
Abstract
We sequenced and annotated the first complete mitochondrial genome (mitogenome) of Ledra auditura (Hemiptera: Cicadellidae: Ledrinae) and reconstructed phylogenetic relationships among 47 species (including 2 outgroup species) on the basis of 3 datasets using maximum likelihood (ML) and Bayesian inference (BI) analyses. The complete L. auditura mitogenome (length, 16,094 bp) comprises 37 genes [13 protein-coding genes (PCGs), 22 tRNAs, and 2 rRNAs], 1 control region, and 2 long non-coding regions. The first long non-coding region (length, 211 bp) is located between tRNA-I and tRNA-Q and the second region (length, 994 bp) between tRNA-S2 and ND1. All PCGs show ATN (Met/Ile) as their start codon and TAR as their stop codon. Except tRNA-S1 (AGN), which lacks the dihydrouridine arm, all tRNAs can fold into the typical cloverleaf secondary structure. The complete L. auditura mitogenome shows a base composition bias of 76.3% A + T (A = 29.9%, T = 46.4%, G = 13.3%, and C = 10.5%), negative AT skew of −0.22, and positive GC skew of 0.12. In ML and BI analyses, L. auditura was clustered with Evacanthus heimianus (Hemiptera: Cicadellidae: Evacanthinae) with strong branch support.
Similar content being viewed by others


Introduction
Ledrinae is a relatively small subfamily within the large and diverse leafhopper family Cicadellidae, which comprises approximately 300 described species of 38 genera divided into 5 tribes. These species are extensively distributed across Australia, Africa, and Southeast Asia1 and primarily inhabit trees and shrubs, except for the grass-feeding Xerophloeini2. Moreover, no Ledrinae species appear to be major vectors of plant diseases3. A few members of Ledrinae show conspicuous ear-like projections on the pronotum and are referred to as eared leafhoppers4. Interest in Ledrinae is often centered on this unique morphology along with its possible implications in the evolutionary history of leafhoppers5,6.
Continuous improvements and advancements in molecular biology techniques have facilitated wide use of high-throughput sequencing for mitochondrial genome (mitogenome) data collection. Complete mitogenomes have been effectively used to understand the evolutionary relationships among insects7,8,9. Since 2016, there has been a drastic increase in the availability of Cicadellidae mitogenome data; however, despite their great diversity, only 40 complete or near-complete Cicadellidae mitogenomes have been reported to date10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27. Therefore, new mitogenomic data will provide support for determining the phylogenetic relationships and evolution of Cicadellidae in the future. Here, using first- and second-generation sequencing, we sequenced the complete mitogenome of Ledra auditura to confirm its phylogenetic relationships and taxonomic status as well as to better understand its mitogenome structure. L. auditura is the first Ledrinae species whose phylogenetic relationships with 40 other leafhoppers and 5 treehoppers have been assessed using maximum likelihood (ML) and Bayesian inference (BI) analyses based on mitogenomes, thereby providing a basis for further molecular research on the related taxa.
Results and Discussion
Genome organization and base composition
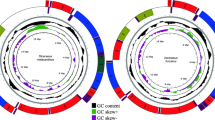
The complete L. auditura mitogenome (GenBank No., MK387845) is 16,094-bp long, which is comparable to the sizes of previously documented mitogenomes of Cicadellidae species, ranging from 15,131 of Trocnadella arisana to 16,811 bp of Parocerus laurifoliae25. The gene order and arrangement of the L. auditura mitogenome are identical to those of other commonly sequenced Hemiptera species8,9,10,11,12,13,14,15,16,17,18,19. A total of 22 genes (9 PCGs and 13 tRNAs) are encoded on the majority strand (J-strand) and 15 (4 PCGs, 9 tRNAs, and 2 rRNAs) on the minority strand (N-strand) (Fig. 1, Table 1). However, there are 2 long non-coding regions in L. auditura in addition to the control region; the first region (length, 211 bp) is located between tRNA-I and tRNA-Q and the second (length, 994 bp) between tRNA-S2 and ND1. The nucleotide composition of the complete mitogenome is as follows: A = 29.9%, T = 46.4%, G = 13.3%, and C = 10.5%. The complete L. auditura mitogenome shows a base composition bias of 76.3% A + T, a negative AT skew of −0.22, and a positive GC skew of 0.12 (Table 2).

Circular map of the Ledra auditura mitogenome.
PCGs and codon usage
The total length of the 13 PCGs is 11,064 bp, and these encode 3,688 amino acids, accounting for 68.7% of the complete L. auditura mitogenome. All PCGs are initially encoded by ATN (Met/Ile). The start codon of 4 genes (ND2, COX2, COX3, and ND6) is ATT, that of 4 other genes (ATP8, ND3, ND4, and ND1) is ATA, that of 1 gene (ND5) is ATC, and that of the remaining 4 genes (COX1, ATP6, ND4L, and Cytb) is ATG. The stop codon of 9 PCGs is the typical TAA and that of 4 (COX1, ATP6, ND3, and Cytb) PCGs is TAG (Table 3).
The base composition of the 13 PCGs is 74.6% A + T (A = 29.3%, T = 45.3%, G = 13.5%, and C = 11.8%), with a negative AT skew (−0.21) and weakly positive GC skew (0.07). The relative synonymous codon usage and codon usage of the 13 PCGs of the L. auditura mitogenome are presented in Fig. 2 (except the stop codons TAA and TAG). Within each codon, the third codon position terminating with A/T is more frequent than that with G/C, thereby resulting in the highest A + T content at the third codon position. The 4 most frequently used codons are Phe (TTT), Leu (TTA), Ile (ATT), and Met (ATA). In addition, codon usage exhibits a high A + T bias that plays a key role in the A + T bias of the entire mitogenome. The codon usage pattern of L. auditura is highly consistent with that of previously reported Cicadellidae species8,9,10,11,12,13,14,15,16,17.

Relative synonymous codon usage and number of codon usage in the Ledra auditura mitogenome Codon families are indicated on the X-axis.
tRNAs and rRNAs
The L. auditura mitogenome comprises the 22 typical tRNAs, with lengths ranging from 61 (Ala, Arg, and Ser1) to 71 (Lys) bp (Table 1). The total length of the 22 tRNAs is 1,408 bp, with 77.3% A + T content. All tRNAs can fold into the typical cloverleaf secondary structure except tRNA-S1, which lacks the dihydrouridine arm, as documented for other Hemiptera species9,28,29. The secondary structure of the 22 tRNAs is presented in Fig. 3.

Predicted secondary structure of the 22 tRNAs in the Ledra auditura mitogenome; dashes (–) indicate Watson–Crick base pairing.
The 16S and 12S rRNA genes in the Cicadellidae mitogenome are highly conserved in terms of their length and secondary structures22,23,24,25. In the L. auditura mitogenome, the 16S rRNA is located between tRNA-L2 and tRNA-V and is 1,160-bp long. The 12S rRNA gene, as identified based on the alignments with Evacanthus heimianus and Idioscopus clypealis13, is located between tRNA-V and the control region and is 721-bp long. In the present study, the hypothetical secondary structures of 2 rRNA genes were drawn using RNA Structure version 5.230, predicted against the known rRNA secondary structures25,31,32. The secondary structure of 16S rRNA in the L. auditura mitogenome comprises 5 domains (domains I, II, IV, V, and VI; domain III is absent, as in other insects) and 43 helices (Fig. 4) and that of 12S rRNA comprises 3 domains (domains I, II and III) and 24 helices (Fig. 5).

Predicted secondary structure of 16S rRNA in the Ledra auditura mitogenome; dashes (–) indicate Watson–Crick base pairing.

Predicted secondary structure of 12S rRNA in the Ledra auditura mitogenome; dashes (–) indicate Watson–Crick base pairing.
Non-coding regions
Although large intergenic regions have been identified in some species, the mitogenomes of most insects are compact33. Usually the long non-coding region is located between 12S rRNA and tRNA-I, which is the control region. In the present study, 3 long non-coding regions (>50 bp) were detected in the L. auditura mitogenome. The first non-coding region (length, 211 bp) is located between tRNA-I and tRNA-Q. The second non-coding region (length, 993 bp) is a repeat region located between tRNA-S2 and ND1. It comprises 2 tandem repeats (Figs 1 and 6): the first repeat sequence is 105-bp long and is repeated 5 times, and the second is 117-bp long and repeated 4 times (Fig. 6). Finally, the third non-coding region, commonly referred to as the control region, is located between 12S rRNA and tRNA-I; it is 721-bp long, which is comparable to that reported in other sequenced leafhoppers, ranging from399 bp of N. cincticeps to 2477 bp of Parocerus laurifoliae. The region shows 91.1% A + T content, and it is the most variable region in the whole mitogenome, with a relatively low pairwise identity. The control region is usually much longer in species with repetitive sequences than in those without repeats. However, there was no association among each repeat unit, the regularity of the occurrence of repetitive sequences, and the significance in the control area, suggesting the need for further research using different methods to resolve this pattern in the future.

Structure of repeat regions in the Ledra auditura mitogenome.
Phylogenetic relationships
ML and BI analyses were used to reconstruct the phylogenetic relationships among 47 species (including 2 outgroup species) based on the following 3 datasets: (1) amino acid sequences of 13 PCGs (3,366 amino acids); (2) nucleotide sequences of 13 PCGs and 2 rRNAs (11,918 bp); (3) the first and second codons of 13 PCGs and complete sequences of 2 rRNAs (8,552 bp). A total of 6 phylogenetic trees (BI-AA, BI-PCGRNA, BI-PCG12RNA, ML-AA, ML-PCGRNA, and ML-PCG12RNA) reconstructed using ML and BI analyses on the 3 datasets are shown in Figs 7, 8 and S1–S5. Previous molecular phylogenetic analyses have suggested that Delocephalinae leafhoppers constitute 1 clade, which has been recovered as the sister group to the other members of Cicadellidae22,23,24,25,26,27. In the present study, the relationships among the 3 clades was consistent with high support in all the trees [clade 1: Membracidae + Megophthalminae; clade 2: Coelidiinae + Iassinae; clade 3: Cicadellinae + (Typhlocybinae + {Evacanthinae + Ledrinae})]; this result is consistent with previously reported phylogenies using partial gene sequences and morphological features34,35,36,37, suggesting that Cicadellidae is paraphyletic with treehoppers, but Cicadellinae subfamilies, including Deltocephalinae, Megophthalminae, Idiocerinae, Typhlocybinae, Cicadellinae, and Coelidiinae are monophyletic, with strong branch support. Within Cicadellidae, the inferred relationship (Iassinae + Coelidiinae) + [Deltophalinae + (Megophthalminae + Idiocerinae)] + [Cicadellinae + (Typhlocybinae + {Evacanthinae + Ledrinae})] was supported with high moderated branch support in 4 phylogenetic trees (BI-PCGRNA, BI-PCG12RNA, ML-PCGRNA and ML-PCG12RNA) (Figs 7, 8, and S3–S5), but Idiocerinae was recovered as the sister clade to Cicadellinae + (Typhlocybinae + (Evacanthinae + Ledrinae)) in BI-AA and (Membracidae + Megophthalminae) + (Coelidiinae + Iassinae) in ML-AA, with low branch support (Figs S1 and S2). Further sampling from different taxonomic units and additional mitogenomic data will provide a better understanding of the phylogenetic and evolutionary relationships among leafhoppers.

Phylogenetic trees of Cicadellidae inferred using maximum likelihood (ML) and MrBayes (BI) analyses based on protein-coding genes and rRNA genes.

Phylogenetic trees of Ledra auditura constructed based on the first and second codons of the 13 PCGs and 2 rRNAs using the GTR + I + G model determined by MrBayes.
Conclusions
In the present study, we successfully sequenced the first complete L. auditura mitogenome in Ledrinae. To the best of our knowledge, this is the first available mitogenome for a species within the subfamily Ledrinae. The mitogenome is 16,094-bp long, ranging between 15,131 bp of Ttocnadella arisana to 16,811 bp of Parocerus laurifoliae. Such variations in mitogenome length can be mainly attributed to difference in control region length25. Consistent with previous observations in Cicadellidae, the sequences of L. auditura mitogenome were highly conserved in terms of gene content, gene size, gene order, base composition, codon usage of PCGs, and RNA secondary structures. Furthermore, there exists a 993-bp-long repeat region between Cytb and ND1, which contains 2 tandem repeats (Figs 1, 6); the first repeat sequence is 105-bp long and repeated 5 times and the second is 117-bp long and repeated 4 times (Fig. 6). Interestingly, the repeat sequences are located within the control region, similar to that reported in previous studies9,25,28. Moreover, we analyzed the mitogenomic features, base composition, codon usage, and phylogenetic relationships of L. auditura. In ML and BI analyses, 40 obvious clusters of leafhoppers were identified, consistent with previous phylogenetic findings based on mitogenome data. While Ledrinae was recovered as a paraphyletic group, it emerged as a sister clade to Tartessinae and Iassinae or Aphrodinae, although with low branch support, and its relationship with other clades remained poorly resolved, as revealed by the ML bootstrap analysis of the concatenated anchored hybrid enrichment nucleotide sequence data set in the study of predecessors35. There were also large variations in results obtained using different datasets; according to transcriptome analyses, Ledrinae was recovered as a monophyletic group with maximum bootstrap support using ML analyses, with relatively low support among Cicadellidae, and the placements of subfamilies relative to one another were not consistent38. Recently, partial mitogenome sequence data were sequenced in leafhoppers, particularly in small groups with few species. Thus, addition of taxa to our small group of mitogenome dataset may help improve the resolution of the still poorly understood relationships among leafhopper lineages. Therefore, the complete mitogenome reported in the present study may provide a basis for further genomic studies of Ledrinae and may be useful for future phylogenetic analyses of Cicadellidae.
Materials and Methods
Sample collection and DNA extraction
L. auditura specimens were collected from Xianheping, Anlong County, Guizhou Province, China (24°58′N, 105°30′E). Live specimens were preserved in 100% ethanol and stored at −20 °C until DNA extraction. Total genomic DNA was extracted from one adult specimen using the DNeasy© Tissue Kit (Qiagen, Germany). Voucher DNA and specimens (GZU-IHC-000252) are deposited at the Institute of Entomology, Guizhou University.
Mitogenome sequencing and assembly
L. auditura mitogenome was sequenced using next-generation sequencing (Illumina HiSeq. 2500 and 2 GB raw data; Berry Genomics, Beijing, China), and 2 sequence fragments were reconfirmed via polymerase chain reaction (PCR) amplification using primers #2 and #3 (Table 3). We used 40 μL genomic DNA for next-generation sequencing and diluted the remaining genome with ddH2O to obtain a concentration of 100 μL for PCR amplification. Primers were designed based on the sequencing results obtained using Primer Premier 6.0 (Premier Biosoft, Palo Alto, CA, USA). PCR was performed using PCR MasterMix (Tiangen Biotech Co., Ltd., Beijing, China) according to the specification manual. The PCG cycling conditions included pre-denaturation at 94 °C for 3 min followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at a suitable temperature for 30 s, elongation at 70 °C for 1 min, and additional elongation at 70 °C for 10 min at the end of all cycles. The annealing temperatures were adjusted according to the melting temperatures of different primers. Table 3 lists primers used in this study. Clean next-generation sequencing results were assembled using Geneious R927 based on the COX1 fragment (sequenced using primer #1; Table 3) of mitochondrial DNA, and the sequencing results obtained via PCR and TA cloning were assembled using the SeqMan program package (DNAStar Inc.; Madison, WI, USA).
Sequence analysis and gene annotation
The assembled mitogenome was initially annotated using the MITOS web server with invertebrate genetic codes39 and then analyzed using Geneious R927 and NCBI BLAST (https://blast.ncbi.nlm.nih.gov). The locations and secondary structures of 22 tRNAs were identified and predicted using tRNAscan-SE version 1.2140 and ARWEN version 1.241. Two rRNA genes were indetified based on the locations of adjacent tRNA genes and then compared with the rRNA genes of other Cicadellidae species. Next, the secondary structures of these rRNAs were predicted based on previously reported models16,17,25. DNASIS version 2.5 (Hitachi Engineering, Tokyo, Japan) and RNA Structure version 5.230 were used to predict helical elements present in variable regions. Strand asymmetry was calculated using the following formulas: AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C)42. Furthermore, base composition and codon usage patterns of PCGs were analyzed using MEGA643. Repeated sequences in the L. auditura mitogenome were identified using the Tandem Repeats Finder tool (http://tandem.bu.edu/trf/trf.html)44. The complete L. auditura mitogenome is deposited in GenBank under the accession number MK387845.
Sequence alignment and phylogenetic analysis
Phylogenetic analysis was based on 45 Cicadellidae species with 2 Fulgoroidea species (Ricania speculum and Peregrinus maidis) selected as outgroups (Table S1). Sequences of 13 PCGs (without stop codons) and 2 rRNA genes were used to analyze the phylogenetic relationships. Each PCG and rRNA sequence was aligned using the MAFFT algorithm in Translator X (http://pc16141.mncn.csic.es/index_v4.html)45,46 and MAFFT v7.0 online server (https://mafft.cbrc.jp/alignment/server/) using the G-INS-i strategy47, respectively. Poorly aligned sequences were eliminated using Gblocks 9.1b (http://www.phylogeny.fr/one_task.cgi?task_type=gblocks)47. Finally, all sequences were assessed and manually corrected using MEGA643.
The alignments of individual genes were concatenated to generate 3 datasets including 13 PCGs and 2 rRNAs: (1) amino acid sequences of 13 PCGs (3,366 amino acids); (2) nucleotide sequences of 13 PCGs and 2 rRNAs (11,918 bp); (3) the first and second codons of 13 PCGs and 2 rRNAs (8,552 bp). ML phylogenetic trees were constructed using IQ-TREE v1.6.348, with the best model for each partition selected under the corrected Akaike Information Criteria using PartitionFinder2 (Table S2)49, and evaluated using the ultrafast bootstrap approximation approach for 10,000 replicates. Furthermore, BI analysis was conducted using MrBayes 3.2.650; following the partition schemes suggested by PartitionFinder, all model parameters were set as unlinked across partitions. Two simultaneous runs with 4 independent Markov chains were performed for 50 million generations, sampling every 100 generations. After the average standard deviation of split frequencies fell below 0.01, the first 25% samples were discarded as burn-in and the remaining trees were used to generate a consensus tree and calculate the posterior probabilities.
References
Jones, J. R. & Deitz, L. L. Phylogeny and systematics of the leafhopper subfamily Ledrinae (Hemiptera: Cicadellidae). Zootaxa. 2186, 1–120 (2009).
Dietrich, C. H. “Guide to the subfamilies of leafhoppers (Cicadellidae)”. Center for Biodiversity. Illinois Natural History Survey-INHS. Illinois. 44 (2006).
Nielson, M. W. The leafhopper vectors of phytopathogenic viruses (Homoptera, Cicadellidae): taxonomy, biology, and virus transmission. No. 1382. US Department of Agriculture (1968).
Nishino, T., Tanahashi, M., Lin, C. P., Koga, R. & Fukatsu, T. Fungal and bacterial endosymbionts of eared leafhoppers of the subfamily Ledrinae (Hemiptera: Cicadellidae). Appl. Entomol. Zool. 51(3), 465–477 (2016).
Ross, H. H. Evolutionary developments in the leafhoppers, the insect family Cicadellidae. Syst. Zool. 6(2), 87–69 (1957).
Evans, J. W. Character selection in systematics with special reference to the classification of leafhoppers (Insecta, Homoptera, Cicadelloidea). Syst. Zool. 7(3), 126–131 (1958).
Li, H. et al. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. P. Roy. Soc. B-Biol. Sci. 284(1862), 20171223 (2017).
Song, N., Cai, W. Z. & Li, H. Deep-level phylogeny of Cicadomorpha inferred from mitochondrial genomes sequenced by NGS. Sci. rep. 7(1), 10429 (2017).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014).
Mao, M., Yang, X. X. & Bennett, G. The complete mitochondrial genome of Entylia carinata (Hemiptera: Membracidae). Mitochondrial DNA Part B. 1(1), 662–663 (2016).
Zhou, N. N., Wang, M. X., Cui, L., Chen, X. X. & Han, B. Y. Complete mitochondrial genome of Empoasca vitis (Hemiptera: Cicadellidae). Mitochondrial DNA Part A. 27(2), 1052–1053 (2016).
Liu, J. H., Sun, C. Y., Long, J. & Guo, J. J. Complete mitogenome of tea green leafhopper, Empoasca onukii (Hemiptera: Cicadellidae) from Anshun, Guizhou Province in China. Mitochondrial DNA Part B. 2(2), 808–809 (2017).
Dai, R. H., Wang, J. J. & Yang, M. F. The complete mitochondrial genome of the leafhopper Idioscopus clypealis (Hemiptera: Cicadellidae: Idiocerinae). Mitochondrial DNA Part B. 3(1), 32–33 (2018).
Song, N., Cai, W. Z. & Li, H. Insufficient power of mitogenomic data in resolving the auchenorrhynchan monophyly. Zool. J. Linn. Soc-Lond. 183(4), 776–790 (2017).
Wu, Y. F., Dai, R. H., Zhan, H. P. & Qu, L. Complete mitochondrial genome of Drabescoides nuchalis (Hemiptera: Cicadellidae). Mitochondrial DNA Part A. 27(5), 3626–3627 (2016).
Wang, J. J., Li, H. & Dai, R. H. Complete mitochondrial genome of Taharana fasciana (Insecta, Hemiptera: Cicadellidae) and comparison with other Cicadellidae insects. Genetica. 145(6), 593–602 (2017).
Wang, J. J., Dai, R. H., Li, H. & Zhan, H. P. Characterization of the complete mitochondrial genome of Japanagallia spinosa and Durgades nigropicta (Hemiptera: Cicadellidae: Megophthalminae). Biochem. Syst. Ecol. 74, 33–41 (2017).
Mao, M., Yang, X. & Bennett, G. The complete mitochondrial genome of Macrosteles quadrilineatus (Hemiptera: Cicadellidae). Mitochondrial DNA Part B. 2(1), 173–175 (2017).
Yu, P. F., Wang, M. X., Cui, L., Chen, X. X. & Han, B. Y. The complete mitochondrial genome of Tambocerus sp. (Hemiptera: Cicadellidae). Mitochondrial DNA Part A. 28(1), 133–134 (2017).
Zhao, X. & Liang, A. P. Complete DNA sequence of the mitochondrial genome of the treehopper Leptobelus gazella (Membracoidea: Hemiptera). Mitochondrial DNA Part A. 27(5), 3318–3319 (2016).
Choudhary, J. S., Naaz, N., Das, B., Bhatt, B. P. & Prabhakar, C. S. Complete mitochondrial genome of Idioscopus nitidulus (Hemiptera: Cicadellidae). Mitochondrial DNA Part B. 3(1), 191–192 (2018).
Du, Y. M., Zhang, C. N., Dietrich, C. H., Zhang, Y. L. & Dai, W. Characterization of the complete mitochondrial genomes of Maiestas dorsalis and Japananus hyalinus (Hemiptera: Cicadellidae) and comparison with other Membracoidea. Sci. Rep. 7(1), 14197 (2017).
Du, Y. M., Dai, W. & Dietrich, C. H. Mitochondrial genomic variation and phylogenetic relationships of three groups in the genus Scaphoideus (Hemiptera: Cicadellidae: Deltocephalinae). Sci. Rep. 7(1), 16908 (2017).
Du, Y. M., Dietrich, C. H. & Dai, W. Complete mitochondrial genome of Macrosteles quadrimaculatus (Matsumura) (Hemiptera: Cicadellidae: Deltocephalinae) with a shared tRNA rearrangement and its phylogenetic implications. Int. J. Bio. Macromol. 122, 1027–1034 (2019).
Wang, J. J., Yang, M. F., Dai, R. H., Li, H. & Wang, X. Y. Characterization and phylogenetic implications of the complete mitochondrial genome of Idiocerinae (Hemiptera: Cicadellidae). Int. J. Bio. Macromol. 120, 2366–2372 (2018).
Wang, J. J., Yang, M. F., Dai, R. H. & Li, H. Complete mitochondrial genome of Evacanthus heimianus (Hemiptera: Cicadellidae: Evacanthinae) from China. Mitochondrial DNA Part B. 4(1), 284–285 (2019).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12), 1647–1649 (2012).
Wang, Y., Chen, J., Jiang, L. Y. & Qiao, G. X. Hemipteran mitochondrial genomes: features, structures and implications for phylogeny. Int. J. Mol. Sci. 16(6), 12382–12404 (2015).
Zhang, B., Ma, C., Edwards, O., Fuller, S. & Kang, L. The mitochondrial genome of the Russian wheat aphid Diuraphis noxia: large repetitive sequences between trnE and trnF in aphids. Gene. 533(1), 253–260 (2014).
Reuter, J. S. & Mathews, D. H. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11(1), 129 (2010).
Wang, Y., Cao, J. J., Li, N., Ma, G. Y. & Li, W. H. The first mitochondrial genome from Scopuridae (Insecta: Plecoptera) reveals structural features and phylogenetic implications. Int. J. Bio. Macromol. 122, 893–902 (2019).
Li, H. et al. The architecture and complete sequence of mitochondrial genome of an assassin bug Agriosphodrus dohrni (Hemiptera: Reduviidae). Int. J. Biol. Sci. 7(6), 792 (2011).
Boore, J. L. Animal mitochondrial genomes. Nucleic acids Res. 27(8), 1767–1780 (1999).
Cryan, J. R., Wiegmann, B. M., Deitz, L. L. & Dietrich, C. H. Phylogeny of the treehoppers (Insecta: Hemiptera: Membracidae): evidence from two nuclear genes. Mol. Phylogenet. Evol. 17(2), 317–334 (2000).
Dietrich, C. H. et al. Anchored hybrid enrichment-based phylogenomics of leafhoppers and treehoppers (Hemiptera: Cicadomorpha: Membracoidea). Insect Syst. Divers. 1(1), 57–72 (2017).
Dietrich, C. H., Rakitov, R. A., Holmes, J. L. & Black, W. C. IV Phylogeny of the major lineages of Membracoidea (Insecta: Hemiptera: Cicadomorpha) based on 28S rDNA sequences. Mol. Phylogenet. Evol. 18(2), 293–305 (2001).
Krishnankutty, S. Systematics and biogeography of leafhoppers in Madagascar. University of Illinois at Urbana-Champaign (2013).
Skinner, R. K. et al. Phylogenomics of Auchenorrhyncha (Insecta: Hemiptera) using transcriptomes: examining controversial relationships via degeneracy coding and interrogation of gene conflict. Syst. Entomol., https://doi.org/10.1111/syen.12381 (2019).
Bernt, M. et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69(2), 313–319 (2013).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33(Suppl_2), W686–W689 (2005).
Laslett, D. & Canbäck, B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 24(2), 172–175 (2007).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41(3), 353–358 (1995).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30(12), 2725–2729 (2013).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27(2), 573–580 (1999).
Abascal, F., Zardoya, R. & Telford, M. J. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38(Suppl_2), W7–W13 (2010).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. (2017).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17(4), 540–552 (2000).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32(1), 268–274 (2014).
Miller, M. A., Pfeiffer, W. & Schwartz, T. “Creating the CIPRES Science Gateway for inference of large phylogenetic trees” in Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, pp. 1–8, 14 Nov 2010.
Nylander, J. A., Ronquist, F., Huelsenbeck, J. P. & Nieves-Aldrey, J. Bayesian phylogenetic analysis of combined data. Syst. Biol. 53(1), 47–67 (2004).
Acknowledgements
This work was supported by the National Natural Science Foundation, China [Grant No. 31672342]; the Program of Excellent Innovation Talents, Guizhou Province, China [Grant No. 20164022]; and the Science and Technology Foundation of Guizhou Province, China [Grant No. J-(2015) 2041].
Author information
Authors and Affiliations
Contributions
D.R.H. and Y.F.M. conceived and designed the experiments; W.J.J., L.D.F. and L.H. analyzed the data; W.J.J. drafted the manuscript; W.J.J., L.D.F., L.H., Y.F.M. and D.R.H. finalized the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, JJ., Li, DF., Li, H. et al. Structural and phylogenetic implications of the complete mitochondrial genome of Ledra auditura. Sci Rep 9, 15746 (2019). https://doi.org/10.1038/s41598-019-52337-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52337-9
- Springer Nature Limited
This article is cited by
-
The complete mitochondrial genome of Morishitium polonicum (Trematoda, Cyclocoelidae) and its phylogenetic implications
Parasitology Research (2023)
-
Description of the whole mitochondrial genome of Bhatia longiradiata (Hemiptera, Cicadellidae, Deltocephalinae: Drabescini) and phylogenetic relationship
Genes & Genomics (2023)
-
A complete sequence of mitochondrial genome of Neolamarckia cadamba and its use for systematic analysis
Scientific Reports (2021)