiri and N. lugens studies on feeding behavior, overwintering strategies, and mechanisms of growth and development. Unveiling the genomic differences between the two species could help the development of more effective biological control methods and more eco-friendly pesticides.

Hi-C contact map of N. muiri. Genome-wide all-by-all Hi-C interaction identified 15 pseudo-chromosome linkage groups of N. muiri genome.

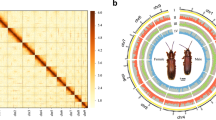
Circos plot of distribution of the genomic elements in N. muiri. The tracks indicate (a) length of the chromosome, (b) distribution of transposable element (TE) density, (c) gene density ranges, and (d) GC density. The densities of TEs, genes, and GC were calculated in 100 kb windows.
Methods
Sample collection and sequencing
N. muiri adults were provided by the College of Life Sciences, Zhejiang Normal University, and reared on their host L. hexandra in climate chambers at 25 °C (±2 °C) under a photoperiod of 16:8 hr (light: dark) with 70% RH (relative humidity). One male and one female were selected for sibling inbreeding for 15 generations. Insects were collected with aspirator and stored in liquid nitrogen. To reduce possible contamination, all adult insects used for genome sequencing had their midguts removed before stored in liquid nitrogen.
For PacBio sequencing, DNA was extracted from about 50 mixed-sex adults individuals. A single-end 20 kb libraries were constructed with SMRTbell Express Template Prep Kit 2.0. 65.77 Gb (126 × coverage) of PacBio HiFi reads were generated from two cells sequenced on the PacBio Sequel IIe platform (Table 1), with a mean read length of 19.59 kb (N50 = 19.61 kb). For Hi-C sequencing, 50 mixed-sex adults individual was used as inputs following previously described standard protocols9 and sequenced on an Illumina NovoSeq6000 platform. 126.46 Gb of paired-end 150 bp Hi-C reads (241 × coverage) were generated (Table 1). For Illumina short-read sequencing, genomic DNA used was extracted from 30 female adults and 30 male adults, respectively. Paired-end libraries with an insert size of 350 bp was constructed by NEBNext Ultra DNA Library Prep Kit (NEB, USA) and sequenced on an Illumina NovoSeq6000 platform. A total 38.50 Gb (73 × coverage) of paired-end 150 bp reads were generated to estimate genome size and identify sex chromosome (Table 1). All the DNA extraction, library construction and sequencing procedures were performed by the Novogene Company (Tianjin, China) according to the manufacturer’s protocols.
Total RNA was isolated from N. muiri at different developmental stages (including eggs, 400 1st-2nd instar nymphs, 150 3rd-5th instar nymphs, 50 mixed-sex adults), using the TRIzol reagent (Thermo Fisher Scientific, USA). Paired-end libraries was constructed by NEBNext Ultra RNA Library Prep Kit (NEB, USA). and sequenced on an Illumina NovoSeq6000 platform by the Novogene Company (Tianjin, China). A total of 31.95 Gb RNA-seq reads were generated (Table 1).
Genome assembly
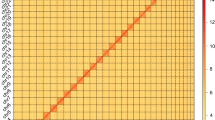
Genome size estimation, heterozygosity, and repetitiveness were analyzed using KMC v3.2.210 and GenomeScope 2.011 with 17-mer frequencies, which estimated a genome size of 528 Mb (Fig. 4). The PacBio HiFi reads were used to produce a draft assembly by Hifiasm 0.19.7-r59812,13 with the”–primary” parameter. Purge_Dups v1.2.314 was used to remove redundant heterozygous contigs from Hifiasm primary assembly, with custom cutoff values (-l 17 -m 80 -h 200). Hi-C reads were mapped to the draft assembly by Chromap v0.2.5-r47315 with the Hi-C preset. YaHS v1.2a116 was used to anchor primary contigs to chromosomes with “-e GATC” parameter. Juicebox v2.1.317 was used to manually correct the errors and remove remaining redundancy. Microbial contamination was detected and removed by NCBI FCS-GX v0.4.018 (r2023-01-24 database) with source organism set to N. muiri (--tax-id 706586). All these contaminants are unanchored contigs that had no interaction with N. muiri chromosomes in Hi-C contact map or had no Hi-C signal, and they were also validated by Blobtoolkit v4.2.119 from blast hits, GC% content and sequencing coverage. BUSCO v5.7.120 was used to assess genome assembly with the insecta_odb10 database. The Hi-C heatmap was generated with HiCExplorer v3.7.221. Genome circos plot was generated by TBtools-II Advanced Circos22. In addition, the mitochondrial genome of N. muiri was assembled by MitoHiFi23 pipeline. The summary of N. muiri genome assembly is showed in Table 2.

The estimated characteristics of N. muiri genome based on Illumina short-read data using 17-mers count histogram. Genome size was estimated to be 528.51 Mb, with a heterozygosity rate of 2.56%.
Genome annotation
Repeat masking was conducted using the EarlGrey pipeline v4.0124, which leverages widely-used tools such as RepeatModeler225 and RepeatMasker26, and integrates automated curation and filtering. The Arthropoda library from the Dfam database v3.727 and RepBase 2018102628 are used in pipeline with “-r Arthropoda”. Putative spurious TE annotations < 100 bp were remove by ‘-m yes’ parameter. The result showed that repeat sequences make up 35.83% of the genome (Table 3). Gene prediction was conducted with the BRAKER v3.0.629 pipeline. RNA-seq reads was aligned to the genome assembly using HISAT v2.2.130 and the open reading frame (ORF) was predicted using StringTie v2.2.131 within BRAKER pipeline as transcriptome-based evidence. Protein sequences of N. lugens were downloaded from NCBI RefSeq database32, and protein sequences of Sogatella furcifera, and Laodelphax striatellus were downloaded from InsectBase V233. These proteins were combined with OrthoDB v1134 Arthropoda clades as homology-based evidence. Genes predicted by AUGUSTUS v3.5.035 and GeneMark-ETP36 were combined by TSEBRA37. The predicted gene sets were functionally annotated using DIAMOND38 BLASTP search against NR database, eggNOG-mapper39,40 web service, InterProScan v5.6541,42 and PANNZER243 web service. Infernal v1.1.544 was used to identify 808 tRNAs, 1 sRNA, 443 rRNAs, 143 snRNAs, and 67 miRNAs based on the alignment with the Rfam45 library (Table 4).
Identification of X chromosome
Previous studies have reported different karyotypes in closely related planthoppers46,47,48,49: The karyotypes of N. lugens are 14 + XY for male and 14 + XX for female. The karyotypes of S. furcifera, L. striatellus, and Nilaparvata bakeri are 14 + X0 for male and 14 + XX for female. Thus the karyotypes of N.muiri was inferred to be 14 + X0 or 14 + XY for male, and 14 + XX for female. We used multiple methods to identify X chromosome and determine whether it is 14 + X0 or 14 + XY for male.
Synteny analysis between N. muiri and N. lugens was carried out by using the TBtools-II v2.07750 with the default parameters. Then chromosomes are renamed and reordered based on synteny analysis. Chromosome 1–14 and X of N. muiri showed conserved synteny with the Chromosome 1–14 and X of N. lugens. No scaffold or contig exhibited synteny with N. lugens Y chromosome. The result is displayed by circos51, which supports our assembly (Fig. 5).

Synteny plot between N. muiri and N. lugens. Chromosome 1–14 and X of N. muiri shows conserved synteny with the Chromosome 1–14 and X of N. lugens.
According to the pipeline of identification methods in sex chromosomes52, Log2(female:male sequencing depth ratio) of X, Y, and autosomes are expected to be around 1, −1, 0. For calculating the sequencing depth ratio between female and male insects, BWA-MEM2 v2.2.153 was used to align reads to the chromosome-level assembly with default parameters. SAM files were converted, sorted and built an index to obtain BAM and BAI files by SAMtools v1.1854. Then, BAM and BAI files were used to analyse chromosomes by sliding windows of 500 bp using BEDTools v2.31.055. In each 500 bp window, we normalized reads numbers, then calculated Log2(female:male ratio) value. Chromosome X has a mean value around 1, and other autosomes have mean value around 0 (Fig. 6). Y chromosome is not detected. Besides, we also tried Redkmer56 pipeline, but no Y chromosome could be assembled. From these results X chromosome was identified and the karyotypes of N.muiri was determined to be 14 + X0 for male and 14 + XX for female.

Identification of X chromosome of N. muiri. (a) average Log2(female:male ratio) per chromosome (b) X chromosome has a Log2(female:male ratio) around 1.
Data Records
PacBio, Illumina, Hi-C and transcriptome sequencing raw data for N. muiri have been deposited in the NCBI Sequence Read Archive with accession number SRP49850357. The genome assembly has been deposited at GenBank under the accession number GCA_039955075.158. It has also been deposited in the Genome Warehouse (GWH) in National Genomics Data Center under accession number GWHESPY0000000059. The gene annotation and TE consensus library are available in Figshare60.
Technical Validation
All Illumina raw reads were filtered by fastp61 by default parameters before further analysis. For assessment of genome assembly, Illumina short reads were mapped to the assembly by BWA-MEM2 v2.2.153 and PacBio HiFi reads were mapped by Minimap2 v2.26-r117562, with mapping rates of 95.3% and 99.8%, respectively. The Hi-C heatmap revealed a well-organized interaction pattern at the chromosomal level, which indirectly confirmed the accuracy of the chromosome assembly. 99.1% and 98.5% BUSCO genes (insecta_odb10) were successfully identified in the genome assembly and annotation, respectively, suggesting a remarkably complete assembly of the N. muiri genome.
Code availability
All software and pipelines used in this study were executed according to the manual and protocols of the published bioinformatic tools. The versions of software and parameters have been described in Methods, and command-line codes used in this study are available in Figshare60.
References
Liu, S. et al. Identification of Nilaparvata lugens and its two sibling species (N. bakeri and N. muiri) by direct multiplex PCR. Journal of economic entomology 111, 2869–2875 (2018).
Lei, H., Liu, G. & Wu, M. Nymphs of Nilaparvata lugens (Stål) Feeding on Leersia hexandra (Swartz) Can Metamorphose into Adults. Chinese Journal of Applied Entomology, 48–35 (1982).
Xu, X., Zhao, J. & Chen, R. Results and Discussion on Rearing Brown Planthopper on Leersia hexandra. Chinese Journal of Applied Entomology, 48–49+17 (1982).
Fu, Q. et al. Presence of a short repeat sequence in internal transcribed spacer (ITS) 1 of the rRNA gene of Sogatella furcifera (Hemiptera: Delphacidae) from geographically different populations in Asia. Applied Entomology and Zoology 47, 95–101 (2012).
Cui, Y., He, J., Luo, J., Lai, F. & Fu, Q. Host plants of Nilaparvata muiri China and N. bakeri (Muir), two sibling species of N. lugens (Stål). Chinese Journal of Rice Science 27, 105–110 (2013).
Ju, L. et al. Population dynamics of Nilaparvata lugens and its two sibling species under black light trap. Chinese Journal of Rice Science 24, 315–319 (2010).
Zhang, X., Zhu, X., Guo, J. & Yin, X. Population Dynamics of Brown Planthopper and Its Sibling Species Under Light Traps from 2019 to 2021 in Luhe, Nanjing. China Plant Protection 42, 29–32 (2022).
Cheng, X. et al. Studies on the migrations of brown planthopper Nilaparvata lugens Stal. Acta Entomologica Sinica, 1–21 (1979).
Belton, J.-M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Kokot, M., Długosz, M. & Deorowicz, S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33, 2759–2761 (2017).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature communications 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170-+ (2021).
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nature Biotechnology 40, 1332-+ (2022).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nature Communications 12 (2021).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39 (2023).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Systems 3, 99–101 (2016).
Astashyn, A. et al. Rapid and sensitive detection of genome contamination at scale with FCS-GX. Genome Biology 25 (2024).
Challis, R., Richards, E., Rajan, J., Cochrane, G. & Blaxter, M. BlobToolKit – Interactive Quality Assessment of Genome Assemblies. G3 Genes|Genomes|Genetics 10, 1361–1374 (2020).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution 38, 4647–4654 (2021).
Ramirez, F. et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nature Communications 9 (2018).
Chen, C., Wu, Y. & Xia, R. A painless way to customize Circos plot: From data preparation to visualization using TBtools. iMeta 1, e35 (2022).
Uliano-Silva, M. et al. MitoHiFi: a python pipeline for mitochondrial genome assembly from PacBio high fidelity reads. Bmc Bioinformatics 24 (2023).
Baril, T., Galbraith, J. & Hayward, A. Earl Grey: a fully automated user-friendly transposable element annotation and analysis pipeline. Molecular Biology and Evolution 41 (2024).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Current Protocols in Bioinformatics 25, 4.10.11–14.10.14 (2009).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mobile DNA 12 (2021).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6 (2015).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Research (2024).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology 37, 907–915 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome biology 20, 1–13 (2019).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic acids research 44, D733–D745 (2016).
Mei, Y. et al. InsectBase 2.0: a comprehensive gene resource for insects. Nucleic Acids Research 50, D1040–D1045 (2022).
Kuznetsov, D. et al. OrthoDB v11: annotation of orthologs in the widest sampling of organismal diversity. Nucleic Acids Research 51, D445–D451 (2023).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-ETP significantly improves the accuracy of automatic annotation oflarge eukaryotic genomes. Genome research 34, 757–768 (2024).
Gabriel, L., Hoff, K. J., Brůna, T., Borodovsky, M. & Stanke, M. TSEBRA: transcript selector for BRAKER. Bmc Bioinformatics 22, 1–12 (2021).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature methods 12, 59–60 (2015).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Molecular Biology and Evolution 38, 5825–5829 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research 47, D309–D314 (2019).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Research 49, D344–D354 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Toronen, P. & Holm, L. PANNZER-A practical tool for protein function prediction. Protein Science 31, 118–128 (2022).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Research 49, D192–D200 (2020).
Noda, H. & Tatewaki, R. Re-examination of chromosomes of three species of rice planthoppers (Homoptera: Delphacidae). Applied Entomology and Zoology 25, 538–540 (1990).
Wang, L. et al. Genome sequence of a rice pest, the white-backed planthopper (Sogatella furcifera). Gigascience 6, giw004 (2017).
Zhu, J. et al. Genome sequence of the small brown planthopper, Laodelphax striatellus. Gigascience 6, gix109 (2017).
Barrion, A. A. & Saxena, R. C. Cytology of Nilaparvata bakeri (Muir), a Grass-Infesting Planthopper (Homoptera: Delphacidae). CYTOLOGIA 50, 577–582 (1985).
Chen, C. et al. TBtools-II: A “one for all, all for one“bioinformatics platform for biological big-data mining. Molecular Plant 16, 1733–1742 (2023).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome research 19, 1639–1645 (2009).
Ma, W. et al. Chromosomal‐level genomes of three rice planthoppers provide new insights into sex chromosome evolution. Molecular ecology resources 21, 226–237 (2021).
Vasimuddin, M., Misra, S., Li, H., Aluru, S. & Ieee. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. IEEE International Parallel and Distributed Processing Symposium (IPDPS). 314–324 (2019).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10 (2021).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Papathanos, P. A. & Windbichler, N. Redkmer: An Assembly-Free Pipeline for the Identification of Abundant and Specific X-Chromosome Target Sequences for X-Shredding by CRISPR Endonucleases. The CRISPR Journal 1, 88–98 (2018).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP498503 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_039955075.1 (2024).
NGDC Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/84800/show (2024).
Cilin, W. Gene annotation, repeat annotation and scripts of Nilaparvata muiri assembly. Figshare. https://doi.org/10.6084/m9.figshare.25592445.v2 (2024).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2, e107 (2023).
Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021).
Ding, J., Hu, C., Fu, Q., He, J. & Xie, M. A colour atlas of commonly encountered delphacids in China rice regions. Zhejiang Sci. Technol. Press 2, 22–26 (2012).
Acknowledgements
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (LGN21C140002).
Author information
Authors and Affiliations
Contributions
Shuhua Liu and Jian Tang conceived and supervised the project; Aiying Wang provided help for insect rearing; Ju Luo and Cilin Wang prepared the samples for PacBio, Illumina, Hi-C and transcriptome sequencing; Cilin Wang performed the bioinformatic analyses; Cilin Wang wrote the manuscript; Shuhua Liu and Guiying Yang revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, C., Luo, J., Wang, A. et al. Chromosome-level genome assembly of the planthopper Nilaparvata muiri. Sci Data 11, 1029 (2024). https://doi.org/10.1038/s41597-024-03870-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03870-4
- Springer Nature Limited